
ABSTRACT
Acute coronary syndrome (ACS) is a term used to describe major cardiovascular diseases, and treatment of in-stent restenosis in patients with ACS remains a major clinical challenge. Further investigation into molecular markers of ACS may aid early diagnosis, and the treatment of ACS and post-treatment recurrence. In the present study, total RNA was extracted from the peripheral blood samples of 3 patients with ACS, 3 patients with percutaneous coronary intervention (PCI)_non-restenosis, 3 patients with PCI_restenosis and 3 healthy controls. Subsequently, RNA library construction and high-throughput sequencing were performed. DESeq2 package in R was used to screen genes that were differentially expressed between the different samples. Moreover, the intersection of the differentially expressed mRNAs (DEmRNAs) and differentially expressed long noncoding RNAs (DElncRNAs) obtained. GeneCodis4.0 was used to perform function enrichment for DEmRNAs, and lncRNA-mRNA co-expression network was constructed. The GSE60993 dataset was utilized for diagnostic analysis, and the aforementioned investigations were verified using in vitro studies. Results of the present study revealed a large number of DEmRNAs and DElncRNAs in the different groups. We selected genes in the top 10 of differential expression and also involved in the co-expression of lncRNA-mRNA for diagnostic analysis in the GSE60993 dataset. The area under curve (AUC) of PDZK1IP1 (0.747), PROK2 (0.769) and LAMP3 (0.725) were all >0.7. These results indicated that the identified mRNAs and lncRNAs may act as potential clinical biomarkers, and more specifically, PDZK1IP1, PROK2 and LAMP3 may act as potential biomarkers for the diagnosis of ACS.
Introduction
Acute coronary syndrome (ACS) describes a range of major cardiovascular diseases, which include acute myocardial infarction and unstable angina [Citation1,Citation2]. The morbidity and mortality rates associated with ACS remain high [Citation3]. Percutaneous coronary intervention (PCI) combined with stent implantation can improve the prognosis of patients with ACS [Citation4]. However, the treatment of in-stent restenosis in patients with ACS remains a major clinical challenge [Citation5]. Thus, further investigation into the expression and interaction of genes associated with ACS and post-treatment in-stent restenosis is of great significance for understanding the molecular mechanisms underlying ACS. Further investigations may provide a novel theoretical basis for the diagnosis and management of ACS.
Although the specific pathological mechanisms underlying ACS remain to be fully elucidated, multiple genes have been reported to be involved in its pathogenesis. mRNA mediates the translation of genetic information from genes into proteins, and plays an important role in disease progression and treatment [Citation6]. Previous studies have demonstrated that abnormal mRNA expression plays an important regulatory role in the progression of ACS. ACS can be caused by inflammatory factors, and macrophage migration inhibitory factor (MIF) is an important regulator of inflammation. Compared with control group, the level of MIF in the serum of patients with ACS was significantly increased [Citation7,Citation8]. Moreover, toll like receptor 2 (TLR2) is a member of the TLR superfamily, which coordinates platelet function when activated. Previous studies have demonstrated that the mRNA expression levels of TLR2 in the platelets of patients with ACS were up-regulated, which may act as a potential biomarker [Citation9]. In vitro, NADH: ubiquinone oxidoreductase subunit C2 (NDUFC2) silencing affected vascular cell viability and angiogenesis. A significant decrease in NDUFC2 expression was detected in ACS, which indicated that NDUFC2 may play an important role in the development of ACS [Citation10]. These studies highlight that differential expression of mRNA plays an important regulatory role in ACS.
Long noncoding RNAs (lncRNAs) are a class of non-coding RNAs ranging in length from ~200 nucleotides to 100 kilobases (kb) and are key components of gene regulatory networks [Citation11,Citation12]. Previous studies have shown that the dysregulation of lncRNA expression can lead to the development of cardiovascular diseases [Citation13]. lncRNA HLA complex group 11 (HCG11) regulates vascular endothelial cell proliferation and angiogenesis through the microRNA (miR/miRNA)-26b-5p/QKI-5 signaling pathway [Citation14]. Moreover, lncRNA antisense non-coding RNA INK4 locus (ANRIL) has become an important risk factor for coronary disease due to its involvement in the regulation of histone methylation [Citation15,Citation16]. Significant changes in the expression of lncRNAs were observed during the development of ACS [Citation2]. Moreover, knockdown of ATPase plasma membrane Ca2+ transporting 1 antisense RNA 1 (ATP2B1-AS1) in mice may inhibit the nuclear factor-κB (NF-κB) pathway through the up-regulation of nuclear factor-kappa-B inhibitor alpha (NFKBIA) expression, thus playing an important role in the regulation of myocardial infarction [Citation17]. miRNAs also play an important regulatory role in the progression of ACS [Citation18]. miRNAs are a class of small non-coding RNAs that are involved in the regulation of gene expression [Citation19]. Serum miRNA-499, miRNA-210 and miRNA-941 have potential diagnostic value in early diagnosis of ACS [Citation20,Citation21]. In addition, lncRNA TCONS_00024652 regulates vascular endothelial cell proliferation and angiogenesis through miRNA-21 [Citation13]. Previous studies have revealed that lncRNAs play an important role in ACS disease progression by regulating mRNAs and miRNAs. However, the expression pattern of lncRNAs and their relative role in ACS require further investigation. Thus, analysis of gene transcriptome data of ACS is of great importance for further diagnosis and the development of novel treatment options.
Transcriptome sequencing (RNAseq) is a well-established method for analyzing the entire transcriptome, and is commonly used to evaluate the differential expression of genes in case-control studies [Citation22]. However, research into the transcriptome of ACS and in-stent restenosis remains limited. Bioinformatics analyses such as functional enrichment analysis, Venn diagrams and correlation network construction are often used to identify key genes in diseases [Citation23–27]. Thus, in order to determine key potential molecular markers for the early diagnosis and treatment of ACS and post-treatment in-stent restenosis, RNA-seq analysis was performed in the present study. The DESeq2 package was subsequently used for the screening of differentially expressed mRNAs (DEmRNAs) and differentially expressed lncRNAs (DElncRNAs). In order to further understand the biological functions of DEmRNAs, functional enrichment analysis was also performed in the present study. In order to identify genes associated with the development of ACS, lncRNA-mRNA co-expression network was developed. Subsequently, we selected the genes in the top 10 of differential expression and also involved in the co-expression of lncRNA-mRNA for diagnostic analysis to further evaluate the reliability of our results. Collectively, the results of the present study may provide novel insights into ACS and post-treatment in-stent restenosis, and may contribute to the development of novel diagnostic and therapeutic targets.
Materials and methods
Patients
The study population included 3 patients with ACS, 3 patients with PCI_non-restenosis (PCI_NR), 3 patients with PCI_restenosis (PCI_Re) and 3 normal healthy controls (NC). Detailed clinical information is displayed in . All patients aged between 56 and 79 years. Patients who met the definition of acute myocardial infarction and unstable angina were included in the ACS group [Citation5]. Detailed exclusion criteria for patients with ACS were as follows: (1) patients who had been administered fish oil or drugs containing n-3 polyunsaturated fatty acids (n-3 PUFA) on admission; (2) patients who had been administered fish oil or received n-3 PUFA treatment following the onset of ACS; (3) patients who exhibited symptoms of active malignant diseases; (4) patients with liver dysfunction (aspartate aminotransferase>100 IU/L, alanine aminotransferase>100 IU/L) and severe renal insufficiency accompanied by hemodialysis. Patients enrolled in the present study initially received interventional treatment, and patients were included in the PCI_Re group following detection of severe in-stent restenosis using coronary angiography [Citation28]. Patients who exhibited signs of restenosis that was not caused by interventional therapy were excluded from the present study. Moreover, patients in which restenosis was not detected following interventional treatment were included in the PCI_NR group. The individuals in the NC group were gender and age matched with the other groups, and they were healthy without any disease.
Table 1. Detailed information of sequencing samples
RNA library construction, sequencing, and rawdata processing
Total RNA was extracted from the peripheral blood samples of the participants. Illumina TruseqTM RNA Sample Prep kit was used to construct the chain specific library. Agilent 2100 BioAnalyzer and ABI StepOnePlus Real-Time PCR System were used to detect the quality of the library. The BGIseq platform was sequenced using the PE100 strategy. Fastp was used for quality control of the original sequencing data. The high quality sequence obtained following quality control was aligned to the human reference genome (GRCh38) in the Ensemble database [Citation29] using the HISAT2 program (https://ccb.jhu.edu/software/hisat2/index.shtml) [Citation30]. Expression of mRNAs and lncRNAs were normalized and outputted using stringtie (http://ccb.jhu.edu/software/stringtie/) [Citation31].
Differential analysis of mRNAs and lncRNAs
The DESeq2 package in R was used to screen genes with significant differences between samples (http://bioconductor.org/packages/DESeq2/) [Citation32]. Firstly, standardize the original read count (mainly to correct the sequencing depth). Then, calculate the probability of hypothesis test (P-value) through the statistical model. Thirdly, multiple hypothesis testing correction (Benjiamini and Hochberg method) was performed to obtain the corrected p value (false discovery rate, FDR). Padj<0.05 and |log2 foldchange| (|log2FC|)>1 were used to the differential expression screening criteria of mRNAs and lncRNAs.
Functional analysis of genes
Biological function enrichment analysis of DEmRNAs was performed. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses were performed using the GeneCodis4.0 database (https://genecodis.genyo.es/) [Citation33]. Pval_adj<0.05 was the screening standard.
Analysis of lncRNA-mRNA co-expression relationship
The correlation between lncRNAs and mRNAs was analyzed using Pearson’s correlation coefficient. P value (P)<0.01 and |correlation coefficient| (|r|)≥0.8 for screening criteria. Then, GO and KEGG functional enrichment analysis were performed on the screened lncRNAs and mRNAs.
Diagnostic analysis
The GSE60993 dataset containing 26 cases of ACS and 7 normal healthy controls was obtained from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) [Citation34], and the RNA sequencing type was mRNA. The aforementioned dataset was used for the diagnostic analysis of identified DEmRNAs. The receiver operating characteristic (ROC) analysis was also performed by using pROC package in R language. The sensitivity and specificity at the cutoffs were determined according to a previous study [Citation35].
RT-qPCR analysis
14 blood samples from 5 patients with ACS, 5 PCI_Re patients and 4 NC were obtained for in vitro validation. Total RNA was extracted from the blood samples using the RNAliquid Ultra-speed Whole Blood (liquid sample) kit (RN2602, Beijing Huitian Oriental Technology Co., Ltd.), according to the manufacturer’s protocols. The cDNA was synthesized using the FastKing cDNA first strand synthesis kit (KR116, TIANGEN), and then stored at −20°C or lower temperature. Real time-polymerase chain reaction (RT-PCR) was performed using SuperReal PreMix Plus (SYBR Green) kit (FP205, TIANGEN). GAPDH and ACTB were the internal reference of mRNA. The relative quantitative analysis of the data was performed by 2−ΔΔCt method [Citation36]. All experimental procedures were approved by The Third Hospital of Hebei Medical University (K2019-012-1).
Statistical analysis
Results are presented as the mean ± standard deviation (SD). One-way ANOVA was used to statistically analyze the in vitro experiments. All experiments were independently repeated at least three times. P < 0.05 was considered to indicate a statistically significant difference.
Results
ACS is one of the most serious cardiovascular diseases. The treatment of in-stent restenosis in ACS patients remains a major clinical challenge. In order to find potential key molecular markers to help early diagnosis and treatment of ACS and post-treatment recurrence, we performed transcriptome sequencing analysis. The study population included 3 patients with ACS, 3 PCI_NR patients, 3 PCI_Re patients and 3 NC. RNA was extracted from blood samples for library construction and high-throughput sequencing. DESeq2 package in R was used to screen genes with differentially expressed between different samples. Subsequently, functional enrichment analysis, lncRNA-mRNA co-expression network construction, diagnostic analysis and in vitro validation were performed. All results indicate that identified mRNAs and lncRNAs may be used as potential clinical biomarkers. In addition, we speculated that PDZK1IP1, PROK2 and LAMP3 may be used as the diagnosis of ACS.
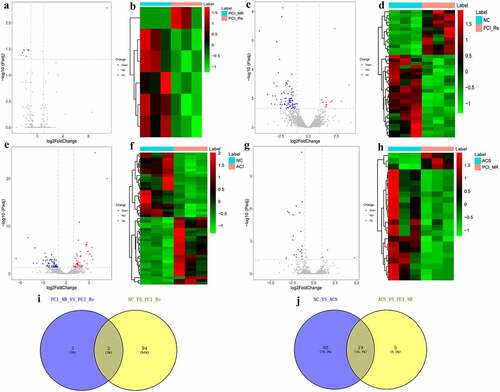
DEmRNAs analysis in PCI_NR/PCI_Re group
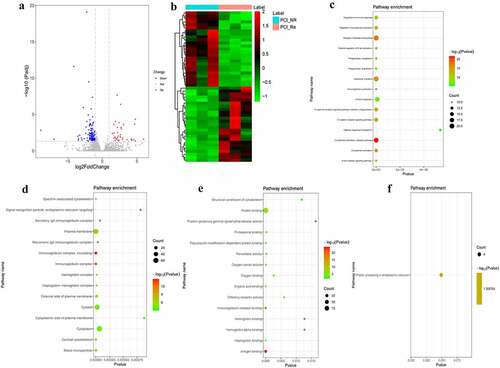
In order to reveal genes associated with in-stent restenosis (recurrence) in patients with ACS, differentially expressed genes were analyzed in the PCI_NR/PCI_Re group. According to the aforementioned screening criteria, there were 170 DEmRNAs (33 up-regulated and 137 down-regulated) in the PCI_NR/PCI_Re group. The volcano map of DEmRNAs is displayed in and a heat map of DEmRNAs is displayed in . To understand the biological functions of DEmRNAs, GO and KEGG functional analyses were performed (Pval_adj<0.05). The biological process (BP), cell composition (CC) and molecular function (MF) enrichment results of the top 15 are displayed in . Results of the KEGG enrichment analysis demonstrated that protein processing in the endoplasmic reticulum was the only significantly enriched signaling pathway ().
Figure 1. DEmRNAs analysis of the PCI_NR/PCI_Re group.
DEmRNAs analysis in NC/PCI_Re group
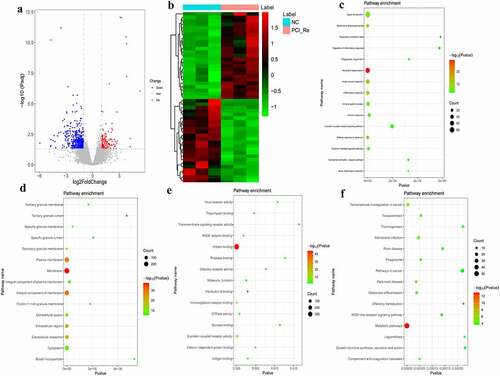
According to the aforementioned screening criteria, there were 635 DEmRNAs (124 up-regulated and 511 down-regulated) in the NC/PCI_Re group. The volcano map of DEmRNAs is displayed in . The heat map of DEmRNAs is displayed in . In order to understand the biological functions of DEmRNAs, GO and KEGG functional analyses were performed (Pval_adj<0.05). The BP, CC and MF enrichment results of the top 15 are displayed in . Results of the KEGG enrichment analysis demonstrated that metabolic pathways and transcriptional misregulation in cancer were significantly enriched signaling pathways ().
Figure 2. DEmRNAs analysis of the NC/PCI_Re group.
DEmRNAs analysis in NC/ACS group
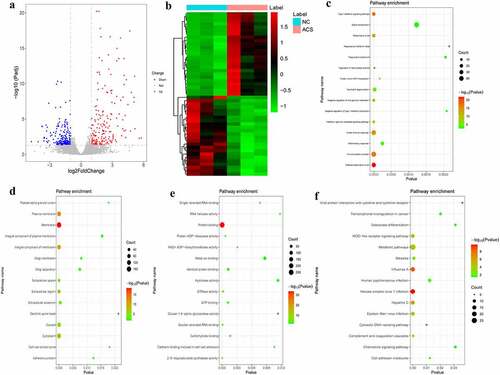
According to the aforementioned screening criteria, there were 408 DEmRNAs (183 up-regulated and 225 down-regulated) in the NC/ACS group. The volcano map of DEmRNAs is displayed in . The heat map of DEmRNAs is displayed in . In order to understand the biological functions of DEmRNAs, GO and KEGG functional analyses were performed (Pval_adj<0.05). The BP, CC and MF enrichment results of the top 15 are displayed in . Results of the KEGG enrichment analysis demonstrated that herpes simplex virus 1 infection and influenza A were significantly enriched signaling pathways ().
Figure 3. DEmRNAs analysis of the NC/ACS group.
DEmRNAs analysis in ACS/PCI_NR group
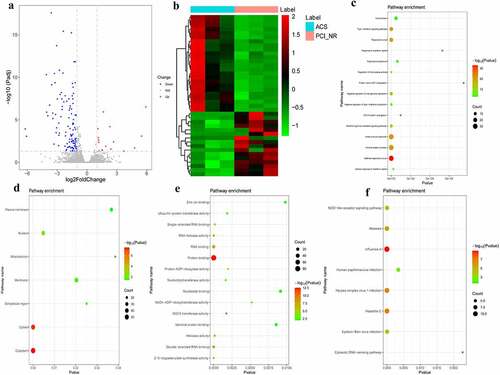
According to the aforementioned screening criteria, there were 127 DEmRNAs (16 up-regulated and 111 down-regulated) in the ACS/PCI group. The volcano map of DEmRNAs is displayed in . The heat map of DEmRNAs is displayed in . In order to understand the biological functions of DEmRNAs, GO and KEGG functional analyses were performed (Pval_adj<0.05). The BP, CC and MF enrichment results of the top 15 are displayed in . Results of the KEGG enrichment analysis demonstrated that influenza A and hepatitis C were significantly enriched signaling pathways ().
Figure 4. DEmRNAs analysis in ACS/PCI_NR group.
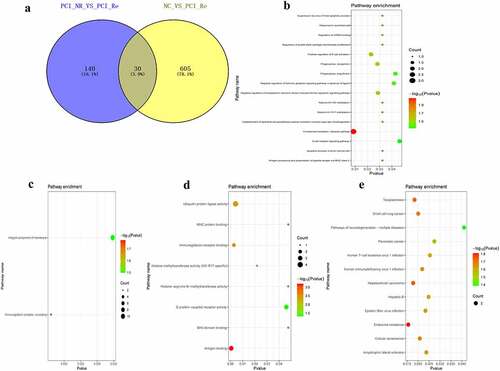
Analysis of DEmRNAs of the intersection of the PCI_NR/PCI_Re and NC/PCI_Re groups
The intersection of DEmRNAs of the PCI_NR/PCI_Re and NC/PCI_Re groups was established, and a total of 30 genes were obtained (). Moreover, the trend in the expression levels of the aforementioned 30 genes was consistent between the two groups. In order to understand the biological functions of intersection DEmRNAs, GO and KEGG functional analyses were performed (Pval_adj<0.05). The BP, CC and MF enrichment results are displayed in . Results of the KEGG enrichment analysis demonstrated that endocrine resistance and hepatocellular carcinoma were significantly enriched signaling pathways ().
Figure 5. Analysis of DEmRNAs in the intersection of the PCI_NR/PCI_Re and NC/PCI_Re groups.
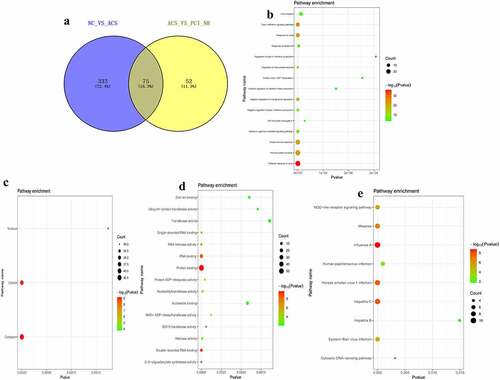
Analysis of DEmRNAs of the intersection of the NC/ACS and ACS/PCI_NR groups
The intersection of DEmRNAs of the NC/ACS and ACS/PCI_NR groups was established, and a total of 75 genes were obtained (). Moreover, the trend in the expression levels of the aforementioned 75 genes was opposite between the two groups. In order to understand the biological functions of intersection DEmRNAs, GO and KEGG functional enrichment analyses were performed (Pval_adj<0.05). The BP, CC and MF enrichment results are displayed in . Results of the KEGG enrichment analysis demonstrated that influenza A and hepatitis C were significantly enriched signaling pathways ().
Figure 6. Analysis of DEmRNAs in the intersection of the NC/ACS and ACS/PCI_NR groups.
Analysis of DElncRNAs in different groups
According to the aforementioned screening criteria, there were 6 DElncRNAs (1 up-regulated and 5 down-regulated) in the PCI_NR/PCI_Re group. The volcano map and heat map of DElncRNAs are displayed in and b. Moreover, there were 97 DElncRNAs (13 up-regulated and 84 down-regulated) in the NC/PCI_Re group. The volcano map and heat map of DElncRNAs are displayed in and d. There were 110 DElncRNAs (47 up-regulated and 63 down-regulated) in the NC/ACS group. The volcano map and heat map of DElncRNAs was shown in and f. Results of the present study also demonstrated that there were 24 DElncRNAs (3 up-regulated and 21 down-regulated) in the ACS/PCI_NR group. The volcano map and heat map of DElncRNAs was shown in and h. The intersection of DElncRNAs of the PCI_NR/PCI_Re and NC/PCI_Re groups was established, and a total of 3 genes were obtained (). Moreover, the trend in the expression levels of the aforementioned 3 genes was consistent between the two groups. The intersection of DElncRNAs of the NC/ACS and ACS/PCI_NR groups was established, and a total of 19 genes were obtained (). The trend in the expression levels of the aforementioned 19 genes was opposite between the two groups.
Figure 7. Analysis of DElncRNAs in different groups.
Co-expression analysis of DEmRNA-DElncRNA in the intersection groups
P < 0.01 and |r|≥0.8 were used as the criteria for determining associations between DElncRNA-DEmRNA pairs. Results of the present study demonstrated that there were 19 DElncRNA-DEmRNA pairs (for example, PROK2-AL035661.1, E2F2-LINC00570) in the PCI_NR/PCI_Re and NC/PCI_Re intersection group (). In the DEmRNA-DElncRNA co-expression network, 15 DEmRNAs and 3 DElncRNAs were included. Among them, E2F2, PDZK1IP1, TRIM10, HBBP1, IGLL5, PROK2 and LINC00570 were the top 10 differentially expressed genes. In order to understand the biological function of DEmRNAs co-represented with DElncRNAs, GO and KEGG functional analyses were performed (Pval_adj<0.05). The BP, CC and MF enrichment results are displayed in . Results of the KEGG enrichment analysis demonstrated that endocrine resistance was the only significantly enriched signaling pathway ().
Figure 8. Co-expression analysis of DEmRNA-DElncRNA.
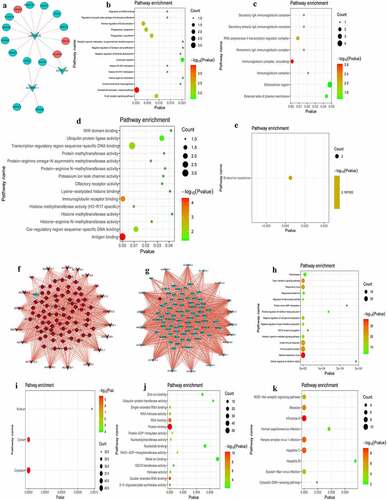
There were 993 DElncRNA-DEmRNA pairs (for example, IFI44-NRIR) in the NC/ACS and ACS/PCI_NR intersection group ( and g). In the DEmRNA-DElncRNA co-expression network, 74 DEmRNAs and 19 DElncRNAs are included. Among them, RNF213, LAMP3, IFI44, IFIT1, IFIT3, IFIT5, REC8, XAF1 and NRIR were the top 10 differentially expressed genes. In order to understand the biological function of DEmRNAs co-represented with DElncRNAs, GO and KEGG functional analysis were performed (Pval_adj<0.05). The BP, CC and MF enrichment results are displayed in . Results of the KEGG enrichment analysis demonstrated that influenza A and hepatitis C were significantly enriched signaling pathways ().
Diagnostic analysis of DEmRNAs
We selected the genes in the top 10 of differential expression and also involved in the co-expression of lncRNA-mRNA for diagnostic analysis in the GSE60993 dataset. Results of the ROC curve analysis demonstrated that only the area under curve (AUC) of PDZK1IP1, PROK2 and LAMP3 were >0.7, at 0.747, 0.769 and 0.725, respectively (). These results indicated that PDZK1IP1, PROK2 and LAMP3 may act as the potential diagnostic genetic biomarkers in ACS.
Figure 9. Diagnostic analysis of PDZK1IP1 (a), PROK2 (b) and LAMP3 (c).

RT-PCR analysis
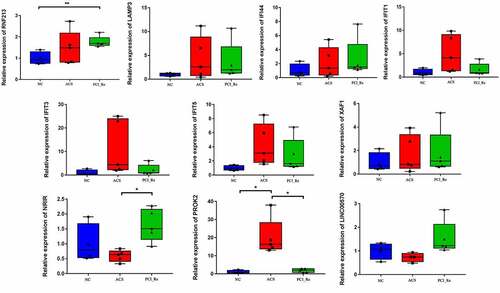
The information of 14 individuals is displayed in . PROK2, LINC00570, RNF213, LAMP3, IFI44, IFIT1, IFIT3, IFIT5, XAF1 and NRIR were selected for RT-PCR validation. All primers used in the present study are shown in . Results of the RT-PCR analysis demonstrated that compared with normal controls, the expressions levels of RNF213, LAMP3, IFI44, IFIT1, IFIT3, IFIT5 and XAF1 were up-regulated in ACS (), which was consistent with the results of RNA-seq. However, the expression levels of NRIR, PROK2 and LINC00570 were not consistent with the results obtained during RNA-seq analysis. Inconsistency in these results may be due to small sample sizes; thus, further investigations are required.
Table 2. The information of 14 individuals for RT-PCR validation
Table 3. Primer sequence in the RT-qPCR
Figure 10. RT-PCR validation of PROK2, LINC00570, RNF213, LAMP3, IFI44, IFIT1, IFIT3, IFIT5, XAF1 and NRIR in blood samples.
Discussion
The mechanisms underlying ACS progression are mediated by multiple molecules [Citation37–40], and numerous previous studies have based their research on the use of public databases or certain molecules for the study of ACS. Notably, ACS and in-stent restenosis have rarely been studied at the level of the transcriptome. The blood transcriptome reflects the state of the disease [Citation41]. However, there are few studies on the identification of novel biomarkers from the peripheral blood transcriptome of patients with ACS. Bioinformatics analysis is a common method to identify potential key molecule biomarkers and signaling pathways in diseases [Citation42–45]. Thus, the present study aimed to identify key molecular markers of ACS and in-stent restenosis from peripheral blood using transcriptome sequencing and bioinformatics analysis. Studying the expression and interaction of ACS and in-stent restenosis-associated genes is of great significance for understanding the molecular mechanisms underlying ACS, and provides a novel theoretical basis for the diagnosis and treatment of ACS.
In the present study, there were 15 DEmRNAs and 3 DElncRNAs in the PCI_NR/PCI_Re and NC/PCI_Re intersection group. We selected genes in the top 10 of differential expression and also involved in the co-expression of lncRNA-mRNA for related discussions. Results of a previous study demonstrated that compared with wild-type mice, E2F transcription factor 2 (E2F2) deficient mice exhibited abnormal blood vessels and elevated blood pressure [Citation46]. E2F2 induces the proliferation of cardiomyocytes, and reduces the expression of pro-apoptotic genes [Citation47,Citation48]. Results of the present study highlighted that the expression of E2F2 was significantly altered, and was co-expressed with LINC00570. Therefore, we hypothesized that E2F2 and LINC00570 play an important regulatory role in the progression of ACS through co-expression regulation of angiogenesis and proliferation of cardiac myocytes. Results of a previous study found demonstrated Prokineticin 2 (PROK2) (also known as PK2) alleviated H9C2 myocardial cell injury induced by hypoxia/reoxygenation (H/R) through the activation of the Akt/mTOR pathway [Citation49]. PROK2 also inhibits high glucose/high palmitic acid-induced apoptosis of cardiomyocytes by inhibiting oxidative stress and autophagosome accumulation [Citation50]. In addition, PROK2 plays a role in angiogenesis and inflammation [Citation51,Citation52]. Through lncRNA-mRNA co-expression network, results of the present study revealed that PROK2 was co-expressed with AL035661.1. Thus, we hypothesized that the roles of PROK2 in angiogenesis, and proliferation and apoptosis of cardiac myocytes may be regulated by AL035661.1, thus affecting the progression and recurrence of ACS. In addition, the AUC of PROK2 was >0.7, which indicated that PROK2 may be a potential diagnostic biomarker of ACS.
Few studies have revealed the role of PDZK1 interacting protein 1 (PDZK1IP1) in cardiovascular diseases; however, PDZK1IP1 acts as a potential biomarker and is abnormally expressed in tumors [Citation53,Citation54]. Tripartite motif containing 10 (TRIM10) is also associated with immune response [Citation55], and regulates cardiac hypertrophy through the PTEN/AKT pathway [Citation56]. Hemoglobin subunit beta pseudogene 1 (HBBP1) plays an important role in erythropoiesis and β-thalassemia [Citation57]. Immunoglobulin lambda like polypeptide 5 (IGLL5) is up-regulated in inflammatory diseases [Citation58,Citation59]. In addition, results of the KEGG enrichment analysis demonstrated that IGLL5 was associated with primary immunodeficiency and chemokine signaling pathways [Citation58]. To the best of our knowledge, the roles of PDZK1IP1, TRIM10, HBBP1 and IGLL5 have not previously been reported in ACS, and the present study may be the first to report the potential molecular regulatory role of the aforementioned genes in ACS. Results of the lncRNA-mRNA co-expression network used in the present study demonstrated that PDZK1IP1, TRIM10, HBBP1 and IGLL5 were co-expressed with LINC00570. However, to the best of our knowledge, the role of LINC00570 is yet to be reported in cardiovascular disease. Further investigation into the roles of PDZK1IP1, TRIM10, HBBP1, IGLL5 and LINC00570 will provide novel insights into the pathogenesis of ACS, and may contribute to the development of potential diagnostic and therapeutic targets for ACS. Notably, results of the present study revealed that the AUC of PDZK1IP1 was >0.7, which indicated that PDZK1IP1 may be a potential diagnostic biomarker of ACS.
Moreover, results of the present study revealed a large number of DEmRNAs and DElncRNAs in the NC_VS_ACS and ACS_VS_PCI_NR intersection group. We selected genes in the top 10 of differential expression and also involved in the co-expression of lncRNA-mRNA for related discussions. Interferon induced protein with tetratricopeptide repeats (IFIT) 1, IFIT3 and IFIT5 belong to the IFIT family of genes [Citation60]. Up-regulation of IFITs expression is critical for cardiomyocyte clearance of coxsackievirus B3 (CVB3) and the prevention of myocarditis [Citation61]. Moreover, increased expression of IFIT1 will promote inflammation, and IFIT1 can be used as a potential target for alleviating atherosclerosis-related diseases [Citation62]. IFIT3 may be a potential therapeutic target for ischemic cardiomyopathy [Citation63], and IFIT1 and IFIT5 are key genes that are used to predict treatment response in patients with microvascular disease in the early stage [Citation64]. Previous studies have demonstrated that IFIT1, IFIT3 and IFIT5 play an important regulatory role in cardiovascular disease. In the present study, the expression levels of IFIT 1, IFIT3 and IFIT5 were significantly altered, and were co-expressed with multiple DElncRNAs. Thus, we hypothesized that IFIT1, IFIT3 and IFIT5 may co-regulate the occurrence of ACS together with related DElncRNAs. Identification of IFIT1, IFIT3 and IFIT5 in the present study contributes to the discovery of potential molecular biomarkers for ACS, and also provides direction for further understanding the pathogenesis of ACS. In addition, results of the KEGG functional enrichment analysis in the present demonstrated that IFIT1 was associated with hepatitis C. Patients with hepatitis C virus (HCV) infection have a greater risk of developing ACS than those without HCV infection [Citation65]. Compared with patients who are not infected with HCV, patients with ACS and HCV infection have increased platelet reactivity, coronary heart disease is more serious, and the prognosis is worse [Citation66]. Notably, successful HCV treatment significantly reduced the incidence of ACS in patients with type 2 diabetes [Citation67]. Therefore, IFIT1 may regulate the progression of ACS by regulating hepatitis C.
Ring finger protein 213 (RNF213) is a susceptibility gene of moyamoya disease and plays an important role in vascular development [Citation68,Citation69]. Overexpression of the vascular endothelial cell-specific RNF213 mutant aggravated the hypoxia-induced PH phenotype (high right ventricular pressure, right ventricular hypertrophy, and pulmonary vascular muscularization) [Citation70]. Notably, results of a previous study demonstrated that the RNF213 p.R4810K variant was significantly associated with coronary artery disease [Citation71]. Moreover, previous studies have also demonstrated that lysosomal associated membrane protein 3 (LAMP3) is expressed in human heart and cardiomyocytes, and that its expression is elevated in dilated cardiomyopathy hearts with severe heart remodeling [Citation72]. Interferon induced protein 44 (IFI44) may be involved in vascular lesions and pathogenesis of systemic sclerosis (SSc) [Citation73], and is also associated with immune and inflammatory diseases [Citation74,Citation75]. REC8 meiotic recombination protein (REC8) inhibits tumor angiogenesis by inhibiting NF-κB mediated expression of vascular endothelial growth factor [Citation76]. XIAP associated factor 1 (XAF1) is a pro-apoptotic factor, and knockdown of XAF1 inhibits the apoptosis of vascular smooth muscle cells induced by interferon (IFN)-gamma [Citation77]. Overexpression of XAF1 induces cell apoptosis and has potential anti-angiogenesis effects [Citation78]. ACS describes a group of severe cardiovascular diseases, and includes acute myocardial infarction and unstable angina [Citation1,Citation2]. Thus, we hypothesized that RNF213, LAMP3, IFI44, REC8 and XAF1 may play key roles in the progression of ACS by regulating vascular lesions. In the present study, the expression levels of RNF213, LAMP3, IFI44, REC8 and XAF1 were significantly altered, and were co-expressed with multiple DElncRNAs. For example, IFI44 and Negative regulator of interferon response (NRIR) was a co-expression pair. NRIR, also known as lncCMPK2 or lncRNA-CMPK2, was significantly up-regulated in SSc monocytes and NRIR expression was associated with the IFN score of patients with SSc [Citation79]. Moreover, NRIR is abnormally expressed in the peripheral blood mononuclear cells of patients with Sjogren’s syndrome, and it is closely associated with the mRNA functions in immune response and cell metastasis [Citation80]. Therefore, we speculated that RNF213, LAMP3, IFI44, REC8 and XAF1 may co-regulate the occurrence of ACS together with related DElncRNAs. Identification of RNF213, LAMP3, IFI44, REC8, XAF1 and NRIR in the present study contributes to the discovery of potential molecular biomarkers for ACS, and also provides direction for further understanding the pathogenesis of ACS. In addition, the AUC of LAMP3 was >0.7, which indicates that LAMP3 may be a potential diagnostic biomarker of ACS.
Dataset GSE60993 was blood transcriptome sequencing data of patients with ACS [Citation41]. Previous studies have identified key genes and signaling pathways associated with ACS progression in the GSE60993 dataset through bioinformatics analysis [Citation81–84]. Numerous previous studies have relied on the use of public databases for analysis. In the present study, patients with ACS, PCI_NR patients, PCI_Re patients and healthy controls were enrolled for transcriptome sequencing analysis, and genes associated with ACS and recurrence following treatment were screened. For further verification, we selected the genes in the top 10 of differential expression and also involved in the co-expression of lncRNA-mRNA for diagnostic analysis in the GSE60993 dataset. Results of the present study revealed that PDZK1IP1, PROK2 and LAMP3 may act as potential biomarkers of ACS and recurrence following treatment, and the expression of these genes may act as biomarkers for early medical intervention of the disease.
However, the present study has limitations. Firstly, the sample sizes used for sequencing and RT-PCR analyses were small. Thus, the sample size of each group must be increased to further verify the results obtained. Moreover, the molecular mechanisms underlying the regulation of the identified genes in ACS and PCI_Re remain to be fully elucidated, and further investigations are required.
Conclusion
In the present study, numerous DEmRNAs and DElncRNAs were identified through transcriptome sequencing. Results of the present study indicated that the identified mRNAs and lncRNAs may be used as potential clinical biomarker for ACS. Although the present study has limitations, ACS transcriptome sequencing analysis and the construction of mRNA-lncRNA co-expression network provide potential targets for the diagnosis and treatment of ACS and PCI_Re, and also provide a novel theoretical basis for future studies. Collectively, results of the present study demonstrated that PDZK1IP1, PROK2 and LAMP3 may act as potential biomarkers of ACS.
Consent for publication
All authors have agreed to the publication of the work.
Ethics approval and consent to participate
All experimental procedures were approved by The Third Hospital of Hebei Medical University (K2019-012-1). The written consent was obtained from the all patients. All participants were informed as to the purpose of this study, and that this study complied with the Declaration of Helsinki.
Highlights
Transcriptome sequencing analysis provides potential biomarkers for the diagnosis and treatment of ACS.
PDZK1IP1, PROK2 and LAMP3 may act as the potential diagnostic genetic biomarkers in ACS.
Identification of potential key genes provides direction for further research.
Availability of data and materials
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. The transcriptome data have been uploaded to Gene Expression Omnibus (accession no. GSE179645).
Authors’ contributions
All authors have made important contributions to data analysis, drafting the article or revising the article.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Wang L, Jin Y. Noncoding RNAs as biomarkers for acute coronary syndrome. BioMed res int. 2020;(2020)(3298696). 10.1155/2020/3298696.
- Lu Y, Meng X, Wang L, et al. Analysis of long non-coding RNA expression profiles identifies functional lncRNAs associated with the progression of acute coronary syndromes. Exp Ther Med. 2018;15(2):1376–1384.
- Makki N, Brennan TM, Girotra S. Acute coronary syndrome. J Intensive Care Med. 2015;30(4):186–200.
- Switaj TL, Christensen SR, Brewer DM. Acute Coronary Syndrome: current Treatment. Am Fam Physician. 2017;95(4):232–240.
- Yagi S, Kondo D, Ise T, et al. Association of decreased docosahexaenoic acid level after statin therapy and low eicosapentaenoic acid level with in-stent restenosis in patients with acute coronary syndrome. J Atheroscler Thromb. 2019;26(3):272–281.
- Uchida S, Perche F, Pichon C, et al. Nanomedicine-based approaches for mRNA delivery. Molecular Pharmaceutics. 2020;17(10):3654–3684.
- Martínez-Fernández DE, Padilla-Gutiérrez JR, Casillas-Muñoz F. Analysis of genetic variation in CD40 and CD40L: relationship with mRNA relative expression and soluble proteins in acute coronary syndrome. Journal of Immunology Research. 2019;(2019):8063983.
- Valdés-Alvarado E, Valle Y, Muñoz-Valle JF. MIF mRNA expression and soluble levels in acute coronary syndrome. Cardiology Research and Practice. 2018;(2018)(9635652). 10.1155/2018/9635652
- Heger LA, Hortmann M, Albrecht M, et al. Inflammation in acute coronary syndrome: expression of TLR2 mRNA is increased in platelets of patients with ACS. PloS One. 2019;14(10):e0224181.
- Raffa S, Chin XLD, Stanzione R, et al. The reduction of NDUFC2 expression is associated with mitochondrial impairment in circulating mononuclear cells of patients with acute coronary syndrome. Int J Cardiol. 2019;286:127–133.
- Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–669.
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81(1):145–166.
- Halimulati M, Duman B, Nijiati J, et al. Long noncoding RNA TCONS_00024652 regulates vascular endothelial cell proliferation and angiogenesis via microRNA-21. Exp Ther Med. 2018;16(4):3309–3316.
- Du J, Han R, Li Y, et al. LncRNA HCG11/miR-26b-5p/QKI5 feedback loop reversed high glucose-induced proliferation and angiogenesis inhibition of HUVECs. Journal of Cellular and Molecular Medicine. 2020;24(24):14231–14246.
- Broadbent HM, Peden JF, Lorkowski S, et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet. 2008;17(6):806–814.
- Yap KL, Li S, Muñoz-Cabello AM, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38(5):662–674.
- Song KY, Zhang XZ, Li F, et al. Silencing of ATP2B1-AS1 contributes to protection against myocardial infarction in mouse via blocking NFKBIA-mediated NF-κB signalling pathway. Journal of Cellular and Molecular Medicine. 2020;24(8):4466–4479.
- Shirvani Samani O, Meder B. microRNA assays for acute coronary syndromes. Diagnosis (Berl). 2016;3(4):183–188.
- Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, et al. An overview of microRNAs: biology, functions, therapeutics, and analysis methods. Journal of Cellular Physiology. 2019;234(5):5451–5465.
- Shalaby SM, El-Shal AS, Shoukry A, et al. Serum miRNA-499 and miRNA-210: a potential role in early diagnosis of acute coronary syndrome. IUBMB Life. 2016;68(8):673–682.
- Bai R, Yang Q, Xi R, et al. miR-941 as a promising biomarker for acute coronary syndrome. BMC Cardiovascular Disorders. 2017;17(1):227.
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63.
- Kumar SU, Kumar DT, Siva R, et al. Integrative bioinformatics approaches to map potential novel genes and pathways involved in ovarian cancer. Front Bioeng Biotechnol. 2019;7:391.
- Wan J, Jiang S, Jiang Y, et al. Data mining and expression analysis of differential lncRNA ADAMTS9-AS1 in prostate cancer. Front Genet. 2019;10:1377.
- Udhaya Kumar S, Thirumal Kumar D, Siva R, et al. Dysregulation of signaling pathways due to differentially expressed genes from the b-cell transcriptomes of systemic lupus erythematosus patients - a bioinformatics approach. Front Bioeng Biotechnol. 2020;8:276.
- Fu D, Zhang B, Yang L, et al. Development of an immune-related risk signature for predicting prognosis in lung squamous cell carcinoma. Front Genet. 2020;11:978.
- Udhaya Kumar S, Thirumal Kumar D, Bithia R, et al. Analysis of differentially expressed genes and molecular pathways in familial hypercholesterolemia involved in atherosclerosis: a systematic and bioinformatics approach. Front Genet. 2020;11:734.
- Li-Sha G, Peng C, Yue-Chun L. Recurrent acute coronary syndrome and restenosis after percutaneous coronary intervention in a patient with idiopathic thrombocytopenic purpura: a case report and literature review. BMC Cardiovasc Disord. 2015;15:101.
- Church DM, Schneider VA, Graves T, et al. Modernizing reference genome assemblies. PLoS Biol. 2011;9(7):e1001091.
- Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Frontiers in Genetics. 2019;37(8):907–915.
- Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology. 2015;33(3):290–295.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Garcia-Moreno A, Lopez-Dominguez R, Ramirez-Mena A, et al. GeneCodis 4: expanding the modular enrichment analysis to regulatory elements. 2021. DOI:10.1101/2021.04.15.439962.
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210.
- Šimundić AM. Measures of diagnostic accuracy: basic definitions. Ejifcc. 2009;19(4):203–211.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408.
- Yu J, Li Y, Leng D, et al. microRNA-3646 serves as a diagnostic marker and mediates the inflammatory response induced by acute coronary syndrome. Bioengineered. 2021;12(1):5632–5640.
- Diao Y, Yin M, Zhang B, et al. Predictive value of N-terminal pro-B-type natriuretic peptide (NT-pro BNP) combined with D-dimer for no-reflow phenomenon in patients with acute coronary syndrome after emergency of percutaneous coronary intervention. Bioengineered. 2021;12(1):8614–8621.
- Liu K, Chen S, Lu R. Identification of important genes related to ferroptosis and hypoxia in acute myocardial infarction based on WGCNA. Bioengineered. 2021;12(1):7950–7963.
- Xie T, Guo J, Jiang Y, et al. Screening differentially expressed proteins of coronary heart disease with congenital cold syndrome based on tandem mass tag (TMT) technology. Bioengineered. 2021;12(1):1338–1350.
- Park HJ, Noh JH, Eun JW, et al. Assessment and diagnostic relevance of novel serum biomarkers for early decision of ST-elevation myocardial infarction. Oncotarget. 2015;6(15):12970–12983.
- Uk S, Rajan B, TK D, et al. Involvement of essential signaling cascades and analysis of gene networks in diabesity. Genes. 2020;11(11). 10.3390/genes11111256
- Udhaya Kumar S, Saleem A, Thirumal Kumar D, et al. A systemic approach to explore the mechanisms of drug resistance and altered signaling cascades in extensively drug-resistant tuberculosis. Adv Protein Chem Struct Biol. 2021;127:343–364
- Udhaya Kumar S, Madhana Priya N, Thirumal Kumar D, et al. An integrative analysis to distinguish between emphysema (EML) and alpha-1 antitrypsin deficiency-related emphysema (ADL)-A systems biology approach. Adv Protein Chem Struct Biol. 2021;127:315–342
- Mishra S, Shah MI, Udhaya Kumar S, et al. Network analysis of transcriptomics data for the prediction and prioritization of membrane-associated biomarkers for idiopathic pulmonary fibrosis (IPF) by bioinformatics approach. Adv Protein Chem Struct Biol. 2021;123:241–273
- Zhou J, Zhu Y, Cheng M, et al. Regulation of vascular contractility and blood pressure by the E2F2 transcription factor. Circulation. 2009;120(13):1213–1221.
- Ebelt H, Zhang Y, Kampke A, et al. E2F2 expression induces proliferation of terminally differentiated cardiomyocytes in vivo. Cardiovasc Res. 2008;80(2):219–226.
- Ebelt H, Hufnagel N, Neuhaus P, et al. Divergent siblings: E2F2 and E2F4 but not E2F1 and E2F3 induce DNA synthesis in cardiomyocytes without activation of apoptosis. Circ Res. 2005;96(5):509–517.
- Su G, Sun G, Liu H, et al. Prokineticin 2 relieves hypoxia/reoxygenation-induced injury through activation of Akt/mTOR pathway in H9c2 cardiomyocytes. Artif Cells Nanomed Biotechnol. 2020;48(1):345–352.
- Yang Z, Wu Y, Wang L. Prokineticin 2 (PK2) rescues cardiomyocytes from high glucose/high palmitic acid-induced damage by regulating the AKT/GSK3β PATHWAY IN VItro. Oxidative Medicine and Cellular Longevity. 2020;2020:3163629.
- Li Y, Wang J, Yu L, et al. Effects of prokineticin 2 on testicular inflammation in rats. Am J Reproduct Immunol. 2018;79(6):e12843.
- Kurebayashi H, Goi T, Shimada M, et al. Prokineticin 2 (PROK2) is an important factor for angiogenesis in colorectal cancer. Oncotarget. 2015;6(28):26242–26251.
- Rivero M, Peinado-Serrano J, Muñoz-Galvan S, et al. MAP17 (PDZK1IP1) and pH2AX are potential predictive biomarkers for rectal cancer treatment efficacy. Oncotarget. 2018;9(68):32958–32971.
- de Miguel-Luken MJ, Chaves-Conde M, de Miguel-Luken V, et al. MAP17 (PDZKIP1) as a novel prognostic biomarker for laryngeal cancer. Oncotarget. 2015;6(14):12625–12636.
- Yang X, Zhang H, Shang J, et al. Comparative analysis of the blood transcriptomes between wolves and dogs. Anim Genet. 2018;49(4):291–302.
- Yang H, Wang XX, Zhou CY, et al. Tripartite motif 10 regulates cardiac hypertrophy by targeting the PTEN/AKT pathway. Journal of Cellular and Molecular Medicine. 2020;24(11):6233–6241.
- Ma Y, Liu S, Gao J, et al. Genome-wide analysis of pseudogenes reveals HBBP1’s human-specific essentiality in erythropoiesis and implication in β-thalassemia. Dev Cell. 2021;56(4):478–493.e411.
- Wu B, He Y, Yang D, et al. Identification of hub genes and therapeutic drugs in rheumatoid arthritis patients. Clin Rheumatol. 2021. DOI:10.1007/s10067-021-05650-6
- Lundmark A, Gerasimcik N, Båge T, et al. Gene expression profiling of periodontitis-affected gingival tissue by spatial transcriptomics. Scientific Reports. 2018;8(1):9370.
- Fensterl V, Sen GC. The ISG56/IFIT1 gene family. J Interferon Cytokine Res. 2011;31(1):71–78.
- Kimura T, Flynn CT, Alirezaei M, et al. Biphasic and cardiomyocyte-specific IFIT activity protects cardiomyocytes from enteroviral infection. PLoS Pathogens. 2019;15(4):e1007674.
- Wang JL, Cai F, XH L, et al. Lipopolysaccharide promotes inflammatory response via enhancing IFIT1 expression in human umbilical vein endothelial cells. DNA Cell Biol. 2020;39(7):1274–1281.
- Yang Y, Yang W, Huo W, et al. Identification of biomarkers for ischemic cardiomyopathy based on microarray data analysis. Cardiol J. 2017;24(3):305–313.
- Ishizu A, Tomaru U, Murai T, et al. Prediction of response to treatment by gene expression profiling of peripheral blood in patients with microscopic polyangiitis. PloS One. 2013;8(5):e63182.
- Tsai MS, Hsu YC, Yu PC, et al. Long-term risk of acute coronary syndrome in hepatitis C virus infected patients without antiviral treatment: a cohort study from an endemic area. Int J Cardiol. 2015;181:27–29.
- Scudiero F, Valenti R, Marcucci R, et al. Platelet reactivity in hepatitis c virus-infected patients on dual antiplatelet therapy for acute coronary syndrome. J Am Heart Assoc. 2020;9(18):e016441.
- Li J, Gordon SC, Rupp LB, et al. Sustained virological response to hepatitis C treatment decreases the incidence of complications associated with type 2 diabetes. Aliment Pharmacol Ther. 2019;49(5):599–608.
- Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PloS One. 2011;6(7):e22542.
- Kobayashi H, Matsuda Y, Hitomi T, et al. Biochemical and functional characterization of RNF213 (Mysterin) R4810K, a susceptibility mutation of moyamoya disease, in angiogenesis in vitro and in vivo. J Am Heart Assoc. 2015;4(7):7.
- Kobayashi H, Kabata R, Kinoshita H, et al. Rare variants in RNF213, a susceptibility gene for moyamoya disease, are found in patients with pulmonary hypertension and aggravate hypoxia-induced pulmonary hypertension in mice. Pulm Circ. 2018;8(3):2045894018778155.
- Morimoto T, Mineharu Y, Ono K, et al. Significant association of RNF213 p.R4810K, a moyamoya susceptibility variant, with coronary artery disease. PloS One. 2017;12(4):e0175649.
- Jiang DS, Yi X, Huo B, et al. The potential role of lysosome-associated membrane protein 3 (LAMP3) on cardiac remodelling. Am J Transl Res. 2016;8(1):37–48.
- Piera-Velazquez S, Mendoza FA, Addya S, et al. Increased expression of interferon regulated and antiviral response genes in CD31+/CD102+ lung microvascular endothelial cells from systemic sclerosis patients with end-stage interstitial lung disease. Clin Exp Rheumatol. 2020.
- DeDiego ML, Nogales A, Martinez-Sobrido L, et al. Interferon-induced protein 44 interacts with cellular FK506-binding protein 5, negatively regulates host antiviral responses, and supports virus replication. mBio. 2019;10(4). DOI:10.1128/mBio.01839-19.
- Rodríguez-Carrio J, López P, Alperi-López M, et al. IRF4 and IRGs delineate clinically relevant gene expression signatures in systemic lupus erythematosus and rheumatoid arthritis. Front Immunol. 2018;9:3085.
- Liu M, Xu W, Su M. REC8 suppresses tumor angiogenesis by inhibition of NF-κB-mediated vascular endothelial growth factor expression in gastric cancer cells. Biological Research. 2020;53(1):41.
- Bai Y, Ahmad U, Wang Y, et al. Interferon-gamma induces X-linked inhibitor of apoptosis-associated factor-1 and Noxa expression and potentiates human vascular smooth muscle cell apoptosis by STAT3 activation. J Biol Chem. 2008;283(11):6832–6842.
- Qiao L, Gu Q, Dai Y, et al. XIAP-associated factor 1 (XAF1) suppresses angiogenesis in mouse endothelial cells. Tumour Biol. 2008;29(2):122–129.
- Mariotti B, Servaas NH, Rossato M, et al. The long non-coding RNA NRIR drives IFN-RESPONSE IN MONOCYTES: IMPLICATION FOR SYSTEMIC SCLEROSIS. Front Immunol. 2019;10:100.
- Peng Y, Luo X, Chen Y, et al. LncRNA and mRNA expression profile of peripheral blood mononuclear cells in primary Sjögren’s syndrome patients. Sci Rep. 2020;10(1):19629.
- Wang Y, Miao L, Tao L, et al. Weighted gene coexpression network analysis identifies the key role associated with acute coronary syndrome. Aging (Albany NY). 2020;12(19):19440–19454.
- Liu Z, Ma C, Gu J, et al. Potential biomarkers of acute myocardial infarction based on weighted gene co-expression network analysis. Biomed Eng Online. 2019;18(1):9.
- Zhang R, Ji Z, Yao Y, et al. Identification of hub genes in unstable atherosclerotic plaque by conjoint analysis of bioinformatics. Life Sci. 2020;262:118517.
- Guo S, Huang Z, Liu X, et al. Biodata mining of differentially expressed genes between acute myocardial infarction and unstable angina based on integrated bioinformatics. Biomed Res Int. 2021;(2021)(5584681). DOI:10.1155/2021/5584681.