
ABSTRACT
Chronic myeloid leukemia (CML) is a malignant myeloproliferative tumor. 2-Methoxyestradiol (2-ME) is an endogenous estrogen metabolite that shows efficacy in human malignancies. Ascorbic acid (AA) possesses antioxidant activity. This study explored the mechanism of 2-ME combined with AA in the apoptosis of CML cells. Firstly, human CML cell lines were treated with 2-ME and AA. The cell viability, apoptosis, reactive oxygen species (ROS), and mitochondrial membrane potential (MMP) were detected. miR-223 expression in CML cells was detected. In addition, CML cells were transfected with miR-223 inhibitor. The binding relationship between miR-223 and FLT3 was verified. Subsequently, the FLT3 was overexpressed or silenced for the function rescue experiment to confirm the role of FLT3 in CML cell apoptosis. The expression levels of key factors of the PI3K/AKT pathway were detected. Finally, xenograft nude mouse models were established for in vivo verification. 2-ME + AA treatment inhibited CML cell viability and promoted apoptosis, elevated ROS content, and reduced MMP. 2-ME + AA treatment promoted miR-223 expression in CML cells. miR-223 targeted FLT3. Moreover, miR-223 inhibitor or FLT3 overexpression partially annulled the effect of 2-ME + AA on CML cells. 2-ME + AA inhibited the PI3K/AKT pathway via the miR-223/FLT3 axis. Furthermore, 2-ME + AA suppressed CML xenograft growth in mice. Collectively, 2-ME + AA promoted miR-223 expression and suppressed FLT3 and the PI3K/AKT pathway, thereby facilitating the apoptosis of CML cells and inhibiting CML xenograft growth in mice.
Graphical abstract
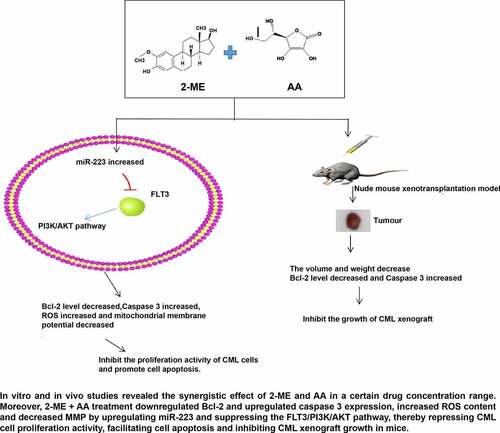
1. Introduction
Chronic myeloid leukemia (CML) is regarded as a clonal myeloproliferative disease that is featured by a chromosomal translocation that produces breakpoint cluster region-Abelson (BCR-ABL) oncogene encoding constitutive kinase activity [Citation1]. BCR-ABL kinase triggers the excessive production and expansion of leukocytes in the bone marrow and eventually crowns out the normal cells in the bone marrow niche [Citation2]. CML is triphasic: most patients are in the chronic phase with easily controllable symptoms, but they may experience an unstable period called acceleration in the absence of effective intervention, which eventually leads to a blast crisis [Citation3]. Up to 50% of CML patients scarcely exhibit obvious symptoms at the chronic phase and can only be diagnosed by blood routine examination such as thrombocytosis and leukocytosis [Citation4]. Although tyrosine kinase inhibitors targeting BCR-ABL possess remarkable efficacy, some patients barely respond or relapse after the initial response, leading to a dismal prognosis [Citation5]. Therein, the elucidation of the CML mechanism and exploration of potent therapeutic agents are of great significance.
Apoptosis is a type of programmed cell death that enables the damaged cells to be removed orderly and effectively [Citation6]. The deregulation of apoptosis is related to unchecked cell proliferation, tumor progression, and drug resistance, and consequently, deregulation of apoptosis is viewed as a hallmark of human malignancies, including CML [Citation7,Citation8]. Deregulation of the apoptotic pathway contributes to the pathophysiology of hematologic malignancies, including CML [Citation9,Citation10]. Therefore, targeting the molecules implicated in apoptotic resistance becomes a promising strategy to restore the sensitivity of CML cells to apoptosis and improve the therapeutic effect [Citation11].
2-methoxyestradiol (2-ME), a bioactive metabolite of 17β-estradiol, has attracted increasing concerns due to its notably anti-tumor activity, and the application of 2-ME has achieved some promising results in clinical studies [Citation12]. The effect of 2-ME on tumor suppression is attributed to the induction of apoptosis and the repression of superoxide dismutase (SOD), and inhibiting SOD causes free radical-mediated mitochondrial membrane damage, eventually resulting in apoptosis [Citation13]. 2-ME induces CML cell apoptosis in a concentration-dependent manner. These findings above indicate that 2-ME bears a broad application prospect in the treatment of CML [Citation14,Citation15]. Ascorbic acid (AA) serves as an electron donor that keeps iron in a ferrous state, thus keeping the full activity of collagen hydroxylase [Citation16]. AA alone or in combination with clinical drugs (such as gefitinib, carboplatin, and paclitaxel) can play a beneficial role in various human tumor models [Citation17]. The recommended intake of AA for healthy people is 120 mg/day and when the daily oral dose of AA is more than 250 mg, the plasma concentration of AA will maintain a stable level of 80 μmol/L; when the required dose of AA exceeds the maximum oral plasma concentration, the plasma concentration of AA can be increased by intravenous injection [Citation18]. The anti-tumor effect of AA involves two prominent mechanisms: oxidative stress induced by hydrogen peroxide and DNA demethylation mediated by TET activation [Citation19]. AA functions as an antioxidant at the physiological concentration, but its therapeutic effect at pharmacological dose seems to be concerned with the pro-oxidant effect, ultimately facilitating tumor cell death [Citation20]. AA combined with menadione bisulfite has been demonstrated to kill CML cells and repress tumor growth in mice [Citation21]. The activity of caspase 3 and the efficacy of nilotinib (a specific drug for CML) can be indirectly evaluated by monitoring the changes of photocurrent induced by AA [Citation22].
However, whether 2-ME combined with AA can produce a therapeutic effect on CML remains unknown. We speculated that 2-ME + AA might promote CML cell apoptosis via miR-223. Through in vitro and in vivo studies, 2-ME + AA treatment was found to downregulate B-cell leukemia/lymphoma-2 (Bcl-2) and upregulate caspase 3 expression, increase reactive oxygen species (ROS) content and decrease mitochondrial membrane potential (MMP) by upregulating miR-223 and suppressing the Fms-like tyrosine kinase 3/phosphatidylinositol-3 kinase/protein kinase B (FLT3/PI3K/AKT) pathway, thereby repressing CML cell proliferation activity, facilitating cell apoptosis and inhibiting CML xenograft growth in mice. This study shall confer a novel theoretical basis for the management of CML.
2. Materials and Methods
2.1. Ethics statement
All conducted animal experiments were approved by the animal ethics committee. Considerable efforts were made to minimize the number of animals and their pains.
2.2. Cell culture
Human CML cells lines (K562 and KCL22) were purchased from American Type Culture Collection (Manassas, VA, USA) and cultured in Roswell Park Memorial Institute (-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (Thermo Fisher) and 1% penicillin-streptomycin (Beyotime, Shanghai, China) at 37°C/5% CO2. The cells were cultured in 2-ME (0, 2, 4, 6, 8 and 10 μM) and AA (0, 25, 50, 62.5, 75, and 87.5 μM). A same amount of dimethyl sulfoxide (DMSO) was used as the control. AA, 2-ME, and DMSO were provided by Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
2.3 (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability under different treatments for 48 h was evaluated using MTT assay to calculate half maximal inhibitory concentration (IC50) value. Growth inhibition = (optical density (OD) control-OD treated sample)/OD control × 100% [Citation23].
The K562 and KCL22 cells were seeded into 96-well plates (8 × 103 cells/well). After 24 h, cells were treated with different concentrations of 2-ME (0, 2, 4, 6, 8 and 10 μM) and AA (0, 25, 50, 62.5, 75 and 87.5 μM). After 48 h, 20 μL MTT (5 mg/mL) solution was dripped into each well for 4 h-incubation at 37°C. Then, the medium was discarded and 150 μL DMSO was supplemented. The plates were shaken in dark conditions for 10 min. The OD value at 490 nm was measured using a microplate reader (Bio-Rad, Hercules, CA, USA). The experiment was replicated 3 times.
2.4. Cell transfection
K562 and KCL22 cells were seeded into 6-well plates (2 × 105 cells/well). After 24 h-incubation, cells were transfected with inhibitor NC, miR-223 inhibitor, pcDNA3.1-NC, pcDNA3.1-FLT3, si-NC, or si-FLT3 (Genechem Co., Ltd., Shanghai, China) (siRNA 50 nM, miRNA-inhibitor 30 nM) using Lipofectamine 2000 (11668–019, Invitrogen, Carlsbad, CA, USA).
The cells were allocated into blank group, DMSO group, 2-ME group, AA group, 2-ME + AA group, 2-ME + AA + inhibitor NC group, 2-ME + AA + miR-223 inhibitor group, 2-ME + AA + pcDNA3.1-NC group, 2-ME + AA + pcDNA3.1-FLT3 group, 2-ME+AA+miR-223 inhibitor+si-NC group, and 2-ME+AA+miR-223 inhibitor+si-FLT3 group. The concentrations of 2-ME and AA were the corresponding IC50 values.
2.5. Flow cytometry
The cells were collected by trypsin detachment and incubated with 100 μL binding buffer, 5 μL Annexin V-fluorescein isothiocyanate, and 5 μL propidium iodide for 10 min. The cells were resuspended in 400 μL binding buffer, and apoptosis rate was detected by FC500 MCL flow cytometer.
2.6. Xenograft nude mouse model
Total 32 male BALB/C nude mice (6-week-old, 20 ± 2 g) provided by Charles River (Beijing, China) were fed in a sterile animal room with 45% humidity at 25°C under 12-h light cycles of 8:00–20:00, with free access to food and water.
Subsequently, the cell suspension of 1 × 107 K562 cells resuspended at 50 mL phosphate-buffered saline (PBS) and 50 mL matrix gel (356234; BD Biosciences, San Jose, CA, USA) was subcutaneously injected into the left dorsal side of BALB/c nude mice. After 48 h, the mice were arbitrarily assigned into the following 4 groups (N = 8 per group): 2-ME group: intravenously injected with 280 mg/kg 2-ME [Citation24]; AA group: intravenously injected with 1 g/kg AA [Citation21]; 2-ME + AA group: intravenously injected with 280 mg/kg 2-ME and 1 g/kg AA; DMSO group: intravenously injected with an equivalent amount of DMSO. After a week, mice were euthanized by intraperitoneally injecting 100 mg/kg pentobarbital sodium. Next, the tumor tissues were removed, weighed, and photographed. The tumor volume was measured with a caliper following the formula: tumor volume = a2 × b × 0.4 (a, the minimum diameter; b, the diameter perpendicular to a). Tissue homogenates were made and then stored at −80°C.
2.7. Detection of reactive oxygen species (ROS)
ROS content in K562 and KCL22 cells was detected by dihydroethidium (DHE) dyeing (Beyotime). The cells were incubated with DHE dyeing at 37°C in dark conditions for 30 min and then observed under the fluorescence microscope (Leica, Wetzlar, Germany).
2.8. Detection of mitochondrial membrane potential (MMP)
MMP was measured by JC-1 staining. The cells or tissue homogenates were incubated with the JC-1 working solution at 37°C in the dark for 30 min. The supernatant was removed, and the cells were washed twice with PBS and observed under the fluorescence microscope (Leica).
2.9. Western blotting
Total protein was extracted from the K562 and KCL22 cells or tissue homogenates using radio-immunoprecipitation lysis buffer (Beyotime) and quantified using the bicinchoninic acid assay kit (Beyotime). The protein was separated by electrophoresis on 8–12% sodium dodecylsulfate-polyacrylamide gel and moved onto the polyvinylidene fluoride membranes, which were blocked with 3% bovine serum albumin, and then incubated with the primary antibodies and horseradish peroxidase (HRP)-labeled secondary antibody (1:5000, A11375/A11374, ThermoFisher). The bands were detected using the image analyzer Tanon 5200 (Shanghai, China). The primary antibodies were as follows: Bcl-2 (1:500, BMS1028, ThermoFisher), caspase 3 (1:500, 43–7800, ThermoFisher), p-PI3K (1:1000, PA5-37820, ThermoFisher), p-AKT (1:1000, PA5-36728, ThermoFisher), and β-actin (1:1000, PA1-183-HRP, ThermoFisher).
2.10. Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol reagent and reversely transcribed into cDNA using M-MLV reverse transcriptase (Clontech, Palo Alto, CA, USA) and PrimeScript® miRNA cDNA synthesis kit (TaKaRa, Dalian, China). RT-qPCR was performed on the instrument (7500, ABI Inc., Foster City, CA, USA), with GAPDH and U6 as internal references. The expressions of miR-223 and FLT3 were calculated based on the 2-ΔΔCT method [Citation25]. The experiment for each sample was repeated 3 times independently. PCR primers are shown in .
Table 1. Primer sequence for RT-qPCR
2.11. Dual-luciferase reporter gene assay
The binding site of miR-223 and FLT3 was predicted through Jefferson online website (https://cm.jefferson.edu/). The wild-type (FLT3-WT) and mutant-type (FLT3-MUT) luciferase plasmids were constructed by cloning the binding sequence and mutant sequence into the luciferase vector pGL3 (Promega Corporation, Madison, WI, USA) respectively. The constructed plasmids were cotransfected with miR-233 mimic or mimic NC into HEK293T cells. The cells were collected and lysed after 48 h, and the luciferase activity was evaluated using the kit (Promega).
2.12. Statistical analysis
Data were analyzed and introduced using SPSS 21.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, USA). Data are expressed as mean ± standard deviation. One-way analysis of variance (ANOVA) was employed for the comparisons among multiple groups, following Tukey’s multiple comparisons test. The p < 0.05 was indicative of statistical significance.
3. Results
We conjectured that 2-ME + AA might facilitate CML cell apoptosis by miR-223. In vitro and in vivo studies revealed that 2-ME + AA reduced Bcl-2 and elevated caspase 3, increased ROS content, and decreased MMP by upregulating miR-223 and inhibiting the FLT3/PI3K/AKT pathway, thus suppressing CML cell proliferation activity, promoting cell apoptosis, and repressing CML xenograft growth in mice.
3.1. 2-ME + AA promoted apoptosis of CML cells
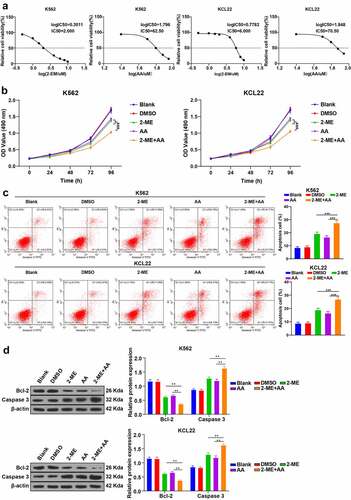
As regulators of the oxidative stress pathway, 2-ME and AA promote apoptosis of CML cells [Citation15,Citation21]. However, the therapeutic effect of 2-ME + AA on CML remains unclear. To explore the function of 2-ME + AA on CML cells, we cultured human CML cell lines (K562 and KCL22) in gradient concentrations of 2-ME and AA, detected the cell viability using MTT assay, and determined the IC50 values of 2-ME (2.00 μM/6.00 μM) and AA (62.50 μM/70.50 μM) in K562 and KCL22 cells for the follow-up experiments ()). The results unveiled that 2-ME + AA treatment significantly reduced cell viability and enhanced apoptosis rate of K562 and KCL22 cells, decreased Bcl-2, and elevated caspase 3 (all p < 0.01; ). Altogether, 2-ME + AA facilitated CML cell apoptosis.
Figure 1. 2-ME combined with AA promotes CML cell apoptosis. K562 and KCL22 cells were treated with 2-ME and AA. (a) Viability of cells treated with different concentrations of 2-ME and AA for 48 h was measured using MTT assay, and IC50 was calculated. IC50 doses of 2-ME and AA were used for subsequent cell treatment. (b) Cell proliferation activity at different time points examined using MTT assay. (c) Cell apoptosis measured using flow cytometry. (d) Levels of Bcl-2 and Caspase 3 measured using Western blotting. Cell experiment was conducted 3 times independently. Data were described as mean ± standard deviation. Data were analyzed using one-way ANOVA, followed by Tukey’s multiple comparison test, **p < 0.01 vs. DMSO group.
3.2. 2-ME + AA promoted apoptosis by increasing ROS and decreasing MMP
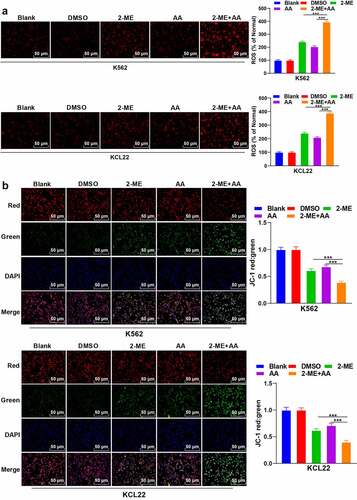
Apoptosis is implicated with the changes of intracellular ROS and MMP. 2-ME can induce the increase of ROS and the loss of MMP [Citation26]. AA increases ROS content and facilitates hepatoma cell apoptosis induced by arsenic trioxide through oxidative pathway [Citation27]. Therefore, we speculated that 2-ME + AA affected apoptosis by influencing ROS and MMP. Then, we measured the levels of ROS and MMP (ΔΨm) in CML cells, and compared with the 2-ME or AA alone group, 2-ME + AA significantly enhanced ROS and reduced MMP (p < 0.001; ). Briefly, 2-ME + AA promoted CML cell apoptosis by increasing ROS and decreasing MMP.
Figure 2. 2-ME combined with AA promotes apoptosis by increasing ROS content and decreasing MMP. K562 and KCL22 cells were treated with 2-ME and AA. (a) Content of ROS. (b) Change of MMP. Cell experiment was conducted 3 times independently. Data were described as mean ± standard deviation and analyzed using one-way ANOVA, followed by Tukey’s multiple comparison test, **p < 0.01.
3.3. 2-ME + AA upregulated intracellular miR-223 expression
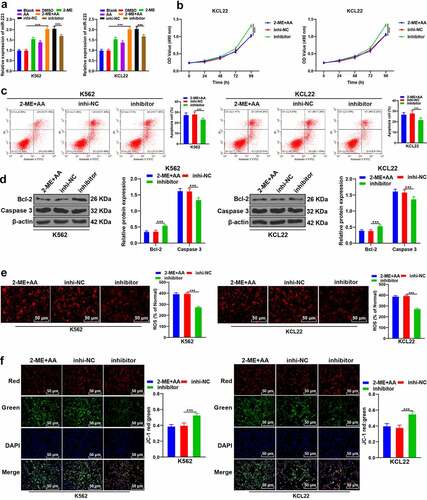
2-ME can relieve pulmonary hypertension induced by chronic intermittent hypoxia by regulating miR-223 [Citation28]. miR-223 expression is under-expressed in CML [Citation29]. Therefore, we speculated that 2-ME + AA regulated the apoptosis of CML cells through miR-223. As RT-qPCR results showed, compared with the 2-ME or AA alone group, 2-ME + AA notably upregulated miR-223 expression in K562 and KCL22 cells (p < 0.001; ). Next, we further confirmed the role of miR-223 in CML by transfecting miR-223 inhibitor into 2-ME + AA-treated K562 and KCL22 cells. RT-qPCR results confirmed the transfection efficiency (p < 0.001; ). miR-223 inhibitor markedly enhanced the proliferation ability and reduced apoptosis rate of CML cells (p < 0.001; ), and elevated Bcl-2 and declined caspase 3 expression (p < 0.001; ). Moreover, the ROS content was decreased and MMP was increased after miR-223 inhibitor transfection (p < 0.001; ). Taken together, 2-ME + AA promoted the apoptosis of CML cells by upregulating miR-223.
Figure 3. 2-ME combined with AA upregulates intracellular miR-223 expression. 2-ME + AA treated K562 and KCL22 cells were delivered with miR-223 inhibitor, with inhibitor NC as control. (a) miR-223 expression tested using RT-qPCR. (b) Cell proliferation at different times measured using MTT assay. (c) Cell apoptosis measured using flow cytometry. (d) Protein levels of Bcl-2 and Caspase 3 measured using Western blotting. (e) Content of ROS. F: Change of MMP. Cell experiment was conducted 3 times independently. Data were described as mean ± standard deviation. Data were analyzed using one-way ANOVA, followed by Tukey’s multiple comparison test, ***p < 0.001. inhi-NC: 2-ME + AA + inhibitor NC; inhibitor: 2-ME + AA + miR-223 inhibitor.
3.4. miR-223 targeted FLT3
To explore the downstream mechanism of miR-223 in promoting CML cell apoptosis, we predicted the targets of miR-223 through the Jefferson website (https://cm.jefferson.edu/). Among them, FLT3 is highly expressed in CML [Citation30]. According to the binding site of miR-223 and FLT3 ()), the dual-luciferase assay was designed and confirmed the binding relation between miR-223 and FLT3 (p < 0.001; )). FLT3 expression in K562 cells was remarkably reduced after 2-ME + AA treatment and increased after further miR-223 inhibitor transfection (p < 0.001; )). Briefly, miR-223 targeted FLT3.
Figure 4. miR-223 targets FLT3. (a) Binding site of miR-223 and FLT3 predicted through Jefferson website. (b) Binding relation between miR-223 and FLT3 verified using dual-luciferase assay. (c) FLT3 expression in K562 and KCL22 cells tested using RT-qPCR. Cell experiment was conducted 3 times independently. Data were described as mean ± standard deviation. Data in panel C were analyzed using one-way ANOVA, and data in panel B were analyzed using independent t test, following Tukey’s multiple comparison test, ***p < 0.001. inhi-NC: 2-ME + AA + inhibitor NC; inhibitor: 2-ME + AA + miR-223 inhibitor.
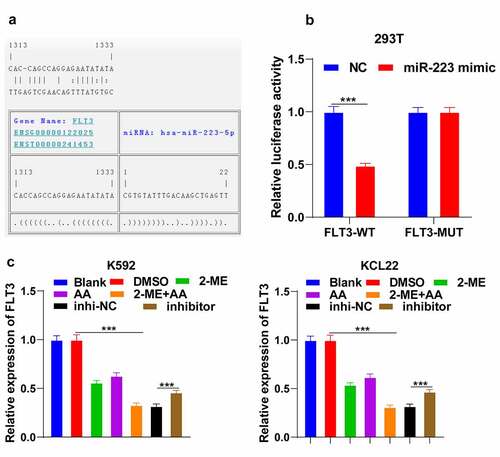
3.5. Upregulation of FLT3 partially reversed the promoting effect of 2-ME + AA on apoptosis of K562 cells
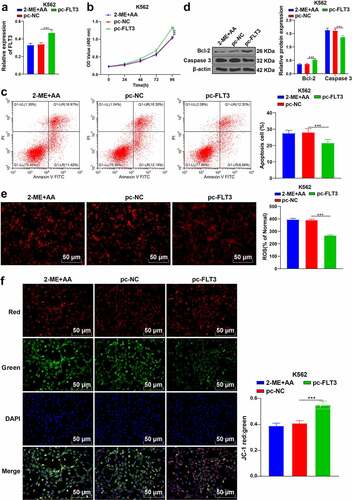
To further determine the function of FLT3 in CML, we successfully upregulated FLT3 expression in K562 cells treated with 2-ME + AA (p < 0.001, )), and found that cell proliferation activity was notably enhanced and apoptosis rate was reduced (p < 0.001; , c)); Bcl-2 expression was elevated and caspase 3 was declined (p < 0.001; )). Additionally, ROS content was decreased and MMP was increased (p < 0.001; ). Taken together, overexpression of FLT3 attenuated the effect of 2-ME + AA on CML cell apoptosis.
Figure 5. Upregulation of FLT3 partially reverses the promoting effect of 2-ME + AA on apoptosis of K562 cells. 2-ME + AA treated K562 cells were delivered with pcDNA3.1-FLT3, with pcDNA3.1-NC as control. (a) FLT3 expression tested using RT-qPCR. (b) Cell proliferation at different times measured using MTT assay. (c) Cell apoptosis measured using flow cytometry. (d) Protein levels of Bcl-2 and Caspase 3 examined using Western blotting. (e) Content of ROS. (f) Change of MMP. Cell experiment was conducted 3 times independently. Data were described as mean ± standard deviation. Data were analyzed using one-way ANOVA, followed by Tukey’s multiple comparison test, ***p < 0.001. pc-NC: 2-ME + AA + pcDNA3.1-NC; pc-FLT3: 2-ME + AA + pcDNA3.1-FLT3.
3.6. 2-ME + AA repressed the PI3K/AKT pathway via the miR-223/FLT3 axis
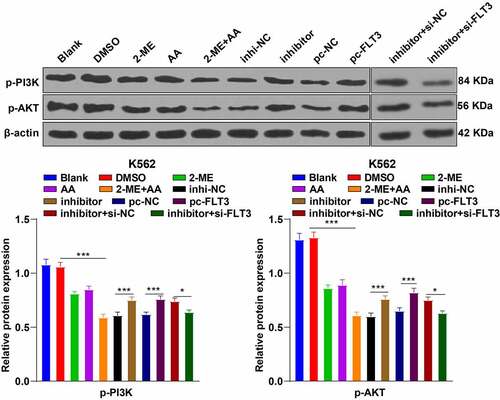
The previous experiments had confirmed that 2-ME + AA promoted K562 cell apoptosis via the miR-233/FLT3 axis. FLT3 mediates the PI3K/Akt pathway transduction in acute myeloid leukemia (AML) [Citation31], and the PI3K/AKT pathway participates in CML as a typical apoptosis-related pathway [Citation32]. Therefore, we speculated that the PI3K/AKT pathway participated in K562 cell apoptosis induced by 2-ME + AA. The results revealed that compared with the 2-ME or AA alone group, 2-ME + AA treatment suppressed the levels of p-PI3K and p-AKT, which was partially annulled by miR-223 inhibitor or pcDNA3.1-FLT3. Finally, K562 cells treated with 2-ME + AA, miR-223 inhibitor, and si-FLT3 exhibited reduced levels of p-PI3K and p-AKT (all p < 0.05; ). In brief, 2-ME + AA promoted K562 cell apoptosis via the miR-233/FLT3/PI3K/AKT axis.
Figure 6. 2-ME combined with AA repressed the PI3K/AKT pathway via the miR-223/FLT3 axis. Protein levels of the PI3K/AKT pathway were tested using Western blotting. Cell experiment was conducted 3 times independently. Data were described as mean ± standard deviation and analyzed using one-way ANOVA and Tukey’s multiple comparison test, ***p < 0.001, *p < 0.05. inhi-NC: 2-ME + AA + inhibitor NC; inhibitor: 2-ME + AA + miR-223 inhibitor; pc-NC: 2-ME + AA + pcDNA3.1-NC; pc-FLT3: 2-ME + AA + pcDNA3.1-FLT3; inhibitor+si-NC: 2-ME + AA + miR-223 inhibitor + si-NC; inhibitor+si-FLT3: 2-ME + AA + miR-223 inhibitor + si-FLT3.
3.7. 2-ME + AA inhibited CML xenograft growth in mice
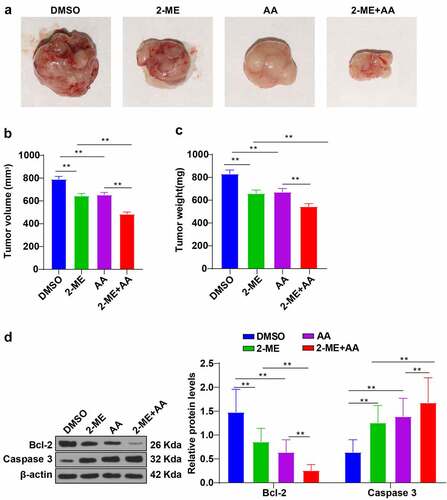
Next, we established xenograft nude mouse models to further verify the role of 2-ME + AA in CML cells. The results unraveled that 2-ME or AA alone notably decreased the tumor volume and weight of mice, and 2-ME + AA treatment augmented these anti-tumor effects (p < 0.01; ). Western blotting revealed that 2-ME or AA alone prominently lowered Bcl-2 and elevated caspase 3 expression, and 2-ME + AA treatment further amplified the changes (p < 0.01; )). Collectively, 2-ME + AA suppressed CML xenograft growth in mice.
Figure 7. 2-ME combined with AA inhibited CML xenograft growth in mice. CML xenograft nude mouse models were established, and then intravenously injected with 2-ME, AA alone, or a combination of 2-ME and AA. (a) Image of mouse tumors. (b) Average tumor volume. (c) Average tumor weight. (d) Protein levels of Bcl-2 and Caspase 3 examined using Western blotting. N = 8, data were presented as mean ± standard deviation. Data among multiple groups were analyzed using one-way ANOVA, followed by Tukey’s multiple comparison test, **p < 0.01.
4. Discussion
Leukemia is common blood cancer, whose treatment usually requires chemotherapy, radiotherapy, and bone marrow transplantation to induce apoptosis, inhibit cell growth and proliferation, thus preventing leukemia progression [Citation33]. Several miRNAs exert inhibitory roles by suppressing cell proliferation and promoting cell apoptosis in AML [Citation34,Citation35]. Prolonging the lifespan of CML patients needs the understanding of molecular pathogenesis and the development of targeted agents [Citation4]. 2-ME represents a promising anti-tumor agent with minimal toxicity [Citation36]. AA exerts synergistic effects on the targeted therapy of chronic lymphoblastic leukemia [Citation37]. This study demonstrated that the combination of 2-ME and AA facilitated apoptosis of CML cells.
2-ME and AA are considered modulators of oxidative stress with substantial involvement in promoting cancer cell apoptosis in CML treatment [Citation15,Citation21]. 2-ME induces leukemia cell apoptosis via free radical-mediated mechanism [Citation38]. 2-ME is not only a microtubule dynamics inhibitor but also a promising anticancer agent [Citation39,Citation40]. 2-ME is relatively nontoxic to normal tissues [Citation41,Citation42]. 2-ME, which preferentially kills tumor cells by inducing accumulation of ROS in cancer cells, is a well-tolerated small molecule and possesses an oral activity, being superior to other leukemia chemotherapeutic agents [Citation43]. High-dose vitamin C was first proposed as a potential anticancer agent by Pauling and Cameron in the 1970s [Citation44]. Recent studies have confirmed the anticancer effect of AA against different human cancers and its selective cytotoxicity in vitro and in vivo [Citation45–47]. In hematologic malignancies, AA is toxic to leukemia cells [Citation48,Citation49] but less harmful to healthy cells [Citation20,Citation50]. The pathogenesis of CML involves the formation of BCR/ABL proteins with abnormal tyrosine kinase activity encoded by the BCR/ABL fusion gene [Citation51,Citation52]. Chromosomal translocation occurs in most CML individuals, and the presence of the Ph chromosome and BCR/ABL fusion gene contribute to drug resistance of leukemia cells to chemotherapy drugs [Citation53]. Although tyrosine kinase inhibitors are effective and widely used in CML treatment [Citation54,Citation55], approximately 25% of patients develop resistance or intolerance, limiting their further use in some patients [Citation56–60]. Therefore, CML therapy requires new, low toxicity, low cost, and relatively safe therapeutic strategies. We have noticed that cancer cells often exhibit poor antioxidant status, thus increasing the likelihood of killing cancer cells by oxidative stress [Citation61–64]. Therefore, we mainly conducted an exploratory study on the role of 2-ME + AA in CML cell (K562/KCL22) apoptosis to find a novel therapeutic approach for the clinical treatment of CML.
This study revealed that 2-ME + AA treatment reduced the proliferation, enhanced the apoptosis of CML cells, and inhibited CML xenograft growth in mice through in vitro and in vivo experiments. Consistently, a previous study has unveiled that AA and α-tocopherol synergistically induce apoptosis of human acute promyelocytic leukemia cells [Citation65]. High ROS level is cytotoxic by causing damages to DNA and mitochondria, thus activating the apoptosis pathway [Citation66]. The main mechanism underlying the anti-tumor effect of AA is the pro-oxidant damage caused by auto-oxidation, which results in the production of cytotoxic hydrogen peroxide (i.e. ROS) [Citation37]. 2-ME-induced apoptosis is implicated with the increased ROS level; 2-ME generates ROS and causes mitochondria damage [Citation15]. Mitochondria-mediated apoptosis is largely modulated by the balance between pro – and anti-apoptotic proteins, and the unbalanced condition may contribute to the disruption of MMP, thereby triggering apoptosis [Citation67]. Therefore, we speculated that 2-ME + AA affected apoptosis by affecting ROS and MMP. The results revealed that 2-ME + AA notably enhanced ROS and reduced MMP. 2-ME treatment leads to the production of ROS and lack of MMP in neuroblastoma cells, implying the mediation of ROS in 2-ME-induced apoptosis [Citation26]. 2-ME preferentially kills leukemia cells by generating ROS, without exerting obvious cytotoxicity to normal lymphocytes [Citation68]. AA triggers AML cell apoptosis through a hydrogen peroxide-mediated mechanism [Citation69]. Briefly, 2-ME + AA facilitated CML cell apoptosis by increasing ROS and decreasing MMP.
Then, we investigated the molecular mechanism of 2-ME + AA in CML cell apoptosis. Hao et al. have indicated that 2-ME can attenuate hypoxic pulmonary hypertension by regulating miR-223 [Citation28]. miR-223 acts a prominent role in the immune system, which is commonly deregulated in hematological malignancy [Citation70]. The miR-223 is notably downregulated in CML cells [Citation29]. BCR-ABL mediates the repression of miR-223 in CML [Citation71]. Therefore, we speculated that 2-ME + AA regulated the apoptosis of CML cells through miR-223. The results revealed that 2-ME + AA treatment prominently upregulated miR-223 in CML cells. miR-223 inhibitor remarkably enhanced proliferation and reduced apoptosis of CML cells. Moreover, miR-223 inhibitor reduced ROS and enhanced MMP. miR-223 reduces proliferation and enhances apoptosis of AML cells [Citation72]. miR-223 silencing results in poor prognosis and disease invasiveness in CML [Citation73]. Briefly, 2-ME + AA promoted the apoptosis of CML cells by upregulating miR-223.
Subsequently, we focused on the downstream mechanism of miR-223 in CML cell apoptosis. The targets of miR-223 were predicted through Jefferson website, among which FLT3 is usually mutated and overexpressed in various hematological malignancies such as CML, myelodysplasia, and mainly AML [Citation30,Citation74,Citation75]. FLT3 is a proto-oncogene implicated in critical hematopoiesis steps, which is primarily expressed in lymphohematopoietic organs including bone marrow, lymph nodes, thymus, liver, and spleen [Citation76]. FLT3 expression was significantly reduced after 2-ME + AA treatment and was increased after miR-223 inhibitor transfection. Briefly, miR-223 targeted FLT3. Overexpression of FLT3 attenuated the effect of 2-ME + AA on CML cell apoptosis. Inhibition of FLT3 facilitates apoptosis of AML cells by promoting degradation of Mcl-1 proteasome [Citation77]. FLT3 contributes to the initiation and progression of AML via the PI3K/AKT signaling [Citation31]. PI3K/AKT pathway functions as a survival-promoting factor for leukemia stem cells, and targeting the PI3K/AKT pathway exhibits pro-apoptotic and anti-proliferative effects on hematological malignancies [Citation78]. Accordingly, our results unveiled that 2-ME + AA treatment restrained the levels of p-PI3K and p-AKT, which was partially reversed by miR-223 inhibitor or pcDNA3.1-FLT3. Additionally, FLT3 silencing reduced p-PI3K and p-AKT levels. Inactivation of the PI3K/AKT pathway retards the progression and invasion of CML [Citation79–81]. 2-ME induces apoptosis and autophagic cell death of glioma cells via the PI3K/AKT pathway in vitro [Citation82]. AA kills thyroid cancer cells by suppressing the PI3K/AKT pathway via ROS-mediated mechanism [Citation83]. In brief, 2-ME + AA promoted CML cell apoptosis via the miR-233/FLT3/PI3K/AKT axis.
To sum up, this study revealed a novel mechanism of 2-ME + AA in the treatment of CML. 2-ME + AA facilitates CML cell apoptosis by inhibiting FLT3 expression and inactivating the PI3K/AKT pathway. Although CML is a very treatable disease with long-term cures approaching over 90%, once CML patients miss the best period of treatment, the number of primordial and immature granulocytes in the body is increased and accumulated, and the disease enters the period of rapid change and becomes increasingly insensitive to the treatment of chemotherapy drugs, and even shows drug resistance. As mentioned in reference 1, the positive expression of FLT3 was as high as 55.9% in CML accelerated phase and blastic phase. It is believed that our research may provide new treatment ideas for CML patients who miss the best treatment period. However, the clinical application of 2-ME + AA and whether miR-223 can be employed as a biomarker for early screening of CML still need further exploration. Moreover, for the study of combination drugs, isobologram analyses should be performed for the combination experiments. A combination index should be given for the drugs. Synergism, additive effect, or antagonism should be indicated according to the combination index. Software such as Calcusyn or Compusyn should be used for the combined analyses, but we did not carry out in-depth research on this aspect, which warrants further researches. In the future, we will investigate the effect of 2-ME + AA on other miRNAs, the other targets of miR-223 in CML, and the specific function of the PI3K/AKT pathway in the treatment of 2-ME + AA on CML.
5. Conclusion
In conclusion, 2-ME + AA lowered Bcl-2 and increased caspase 3, elevated ROS content, and reduced MMP by upregulating miR-223 and repressing the FLT3/PI3K/AKT pathway, thereby facilitating CML cell apoptosis and inhibiting CML xenograft growth in mice.
Authors’ contributions
SWZ and HHY are the guarantors of the integrity of the entire study, SWZ and HHY contributed to the study concepts, study design, and definition of intellectual content and contributed to the literature research; JZL contributed to the manuscript preparation; JRF contributed to data acquisition; JCC contributed to the data analysis and statistical analysis. All authors read and approved the final manuscript.
Availability of data and materials
All the data generated or analyzed during this study are included in this published article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Zhou H, Xu R. Leukemia stem cells: the root of chronic myeloid leukemia. Protein Cell. 2015;6(6):403–412.
- Burslem GM, Schultz AR, Bondeson DP, et al. Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 2019;79:4744–4753.
- Bennour A, Saad A, Sennana H. Chronic myeloid leukemia: relevance of cytogenetic and molecular assays. Crit Rev Oncol Hematol. 2016;97:263–274.
- Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385:1447–1459.
- Bavaro L, Martelli M, Cavo M, et al. Mechanisms of disease progression and resistance to tyrosine kinase inhibitor therapy in chronic myeloid leukemia: an update. Int J Mol Sci. 2019;20. DOI:10.3390/ijms20246141.
- Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758.
- Ngoi NYL, Choong C, Lee J, et al. Targeting mitochondrial apoptosis to overcome treatment resistance in cancer. Cancers (Basel). 2020;12. DOI:10.3390/cancers12030574.
- Pistritto G, Trisciuoglio D, Ceci C, et al. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (Albany NY). 2016;8:603–619.
- Valentin R, Grabow S, Davids MS. The rise of apoptosis: targeting apoptosis in hematologic malignancies. Blood. 2018;132:1248–1264.
- Yalniz FF, Wierda WG. Targeting BCL2 in chronic lymphocytic leukemia and other hematologic malignancies. Drugs. 2019;79(12):1287–1304.
- Fulda S. Targeting apoptosis for anticancer therapy. Semin Cancer Biol. 2015;31:84–88.
- Mueck AO, Seeger H. 2-Methoxyestradiol–biology and mechanism of action. Steroids. 2010;75(10):625–631.
- Leung K. 2-[methyl-(11)C]Methoxyestradiol. Molecular imaging and contrast agent database (MICAD). Bethesda (MD): National Center for Biotechnology Information (US); 2004-2013.
- Li DL, Zhang J, Zhang WJ, et al. Effects of 2-methoxyestradiol on the expression of caspase-3 and survivin in chronic myelocytic leukemia K562 cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2009;17:335–339.
- She MR, Li JG, Guo KY, et al. Requirement of reactive oxygen species generation in apoptosis of leukemia cells induced by 2-methoxyestradiol. Acta Pharmacol Sin. 2007;28:1037–1044.
- Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826:443–457.
- Blaszczak W, Barczak W, Masternak J, et al. Vitamin C as a modulator of the response to cancer therapy. Molecules. 2019;24. DOI:10.3390/molecules24030453.
- Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93:3704–3709.
- Shenoy N, Creagan E, Witzig T, et al. Ascorbic acid in cancer treatment: let the phoenix fly. Cancer Cell. 2018;34:700–706.
- Chen Q, Espey MG, Sun AY, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104:8749–8754.
- Verrax J, Stockis J, Tison A, et al. Oxidative stress by ascorbate/menadione association kills K562 human chronic myelogenous leukaemia cells and inhibits its tumour growth in nude mice. Biochem Pharmacol. 2006;72:671–680.
- Zhou S, Wang Y, Zhao M, et al. CdSeTe@CdS@ZnS quantum-dot-sensitized macroporous Tio2 film: a multisignal-amplified photoelectrochemical platform. Chemphyschem. 2015;16:2826–2835.
- Aalami AH, Mesgari M, Sahebkar A. Synthesis and characterization of green zinc oxide nanoparticles with antiproliferative effects through apoptosis induction and microrna modulation in breast cancer cells. Bioinorg Chem Appl. 2020;2020:8817110.
- Gerby B, Veiga DF, Krosl J, et al. High-throughput screening in niche-based assay identifies compounds to target preleukemic stem cells. J Clin Invest. 2016;126:4569–4584.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408.
- Zhang Q, Ma Y, Cheng YF, et al. Involvement of reactive oxygen species in 2-methoxyestradiol-induced apoptosis in human neuroblastoma cells. Cancer Lett. 2011;313:201–210.
- Li JJ, Tang Q, Li Y, et al. Role of oxidative stress in the apoptosis of hepatocellular carcinoma induced by combination of arsenic trioxide and ascorbic acid. Acta Pharmacol Sin. 2006;27:1078–1084.
- Hao S, Jiang L, Fu C, et al. 2-Methoxyestradiol attenuates chronic-intermittent-hypoxia-induced pulmonary hypertension through regulating microRNA-223. J Cell Physiol. 2019;234:6324–6335.
- Agatheeswaran S, Chakraborty S. MEF2C and CEBPA: possible co-regulators in chronic myeloid leukemia disease progression. Int J Biochem Cell Biol. 2016;77:165–170.
- Hattori A, McSkimming D, Kannan N, et al. RNA binding protein MSI2 positively regulates FLT3 expression in myeloid leukemia. Leuk Res. 2017;54:47–54.
- Zhong Y, Qiu RZ, Sun SL, et al. Small-molecule fms-like tyrosine kinase 3 inhibitors: an attractive and efficient method for the treatment of acute myeloid leukemia. J Med Chem. 2020;63:12403–12428.
- Zhou X, Ma X, Sun H, et al. Let-7b regulates the Adriamycin resistance of chronic myelogenous leukemia by targeting AURKB in K562/ADM cells. Leuk Lymphoma. 2020;61:3451–3459.
- Zhan H, Bai Y, Lv Y, et al. Pharmacological mechanism of mylabris in the treatment of leukemia based on bioinformatics and systematic pharmacology. Bioengineered. 2021;12:3229–3239.
- Wang G, Yu X, Xia J, et al. MicroRNA-9 restrains the sharp increase and boost apoptosis of human acute myeloid leukemia cells by adjusting the Hippo/YAP signaling pathway. Bioengineered. 2021;12:2906–2914.
- Zhou H, Jia X, Yang F, et al. miR-148a-3p suppresses the progression of acute myeloid leukemia via targeting cyclin-dependent kinase 6 (CDK6). Bioengineered. 2021;12:4508–4519.
- Verenich S, Gerk PM. Therapeutic promises of 2-methoxyestradiol and its drug disposition challenges. Mol Pharm. 2010;7:2030–2039.
- Darwiche W, Gomila C, Ouled-Haddou H, et al. Ascorbic acid (vitamin C) synergistically enhances the therapeutic effect of targeted therapy in chronic lymphocytic leukemia. J Exp Clin Cancer Res. 2020;39:228.
- Zhou Y, Hileman EO, Plunkett W, et al. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood. 2003;101:4098–4104.
- Lin H, Jiang X, Zhu H, et al. 2ME2 inhibits the activated hypoxia-inducible pathways by cabozantinib and enhances its efficacy against medullary thyroid carcinoma. Tumour Biol. 2016;37:381–391.
- Lin HL, Liu TY, Chau GY, et al. Comparison of 2-methoxyestradiol-induced, docetaxel-induced, and paclitaxel-induced apoptosis in hepatoma cells and its correlation with reactive oxygen species. Cancer. 2000;89:983–994.
- Huang P, Feng L, Oldham EA, et al. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395.
- Pani G, Colavitti R, Bedogni B, et al. Mitochondrial superoxide dismutase: a promising target for new anticancer therapies. Curr Med Chem. 2004;11:1299–1308.
- Chen YY, Yeh CH, So EC, et al. Anticancer drug 2-methoxyestradiol protects against renal ischemia/reperfusion injury by reducing inflammatory cytokines expression. Biomed Res Int. 2014;2014:431524.
- Cameron E, Pauling L, Leibovitz B. Ascorbic acid and cancer: a review. Cancer Res. 1979;39:663–681.
- Di Tano M, Raucci F, Vernieri C, et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat Commun. 2020;11:2332.
- Ngo B, Van Riper JM, Cantley LC, et al. Targeting cancer vulnerabilities with high-dose vitamin C. Nat Rev Cancer. 2019;19:271–282.
- Schoenfeld JD, Sibenaller ZA, Mapuskar KA, et al. O2(-) and H2O2-mediated disruption of fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell. 2017;32:268.
- Mastrangelo D, Massai L, Lo Coco F, et al. Cytotoxic effects of high concentrations of sodium ascorbate on human myeloid cell lines. Ann Hematol. 2015;94:1807–1816.
- Park CH, Amare M, Savin MA, et al. Growth suppression of human leukemic cells in vitro by L-ascorbic acid. Cancer Res. 1980;40:1062–1065.
- Chen Q, Espey MG, Krishna MC, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102:13604–13609.
- Bartram CR, de Klein A, Hagemeijer A, et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306:277–280.
- Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183.
- Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26:428–442.
- Ben-Neriah Y, Daley GQ, Mes-Masson AM, et al. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–214.
- Soverini S, Mancini M, Bavaro L, et al. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17:49.
- Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8:1018–1029.
- Bhaskar A, Raturi K, Dang S, et al. Current perspectives on the therapeutic aspects of chronic myelogenous leukemia. Expert Opin Ther Pat. 2014;24:1117–1127.
- Braun TP, Eide CA, Druker BJ. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell. 2020;37:530–542.
- Jiang G, Huang Z, Yuan Y, et al. Intracellular delivery of anti-BCR/ABL antibody by PLGA nanoparticles suppresses the oncogenesis of chronic myeloid leukemia cells. J Hematol Oncol. 2021;14:139.
- Rosti G, Castagnetti F, Gugliotta G, et al. Tyrosine kinase inhibitors in chronic myeloid leukaemia: which, when, for whom? Nat Rev Clin Oncol. 2017;14:141–154.
- Oberley TD, Oberley LW. Antioxidant enzyme levels in cancer. Histol Histopathol. 1997;12:525–535.
- Sun Y, Oberley LW, Elwell JH, et al. Antioxidant enzyme activities in normal and transformed mouse liver cells. Int J Cancer. 1989;44:1028–1033.
- Verrax J, Vanbever S, Stockis J, et al. Role of glycolysis inhibition and poly(ADP-ribose) polymerase activation in necrotic-like cell death caused by ascorbate/menadione-induced oxidative stress in K562 human chronic myelogenous leukemic cells. Int J Cancer. 2007;120:1192–1197.
- Yang J, Lam EW, Hammad HM, et al. Antioxidant enzyme levels in oral squamous cell carcinoma and normal human oral epithelium. J Oral Pathol Med. 2002;31:71–77.
- Vineetha RC, Hariharan S, Jaleel A, et al. L-ascorbic acid and alpha-tocopherol synergistically triggers apoptosis inducing antileukemic effects of arsenic trioxide via oxidative stress in human acute promyelocytic leukemia cells. Front Oncol. 2020;10:65.
- Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–721.
- Yun X, Rao W, Xiao C, et al. Apoptosis of leukemia K562 and Molt-4 cells induced by emamectin benzoate involving mitochondrial membrane potential loss and intracellular Ca(2+) modulation. Environ Toxicol Pharmacol. 2017;52:280–287.
- Hileman EO, Liu J, Albitar M, et al. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219.
- Park S, Han SS, Park CH, et al. L-Ascorbic acid induces apoptosis in acute myeloid leukemia cells via hydrogen peroxide-mediated mechanisms. Int J Biochem Cell Biol. 2004;36:2180–2195.
- Haneklaus M, Gerlic M, O’Neill LA, et al. miR-223: infection, inflammation and cancer. J Intern Med. 2013;274:215–226.
- Agatheeswaran S, Singh S, Biswas S, et al. BCR-ABL mediated repression of miR-223 results in the activation of MEF2C and PTBP2 in chronic myeloid leukemia. Leukemia. 2013;27:1578–1580.
- Xiao Y, Su C. Deng T. miR-223 decreases cell proliferation and enhances cell apoptosis in acute myeloid leukemia via targeting FBXW7. Oncol Lett. 2016;12:3531–3536.
- Stamatopoulos B, Meuleman N, Haibe-Kains B, et al. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood. 2009;113:5237–5245.
- Grunwald MR, Levis MJ. FLT3 inhibitors for acute myeloid leukemia: a review of their efficacy and mechanisms of resistance. Int J Hematol. 2013;97:683–694.
- Molina-Aguilar R, Montiel-Cervantes LA, Anguiano-Penaloza SV, et al. gammadelta T Cells Number, CD200, and Flt3 expression is associated with higher progression free survival in patients with chronic myeloid leukemia. Arch Med Res. 2020;51:194–203.
- Li L, Piloto O, Kim KT, et al. FLT3/ITD expression increases expansion, survival and entry into cell cycle of human haematopoietic stem/progenitor cells. Br J Haematol. 2007;137:64–75.
- Kapoor S, Natarajan K, Baldwin PR, et al. Concurrent inhibition of pim and flt3 kinases enhances apoptosis of FLT3-ITD acute myeloid leukemia cells through increased Mcl-1 proteasomal degradation. Clin Cancer Res. 2018;24:234–247.
- Bertacchini J, Heidari N, Mediani L, et al. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. 2015;72:2337–2347.
- Li L, Qi Y, Ma X, et al. TRIM22 knockdown suppresses chronic myeloid leukemia via inhibiting PI3K/Akt/mTOR signaling pathway. Cell Biol Int. 2018;42:1192–1199.
- Tu YX, Wang SB, Fu LQ, et al. Ovatodiolide targets chronic myeloid leukemia stem cells by epigenetically upregulating hsa-miR-155, suppressing the BCR-ABL fusion gene and dysregulating the PI3K/AKT/mTOR pathway. Oncotarget. 2018;9:3267–3277.
- Zheng Q, Peng X, Yu H. Local anesthetic drug inhibits growth and survival in chronic myeloid leukemia through suppressing PI3K/Akt/mTOR. Am J Med Sci. 2018;355:266–273.
- Kirches E, Warich-Kirches M. 2-methoxyestradiol as a potential cytostatic drug in gliomas? Anticancer Agents Med Chem. 2009;9:55–65.
- Su X, Shen Z, Yang Q, et al. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics. 2019;9:4461–4473.