
ABSTRACT
Lapatinib (LAP) is an important anti-cancer drug and is frequently alongside doxorubicin (DOX) as a combination therapy for better anti-cancer efficacy. However, many studies have reported that LAP in combination with DOX may induce highly cardiotoxicity. Accordingly, we aimed to explore the potential mechanism involved in the synergistic effect of LAP in DOX-induced cardiotoxicity. Here, cell counting kit-8 was used to detect cell viability and lactate dehydrogenase measurement was performed to assess cell injury. Cell apoptosis was evaluated by TUNEL assay and western blot assay. Mitochondrial dysfunction was identified by JC-1 assay, adenosine triphosphate (ATP) and Cytochrome C. Moreover, the activity of ROS, SOD, CAT and GSH were measured to elucidate oxidative stress level. Ferroptosis was examined by levels of Fe2+, GPX4 and ASCL4. Expressions of PI3K/AKT signaling were identified by western blot assay. The results revealed that LAP inhibited the cell viability and exacerbated cell injury induced by Dox, as well as increased cell apoptosis. LAP aggravated DOX-induced mitochondria damage by changed mitochondrial membrane potential, decreased ATP and increased level of Cytochrome C. In addition, the combination of LAP and DOX induced oxidative stress and ferroptosis in H9c2 cells. The activation of PI3K/AKT signaling reversed the detrimental effects of LAP on DOX-induced H9c2 cells. The data in this study showed for the first time that LAP aggravated Dox-induced cardiotoxicity by promoting oxidative stress and ferroptosis in cardiomyocytes via PI3K/AKT-mediated mitochondrial dysfunction, suggesting that PI3K/AKT activation is a promising cardioprotective strategy for DOX /LAX combination therapies.
Introduction
Doxorubicin (DOX) is a kind of anthracyclines and has been widely used as an effective antitumor drug [Citation1]. However, the clinical application of DOX is limited because of various side effects, especially its severe dose-dependent and cumulative cardiotoxicity [Citation2]. DOX may induce acute or chronic cardiomyopathy, among which acute cardiotoxicity is unfrequent but chronic cardiactoxicity is dose-dependent, resulting in cardiac dysfunction, cardiomyopathy, heart failure and eventually death [Citation3,Citation4]. Although in-depth study on DOX-induced cardiotoxicity have proceeded for many years, the underlying mechanisms responsible for DOX-induced cardiotoxicity are still not understood [Citation5].
Lapatinib (LAP) is an anticancer drug, which is a tyrosine kinase inhibitor (). LAP is regarded as a nice alternative to trastuzumab when drug resistance emerges in patients with metastatic human epidermal growth factor receptor 2 (HER2)-positive breast cancer [Citation6]. LAP is usually used with the application of anthracyclines (ANT) including DOX, as a combination therapy for enhanced anti-tumor efficacy [Citation7]. According to the clinical data available so far, LAP may cause adverse reactions in cardiovascular system [Citation8]. Besides, LAP in combination with DOX may highly damage cardiac myocytes. LAP alone did not significantly cause myocyte injury at pharmacological concentrations [Citation9]. However, DOX-induced myocyte damage was greatly increased by the addition of low concentrations of LAP [Citation10]. It has been shown that LAP synergistically increased DOX toxicity in a time- and dose-dependent manner in human pluripotent stem cell-derived cardiomyocytes through iNOS signaling [Citation11]. Thus, understanding the molecular mechanisms underlying cardiactoxicity of LAP/DOX combination and developing preventive strategies and effective treatments against LAP/DOX combination-induced cardiotoxicity are urgent.
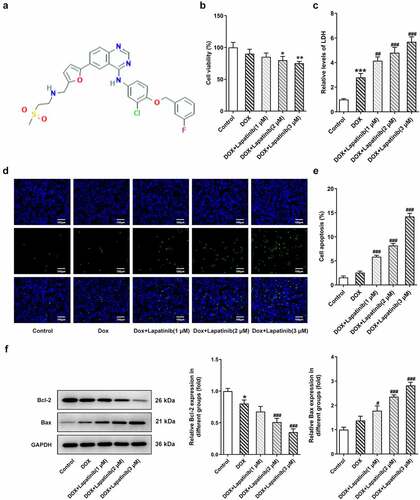
Figure 1. Effect of LAP on DOX-induced cytotoxicity in H9c2 cells. H9c2 cells were treated with 1 μM DOX with or without 1–3 μM LAP for 24 h. A, Chemical structure of Lapatinib. B, Cytotoxicity effect of LAP on H9c2 cells. C, LDH level in DOX-induced cells with or without different doses of LAP. D and E, Cell apoptosis was detected by TUNEL assay. Original magnification: x200. F, Protein levels of Bcl-2 and Bax were measured by western blot assay. LAP: Lapatinib; DOX: Doxorubicin; LDH: Lactate dehydrogenase. Data are expressed as mean ± SD calculated from three independent experiments. *P < 0.05, ***P < 0.001 versus control. #P < 0.05, ##P < 0.01, ###P < 0.001 versus DOX
In this study, we aims to investigate the role of LAP in DOX-induced cardiotoxicity and its possible mechanism and we hypothesized that LAP plays a reinforced role in DOX-induced cardiomyocyte injury. We found that different doses of LAP aggravated Dox-induced cell injury, apoptosis, mitochondrial dysfunction, oxidative stress and ferroptosis by repressing the PI3K/Akt signaling pathway. Conversely, 740Y-P, an agonist of PI3K/Akt signaling pathway, alleviate Dox-induced cardiotoxicity. The findings of this study might put forward a novel fundamental insight into how LAP exacerbates Dox-induced cardiotoxicity and provide a potential target for intervention against cardiotoxicity induced Dox treatment.
Materials and methods
Cell culture and treatment
Rat cardiomyoblast cell line H9c2 was obtained from Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), 100 IU/mL penicillin and 100 μg/mL streptomycin in a humidified 5% CO2 atmosphere at 37°C. To assess the effects of LAP on DOX-induced cardiotoxicity, H9c2 cells were treated with 1 μM Dox or co-treated with Dox (1 μM) plus LAP (1–3 μM) for 24 h, respectively. In pathway activation tests, H9c2 cells were pretreated with 30 μM 740Y-P (PI3K activator) for 1 h and then incubated with 1 μM Dox or co-incubated with Dox (1 μM) plus LAP (1–3 μM) for 24 h before analysis [Citation12].
Cell viability
Cell viability was measured using the cell counting kit-8 (CCK-8) in accordance with the manufacturer’s instructions [Citation13]. Cells were seeded at a density of 5 × 103 cells/well in a 96-well plate and treated with 1 μM Dox in the presence and absence of 1, 2, 3 μM LAP for 24 h. After that, 10 µl CCK-8 solution was added to each well and cells were incubated at 37°C for 2 h. The absorbance at 450 nm was measured by a microplate Reader (Bio-Rad, La Jolla, CA, USA). Each group was repeated six times and all experiments were done in triplicate.
Measurement of Lactate dehydrogenase (LDH) activity
Myocardial cytotoxicity was detected by using a LDH assay kit (Nanjing Jiancheng Biotechnology, Nanjing, China) [Citation14]. H9c2 cells were seeded (1x104 cells/well) in 96-well plates and were treated with 1 μM Dox or co-treated with Dox (1 μM) plus LAP (1, 2, 3 μM) for 24 h. At the end of the treatment, the cell culture media were harvested and the LDH activity was detected at 530 nm using a microplate reader (Benchmark; Bio-Rad Laboratories, Inc.).
Terminal deoxynucleotidyl transferasemediated dUTP nick end-labeling (TUNEL)
TUNEL assay was performed to evaluate H9c2 cell apoptosis [Citation15]. After treatment with Dox or Dox plus LAP, cells were washed and fixed with 4% paraformaldehyde at room temperature. After incubation with proteinase K for 15 min, cells were placed in 3% H2O2 for 15 min at room temperature and treated by TUNEL detection kit. After incubation, cells were co-labeled with 1 μg/ml DAPI working solution for 10 min. The labeled H9c2 cells were visualized by a fluorescence microscope (Olympus BX53, Japan).
DCFH-DA staining
To identify the ROS production, a fluorescenceprobe 2, 7-dichlorofluorescin diacetate kit (DCFH-DA, Sigma) was used [Citation16]. H9c2 cells after the experiment procedures were washed with PBS and incubated with DCFH-DA without light for 30 min at 37°C. The cells were then washed with PBS three times and the fluorescence intensity was detected by using a fluorescence microscope (Thermo Fisher Scientific, Waltham, MA, USA) at an excitation and an emission wavelength at 485 and 520 nm, respectively.
Detection of SOD, CAT and GSH level
H9c2 cells were seeded in six-well plates at a density of 4 × 105 cells/well. Following treatment of Dox or co-treatment with Dox plus LAP, the activity of SOD, CAT and GSH in cell media was analyzed with a SOD kit from Beyotime Institute of Biotechnology (Jiangsu, China). The absorbance at 532 nm was recorded using a microplate reader (Benchmark; Bio-Rad Laboratories, Inc.) [Citation17].
Evaluation of mitochondrial membrane motential (MMP)
Cationic dye 5, 5ʹ, 6, 6ʹ-tetrachloro-1, 1ʹ, 3, 3-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1, Sigma-Aldrich, MO, USA) staining was conducted to assess mitochondrial membrane motential (MMP) [Citation18]. Briefly, after treatment with Dox or Dox plus LAP in the presence or absence of 30 μM 740Y-P, H9c2 cells were collected and incubated with JC-1 for 20 min at 37°C, and were then detected with fluorescence microscope. Red fluorescence represented a potential-dependent aggregation in the mitochondria, reflecting ΔΨm. Green emission of the dye represented the monomeric form of JC-1, appearing in the cytosol after mitochondrial membrane depolarization. The wavelengths of excitation and emission were 514 nm and 529 nm for detection of monomeric form of JC-1, while 585 nm and 590 nm were used to detect aggregation of JC-1.
Measurement of Adenosine Triphosphate (ATP) level and Fe2+
The ATP level was evaluated by using the CellTiter-Glo Luminescent Assay Kit (Promega K.K., Tokyo, Japan) according to the manufacturer’s protocol [Citation19]. In addition, Fe2+ level was measured using kits from Abcam (product code ab83366, 593 nm) according to the manufacturer’s instructions.
Western blot analysis
The total proteins from H9c2 cells were extracted using RIPA buffer (Bio-Rad, CA, USA). The protein concentration was determined with Detergent Compatible Bradford Protein Assay Kit (Beyotime, Shanghai, China). The samples were separated by 10% SDS-PAGE (Bio-Rad, Hercules, CA) and transferred electrophoretically onto PVDF membranes (Millipore, USA). Then the membranes were incubated at 4°C overnight with the primary antibodies targeting: Bcl-2 (1:2000; ab182858, Abcam), Bax (1:1000; ab32503, Abcam), Cytochrome C (1:5000; ab133504, Abcam), GPX4 (1:1000; ab125066, Abcam), ASCL4 (1:1000; ab81150, Abcam), PI3K(1:1000; #4292, Cell Signaling Technology), AKT (1:1000; #9272, Cell Signaling Technology); p-PI3K (1:1000; #17,366, Cell Signaling Technology), p-AKT (1:1000; #9611, Cell Signaling Technology) and GAPDH (1:2500; ab9485, Abcam), and then incubated with the corresponding HRP-conjugated secondary antibody (1:2000; ab6721, Abcam) for 2 hours at room temperature. The bands were detected by using an ECL detection system (Beyotime Institute of Biotechnology, China) and the density of the bands was determined using ImageJ software (NIH, Bethesda, MD, USA) [Citation20].
Statistical analysis
The statistical analysis was performed using Graphpad Prism 8 (GraphPad Software, Inc.). All data were reported as mean ± standard deviation. The differences between groups were examined using one-way ANOVA followed by Bonferroni post hoc test. P values of less than 0.05 were considered statistically significant.
Results
In this study, we investigate the effects and the potential mechanism of LAP in DOX-induced H9c2 cells. The data revealed the administration of LAP caused the increased DOX-induced cardiotoxicity and potentiated mitochondria damage. In addition, LAP treatment exacerbate DOX-stimulated oxidative stress and ferroptosis in H9c2 cells. Mechanistic investigations showed that LAP in combination with DOX induced elevated level of oxidative stress and ferroptosis in H9c2 cells through inhibition of the PI3K/AKT pathway. Overall, our study presented a novel mechanism involved in ferroptosis for the synergistic cardiotoxicity of LAP-plus-DOX combination therapy, which may be associated with the PI3K/Akt signaling. These findings provided a better understanding of the synergistic toxicity of LAP-plus-DOX combined treatment.
LAP results in increased cytotoxicity in DOX-induced H9c2 cells in a dose-dependent manner
To investigate the effects of LAP on cardiac myocytes following DOX treatment, cell viability of H9c2 cells was measured firstly. As shown in , 1 μM DOX inhibited cell viability, and LAP further resulted in a dose-dependent decrease in cell viability. Additionally, the results from LDH activity experiment showed that the LDH level was dramatically increased by DOX treatment. LAP exacerbated the production of LDH under exposure of H9c2 cells to DOX for 24 h (). Subsequently, we detected the effects of LAP-plus-DOX combined treatment on cell apoptosis. As presented in and E, DOX exposure increased the rate of cell apoptosis, and the combined treatment of LAP-plus-DOX promoted more cell apoptosis when compared with DOX alone. Expectedly, western blot assay results revealed that LAP-plus-DOX combined treatment reduced the Bcl-2 level while enhanced the Bax level in H9c2 cells (), suggesting that LAP reinforced DOX-induced toxicity in cardiomyocytes.
LAP-plus-DOX combined treatment potentiates mitochondria damage in H9c2 cells
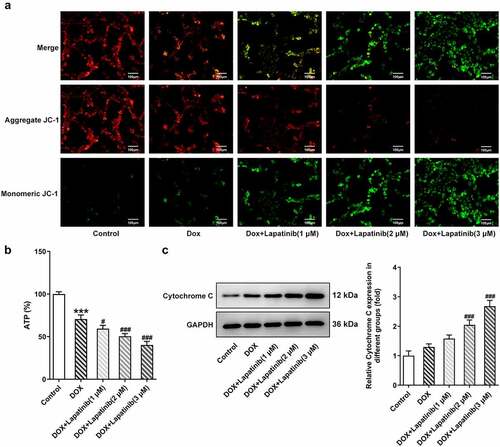
It has been reported that tyrosine-kinase inhibitors, including LAP, affect normal mitochondria structure and function [Citation21,Citation22]. With this in mind, we evaluated mitochondria injury in H9c2 cells. As shown in , aggregated JC-1 in mitochondria of normal cardiomyocytes exhibited red fluorescence, and exposure to 1 μM DOX increased dissipation of ΔΨm, with increased green fluorescence (monomeric JC-1) in mitochondria. In addition, combined treatment (LAP-plus-DOX) facilitated DOX-induced dissipation of ΔΨm in a concentration-dependent manner. Furthermore, the ATP level was found to be decreased in the DOX group compared to the control group, and a less level of ATP was shown in the combination group, as compared to the DOX group (). Also, an increase in the level of Cytochrome C was observed in H9c2 cells induced with DOX for 24 h in contrast to the control group. As expected, LAP treatment for 24 h after DOX exposure significantly stimulated the production of Cytochrome C (). These results indicate that LAP synergistically enhances DOX-induced toxicity by inducing mitochondrial dysfunction.
Figure 2. Effect of LAP on DOX-induced mitochondria damage in H9c2 cells. H9c2 cells were treated with 1 μM DOXwith or without 1–3 μM LAP for 24 h. A, MMP was identified by JC-1 staining. Red fluorescence represents the mitochondrial aggregate form of JC-1, indicating intact mitochondrial membrane potential. Green fluorescence represents the monomeric form of JC-1, indicating dissipation of ΔΨm. Original magnification: x200. B, ATP level in DOX-induced cells with or without different doses of LAP. C, Protein level of Cytochrome C was measured by western blot assay. LAP: Lapatinib; DOX: Doxorubicin; MMP: mitochondrial membrane potential. Data are expressed as mean ± SD calculated from three independent experiments. ***P < 0.001 versus control. #P < 0.05, ###P < 0.001 versus DOX
LAP aggravates DOX-induced oxidative stress and ferroptosis in H9c2 cells
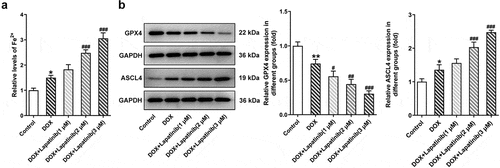
Mitochondria is the primary organelle that takes the center role in substance and energy metabolism, and iron metabolism in iron catabolic, anabolic and utilization pathways, which are associated with the process of oxidative stress and ferroptosis [Citation23]. Thus, we explore effects of mitochondrial dysfunction induced by LAP treatment on oxidative stress and ferroptosis in H9c2 cells. As shown in , the number of stained cells was remarkably increased following exposure of H9c2 cells to DOX for 24 h. Besides, LAP-plus-DOX caused an increase in ROS level compared with the treatment of DOX alone in a dose-dependent manner. Moreover, DOX induction significantly suppressed the generations of SOD, CAT and GSH compared to the control group, and the inhibitory effect was more obvious upon supplementation with LAP (). Then, ferroptosis in H9c2 cells was identified with detection of F2+ and western blot analysis. As depicted in , the content of F2+ in cells treated with DOX was increased and was further elevated in LAP-plus-DOX combined treatment. Similarly, data from western blotting demonstrated that, GPX4 level in DOX-stimulated cells, compared with that in the control cells, was greatly reduced while ASCL4 level was increased in a dose-dependent manner. In addition, exposure to LAP reduced DOX-induced decreased GPX4 level but increased ASCL4 level to a higher degree (). These findings suggest that LAP improves DOX-induced cell injury by aggravating oxidative stress and ferroptosis in H9c2 cells.
Figure 3. Effect of LAP on DOX-induced oxidative stress in H9c2 cells. H9c2 cells were treated with 1 μM DOXwith or without 1–3 μM LAP for 24 h. A, ROS level was detected by DCFH-DA staining. Original magnification: x200. The activity of SOD (b), CAT (c) and GSH (d) was assessed in DOX-induced cells with or without different doses of LAP. LAP: Lapatinib; DOX: Doxorubicin; ROS: Reactive oxygen species; SOD: superoxide dismutase; CAT: Catalase; GSH: Glutathione. Data are expressed as mean ± SD calculated from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus control. #P < 0.05, ##P < 0.01, ###P < 0.001 versus DOX
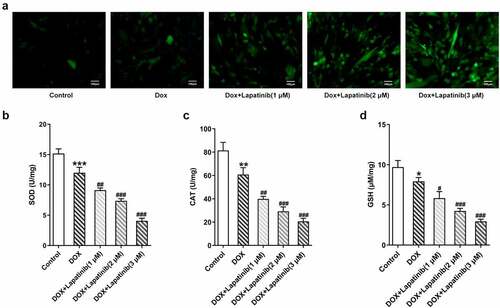
Figure 4. Effect of LAP on DOX-induced ferroptosis in H9c2 cells. H9c2 cells were treated with 1 μM DOXwith or without 1–3 μM LAP for 24 h. A, The level of Fe2+ in DOX-induced cells with or without different doses of LAP. B, Protein levels of GPX4 and ASCL4 were evaluated by western blot assay. LAP: Lapatinib; DOX: Doxorubicin; GPX4: Glutathione peroxidase 4; ASCL4: acyl-CoA synthetase long-chain family member 4. Data are expressed as mean ± SD calculated from three independent experiments. *P < 0.05, **P < 0.01 versus control. #P < 0.05, ##P < 0.01, ###P < 0.001 versus DOX
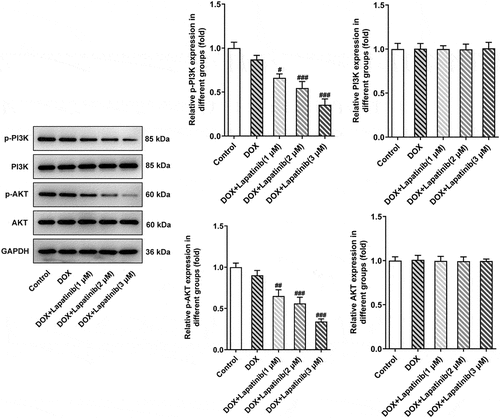
PI3K/AKT signaling pathway is inhibited by LAP upon exposure of H9c2 cells to DOX
To further understand the mechanism responsible for the synergistic toxicity of LAP-plus-DOX combined treatment, we detected the expression alteration of PI3K/AKT signaling after combining LAP-plus-DOX. According to , DOX alone restrained the phosphorylation of both PI3K and AKT, and LAP-plus-DOX combined treatment caused the lower expressions of p-PI3K and p-AKT compared with the DPX group. However, the total protein levels of PI3K and AKT were not affected significantly by DOX alone or combination of LAP-plus-DOX, suggesting that the treatment of LAP may potentiate DOX-induced cardiomyocyte injury and cytotoxicity via blocking the PI3K/AKT signaling pathway.
Figure 5. Effect of LAP on PI3K/AKT signaling pathway in DOX-induced H9c2 cells. H9c2 cells were treated with 1 μM DOXwith or without 1–3 μM LAP for 24 h. Protein levels of p-PI3K, PI3K, p-AKT and AKT were detected by western blot analysis. Data are expressed as mean ± SD calculated from three independent experiments. #P < 0.05, ##P < 0.01, ###P < 0.001 versus DOX
LAP in combination with DOX promotes mitochondria damage in H9c2 cells through inhibition of PI3K/AKT pathway
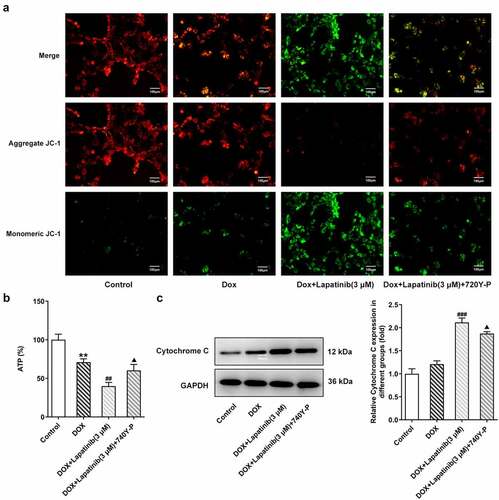
To investigate the role of PI3K/AKT in regulation of LAP in combination with DOX in H9c2 cells, 30 μM of 740Y-P was used to activate the PI3K/AKT pathway. As shown in , LAP-plus-DOX combined treatment significantly reduced aggregate JC-1 whereas increased monomeric JC-1, but 740Y-P abated the dissipation of ΔΨm, showing more red fluorescence and less green fluorescence. Additionally, the ATP level was undermined by combining LAP with DOX, while administration of 740Y-P reversed the decreased level of ATP in cells co-treated with LAP with DOX (). Subsequently, western blot assay revealed an increase in Cytochrome C level was caused by DOX and combination of LAP with DOX, but treatment of 740Y-P rehabilitated the level of Cytochrome C (). Thus, our results indicated that PI3K/AKT pathway is associated with mitochondria injury induced by LAP-plus-DOX combined treatment in H9c2 cells.
Figure 6. Effects of the activation of PI3K/AKT pathway on mitochondria damage in H9c2 cells with combined treatment of LAP-plus-DOX. 30 μM of 740Y-P was added to H9c2 cells with combined treatment of LAP-plus-DOX. A, MMP was identified by JC-1 staining. Original magnification: x200. B, ATP level in DOX/LAP-induced cells with or without 740Y-P. C, Protein level of Cytochrome C was measured by western blot assay. LAP: Lapatinib; DOX: Doxorubicin; MMP: mitochondrial membrane potential. Data are expressed as mean ± SD calculated from three independent experiments. **P < 0.01 versus control. ##P < 0.01, ###P < 0.001 versus DOX. ΔP < 0.05 versus DOX+Lapatinib (3 μM)
LAP in combination with DOX exacerbates oxidative stress and ferroptosis in H9c2 cells induced by DOX through inhibition of PI3K/AKT pathway
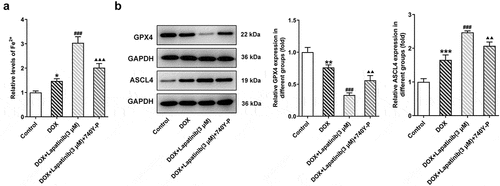
Finally, we investigated the role of PI3K/AKT pathway in oxidative stress and ferroptosis upon LAP-plus-DOX treatment. As illustrated in , following treatment with DOX and LAP, the ROS production was significantly increased but this effect was reversed by 740Y-P treatment. The levels of SOD, CAT and GSH were significantly abolished in the DOX group, especially in the combination group. However, the 740Y-P group showed the inverse results (). In addition, the content of F2+, compared with the DOX group, was revealed to be elevated by LAP-plus-DOX combined treatment, while the level was declining after 740Y-P was administrated (). Moreover, GPX4 expression was decreased and ASCL4 level was higher following DOX treatment or the combination therapy of LAP and DOX, but it was observed that 740Y-P preconditioning enhanced GPX4 expression while decreased ASCL4 expression (). The data indicate that LAP facilitates oxidative stress and ferroptosis in DOX-induced H9c2 cells through blocking the PI3K/AKT pathway.
Figure 7. Effects of the activation of PI3K/AKT pathway on oxidative stress in H9c2 cells with combined treatment of LAP-plus-DOX. 30 μM of 740Y-P was added to H9c2 cells with combined treatment of LAP-plus-DOX. A, ROS level was detected by DCFH-DA staining. Original magnification: x200. The activity of SOD (b), CAT (c) and GSH (d) was assessed in DOX/LAP-induced cells with or without 740Y-P. LAP: Lapatinib; DOX: Doxorubicin; ROS: Reactive oxygen species; SOD: superoxide dismutase; CAT: Catalase; GSH: Glutathione. Data are expressed as mean ± SD calculated from three independent experiments. **P < 0.01 versus control. ###P < 0.001 versus DOX. ΔΔΔP < 0.001 versus DOX+Lapatinib (3 μM)
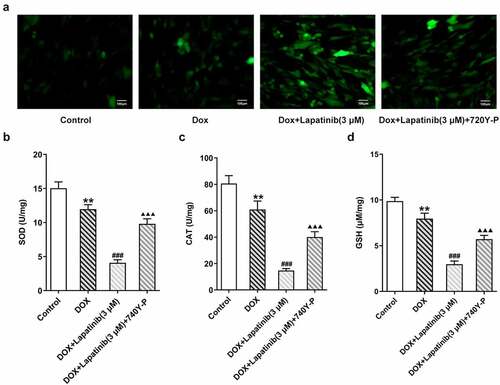
Figure 8. Effects of the activation of PI3K/AKT pathway on ferroptosis in H9c2 cells with combined treatment of LAP-plus-DOX. 30 μM of 740Y-P was added to H9c2 cells with combined treatment of LAP-plus-DOX. A, The level of Fe2+ in DOX/LAP-induced cells with or without 740Y-P. B, Protein levels of GPX4 and ASCL4 were evaluated by western blot assay. LAP: Lapatinib; DOX: Doxorubicin; GPX4: Glutathione peroxidase 4; ASCL4: acyl-CoA synthetase long-chain family member 4. Data are expressed as mean ± SD calculated from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus control. ###P < 0.001 versus DOX. ΔΔP < 0.01, ΔΔΔP < 0.001 versus DOX+Lapatinib (3 μM)
Discussion
In the present study, we examined the role of LAP in DOX-induced cardiotoxicity and the mechanism behind the synergistic cardiactoxicity of LAP-plus-DOX combined treatment. Compared to DOX alone, LAP in combination with DOX could increase the cytotoxicity and apoptotic death of H9c2 cells, and DOX-induced ΔΨm dissipation reinforced mitochondrial damage in myocardial cells, with the decreased ATP level and increased content of Cytochrome C. LAP also facilitated severe oxidative stress and ferroptosis, by regulating the activation of PI3K/AKT signaling pathway. The effect of pro-apoptosis, promoting mitochondrial dysfunction, oxidation, and ferroptosis were leading to LAP’s synergistic cytotoxic effect.
Although better anti-tumor effects exist upon combined treatment with LAP-plus-DOX, the cardiactoxicity due to combination therapy cannot be ignored [Citation24]. Mitochondria is the major target organelle of DOX-induced toxicity in cardiomyocytes [Citation25]. DOX has an accumulation in mitochondria, resulting in the greater mitochondrial concentration of DOX than the simultaneous serum concentration from clinical data [Citation26]. DOX used the enzymatic-related mechanism in mitochondrial respiratory chain and promotes the accumulation of ROS, which caused primarily DOX-induced cardiotoxicity [Citation27]. As reported by Childs et al., generation of ROS induced by DOX in mitochondrial leads to loss in mitochondrial membrane potential (MMP), direct activation of the mitochondrial permeability transition pore (MPTP), and the release of Cytochrome C [Citation28]. In addition, sufficient ATP production for sustaining normal cardiac contraction is needed fue to the high-energy-demand nature of the cardiac muscle requires [Citation29]. Wu et al. demonstrated that Dox significantly damaged mitochondrial function and ATP production in male rats [Citation30]. In this study, LAP treatment significantly potentiated DOX-induced H9c2 cell toxicity by increased LDH level and improved cell apoptosis, and caused mitochondria damage with dissipating MMP, reducing ATP level and enhancing Cytochrome C.
Cardiac cells show low antioxidant enzyme activity and are regarded as a main target for Dox-induced oxidative stress [Citation31]. In our study, exposure of H9c2 to Dox accelerated ROS production, and inhibited the activities of SOD, CAT and GSH which have strong anti-oxidative capacities. Meanwhile, LAP administration promoted the production of ROS whereas restrained the expressions of SOD, CAT and GSH, suggesting the acceleration of LAP in DOX-treated oxidative stress in cardiomyocytes, which was in line with previous results [Citation4,Citation32].
ROS generation is considered as one of the keys in regulating the process of ferroptosis [Citation33]. Ferroptosis is a novel iron-dependent regulated cell death (RCD) upon intracellular and occurs as a consequence of lethal lipid oxidation [Citation34–36]. Ferroptosis can be induced by the small molecule erastin, causing the inactivation of the glutathione peroxidase 4 (GPx4). Inactivation of GPx4 lost the ability to regulate the production of ROS, laeding to the accumulation of lipid peroxidation and eventually cell death [Citation37]. Acyl-CoA synthetase long-chain family member 4 (ACSL4) is an enzyme involved in the activation of polyunsaturated fatty acids and found to contribute to ferroptosis [Citation38]. ACSL4 expression has been used as a meaningful biomarker for monitoring ferroptosis [Citation39]. Furthermore, it has reported that DOX treatment induced features of typical ferroptotic cell death in cardiomyocytes of murine models of DOX induced cardiomyopathy [Citation40]. Besides, Ma et al. revealed that treatment with LAP synergistically induced ferroptosis in siramesine-treated breast cancer cells [Citation41]. Here, DOX treatment induced the production of Fe2+, reduced the protein expression of GPX4 but enhanced the acyl-CoA synthetase long-chain family member 4 (ASCL4) expression. LAP-plus-DOX combined treatment caused higher level of Fe2+, inhibitory expression of GPX4 and elevation of ASCL4 level, indicating LAP strengthened DOX-induced ferroptosis in cardiomyocytes.
Previous studies have showed that PI3K/Akt signaling is a key pathway of mitochondrial apoptosis induced by DOX [Citation42]. Li et al. also found that LAP can effectively inhibit PI3K/Akt signal transduction in non-small cell lung cancer [Citation43]. Thus we speculated the synergistic interaction of LAP and DOX may be associated with the PI3K/Akt signaling pathway. In the current study, 30 μM of 740Y-P was used as an agonist of PI3K/Akt pathway and added into DOX-induced or LAP-plus-DOX induced cells. We found that 740Y-P treatment significantly reversed the effects of LAP on mitochondrial dysfunction, oxidative stress and ferroptosis upon DOX induction. In addition, the effects of LAP explored in this study was at the cellular level, and we will verify our results in animal experiments and clinical trials in further study.
Conclusion
In conclusion, the results presented a novel mechanism involved in ferroptosis for the synergistic cardiotoxicity of LAP-plus-DOX combination therapy and these pernicious effects may be associated with the PI3K/Akt signaling, which contributes to our understanding of the mechanism behind the synergistic toxicity of LAP-plus-DOX combined treatment and provides targets for cardiactoxicity induced by LAP-plus-DOX combination therapy.
Authors’ contributions
LS and XL designed the research, drafted and revised the manuscript. LS, HW, DX and SY performed the experiments. HW and LZ searched the literature and analyzed the data. XL guided the experiments. All authors read and approved the final manuscript.
Highlights
Lapatinib increases doxorubicin-induced cytotoxicity and mitochondria damage in cardiomyocytes.
Lapatinib potentiates oxidative stress and ferroptosis in doxorubicin-induced cardiomyocytes.
Lapatinib blocks the PI3K/Akt signaling pathway in doxorubicin-induced cardiomyocytes.
Lapatinib regulates doxorubicin-induced cardiomyocytes through inhibiting the PI3K/Akt signaling pathway.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Carvalho FS, Burgeiro A, Garcia R, et al. Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. 2014;34(1):106–135.
- Shabalala S, Muller CJF, Louw J, et al. Polyphenols, autophagy and doxorubicin-induced cardiotoxicity. Life Sci. 2017;180:160–170.
- Deng J, Huang M, Wu H. Protective effect of limonin against doxorubicin-induced cardiotoxicity via activating nuclear factor - like 2 and Sirtuin 2 signaling pathways. Bioengineered. 2021;12(1):7975–7984.
- Songbo M, Lang H, Xinyong C, et al. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol Lett. 2019;307:41–48.
- Wenningmann N, Knapp M, Ande A, et al. Insights into doxorubicin-induced cardiotoxicity: molecular mechanisms, preventive strategies, and early monitoring. Mol Pharmacol. 2019;96(2):219–232.
- D’Amato V, Raimondo L, Formisano L, et al. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev. 2015;41(10):877–883.
- Bonnefoi H, Jacot W, Saghatchian M, et al. Neoadjuvant treatment with docetaxel plus lapatinib, trastuzumab, or both followed by an anthracycline-based chemotherapy in HER2-positive breast cancer: results of the randomised phase II EORTC 10054 study. Ann Oncol. 2015;26(2):325–332.
- Hasinoff BB, Patel D, Wu X. The dual-targeted HER1/HER2 tyrosine kinase inhibitor lapatinib strongly potentiates the cardiac myocyte-damaging effects of doxorubicin. Cardiovasc Toxicol. 2013;13(1):33–47.
- Yang B, Papoian T. Tyrosine kinase inhibitor (TKI)-induced cardiotoxicity: approaches to narrow the gaps between preclinical safety evaluation and clinical outcome. J Appl Toxicol. 2012;32(12):945–951.
- Wang H, Li F, Du C, et al. Doxorubicin and lapatinib combination nanomedicine for treating resistant breast cancer. Mol Pharm. 2014;11(8):2600–2611.
- Hsu WT, Huang CY, Yen CYT, et al. The HER2 inhibitor lapatinib potentiates doxorubicin-induced cardiotoxicity through iNOS signaling. Theranostics. 2018;8:3176–3188.
- Zhao L, Qi Y, Xu L, et al. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018;15:284–296.
- Jiang Y, Zhang Q. Catalpol ameliorates doxorubicin-induced inflammation and oxidative stress in H9C2 cells through PPAR-γ activation. Exp Ther Med. 2020;20:1003–1011.
- Zhao L, Tao X, Qi Y, et al. Protective effect of dioscin against doxorubicin-induced cardiotoxicity via adjusting microRNA-140-5p-mediated myocardial oxidative stress. Redox Biol. 2018;16:189–198.
- Li HR, Wang C, Sun P, et al. Melatonin attenuates doxorubicin-induced cardiotoxicity through preservation of YAP expression. J Cell Mol Med. 2020;24:3634–3646.
- Vishnu KV, Ajeesh Kumar KK, Chatterjee NS, et al. Sardine oil loaded vanillic acid grafted chitosan microparticles, a new functional food ingredient: attenuates myocardial oxidative stress and apoptosis in cardiomyoblast cell lines (H9c2). Cell Stress Chaperones. 2018;23(2):213–222.
- Subbarao RB, Ok SH, Lee SH, et al. Lipid emulsion inhibits the late apoptosis/cardiotoxicity induced by doxorubicin in rat cardiomyoblasts. Cells. 2018;7:144.
- Helal M, Alcorn J, Bandy B. Doxorubicin cytotoxicity in differentiated H9c2 cardiomyocytes: evidence for acute mitochondrial superoxide generation. Cardiovasc Toxicol. 2021;21(2):152–161.
- Xia Y, Chen Z, Chen A, et al. LCZ696 improves cardiac function via alleviating Drp1-mediated mitochondrial dysfunction in mice with doxorubicin-induced dilated cardiomyopathy. J Mol Cell Cardiol. 2017;108:138–148.
- Yu W, Qin X, Zhang Y, et al. Curcumin suppresses doxorubicin-induced cardiomyocyte pyroptosis via a PI3K/Akt/mTOR-dependent manner. Cardiovasc Diagn Ther. 2020;10(4):752–769.
- Paech F, Bouitbir J, Krähenbühl S. Hepatocellular toxicity associated with tyrosine kinase inhibitors: mitochondrial damage and inhibition of glycolysis. Front Pharmacol. 2017;8:367.
- Rodríguez-Hernández MA, de la Cruz-Ojeda P, López-Grueso MJ, et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020;36:101510.
- Wang H, Liu C, Zhao Y, et al. Mitochondria regulation in ferroptosis. Eur J Cell Biol. 2020;99(1):151058.
- Raschi E, Vasina V, Ursino MG, et al. Anticancer drugs and cardiotoxicity: insights and perspectives in the era of targeted therapy. Pharmacol Ther. 2010;125(2):196–218.
- Shi W, Deng H, Zhang J, et al. Mitochondria-targeting small molecules effectively prevent cardiotoxicity induced by doxorubicin. Molecules. 2018;23(6):1486.
- Tadokoro T, Ikeda M, Ide T, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5. DOI:10.1172/jci.insight.132747
- Shadle SE, Bammel BP, Cusack BJ, et al. Daunorubicin cardiotoxicity: evidence for the importance of the quinone moiety in a free-radical-independent mechanism. Biochem Pharmacol. 2000;60:1435–1444.
- Childs AC, Phaneuf SL, Dirks AJ, et al. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2: baxratio. Cancer Res. 2002;62:4592–4598.
- Tewari SG, Bugenhagen SM, Palmer BM, et al. Dynamics of cross-bridge cycling, ATP hydrolysis, force generation, and deformation in cardiac muscle. J Mol Cell Cardiol. 2016;96:11–25.
- Wu R, Wang HL, Yu HL, et al. Doxorubicin toxicity changes myocardial energy metabolism in rats. Chem Biol Interact. 2016;244:149–158.
- Zhang HS, Wang SQ. Nrf2 is involved in the effect of tanshinone IIA on intracellular redox status in human aortic smooth muscle cells. Biochem Pharmacol. 2007;73:1358–1366.
- Segredo MP, Salvadori DM, Rocha NS, et al. Oxidative stress on cardiotoxicity after treatment with single and multiple doses of doxorubicin. Hum Exp Toxicol. 2014;33:748–760.
- Mou Y, Wang J, Wu J, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34.
- Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379.
- Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285.
- Zheng B, Zhou X, Pang L, et al. Baicalin suppresses autophagy-dependent ferroptosis in early brain injury after subarachnoid hemorrhage. Bioengineered. 2021;12(1):7794–7804.
- Alim I, Caulfield JT, Chen Y, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262–1279. e1225.
- Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343.
- Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98.
- Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680.
- Ma S, Henson ES, Chen Y, et al. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307.
- Jia Y, Zuo D, Li Z, et al. Astragaloside IV inhibits doxorubicin-induced cardiomyocyte apoptosis mediated by mitochondrial apoptotic pathway via activating the PI3K/Akt pathway. Chem Pharm Bull (Tokyo). 2014;62:45–53.
- Li B, Ding CM, Li YX, et al. Over-regulation of microRNA-133b inhibits cell proliferation of cisplatin-induced non-small cell lung cancer cells through PI3K/Akt and JAK2/STAT3 signaling pathway by targeting EGFR. Oncol Rep. 2018;39:1227–1234.