
ABSTRACT
Reperfusion therapy after acute myocardial infarction can induce myocardial ischemia-reperfusion injury (IRI). Novel evidence has illustrated that N6-methyladenosine (m6A) modification modulates the myocardial IRI progression. Here, our study focuses on the role of m6A methyltransferase fat mass and obesity-associated protein (FTO) in myocardial ischemia/reoxygenation injury and explores potential regulatory mechanisms. Results discovered that FTO down-expressed in myocardial IRI mice and hypoxia/reoxygenation (H/R)-induced cardiomyocytes. Functionally, FTO overexpression attenuated the H/R-induced apoptosis and inflammation of cardiomyocytes. Mechanistically, methylated RNA immunoprecipitation quantitative polymerase chain reaction (MeRIP-qPCR) assay and RIP assay revealed that Yap1 mRNA acted as the target of FTO in cardiomyocytes. Moreover, FTO uninstalled the methylation of Yap1 mRNA, and enforced the stability of Yap1 mRNA. Taken together, our study reveals the role of FTO in H/R-induced myocardial cell injury via m6A-dependent manner, which may provide a new approach to improve myocardial IRI.
1. Introduction
Myocardial ischemia-reperfusion injury (IRI) is one of the most common clinical ischemic heart disease pathological conditions, especially percutaneous coronary intervention (PCI) after acute myocardial infarction (AMI). Myocardial IRI acts as one of the main causes of treatment failure and deterioration of AMI [Citation1,Citation2]. Myocardial IRI occurs when blood flow stops suddenly, leading to heart muscle death. Myocardial IRI is caused by the restoration of blood supply after hypoxia or a long-term period of ischemia [Citation3]. Severe myocardial damage after AMI results in ventricular remodeling, which is closely related to heart failure [Citation4]. The lack of clarity on the etiology of myocardial IRI leads to our inaccurate treatment. Thus, uncovering the mechanisms of hypoxia-induced dysfunction of cardiomyocytes is critical for effective clinical therapy of myocardial IRI.
N6-methyladenosine (m6A) is the most abundant posttranscriptional modification of mRNAs and ncRNAs, which controls RNA fate, including RNA splicing, stabilization, and nuclear export [Citation5,Citation6]. Recently, numerous literature studies have illustrated the role of m6A-modified mRNAs in human heart disease progression [Citation7]. For example, methyltransferase-like 3 (METTL3) is downregulated in mice ischemia-reperfusion injury myocardial tissues and hypoxia/reoxygenation (H/R) cardiomyocytes, moreover, the upregulation of METTL3 attenuates the IRI and H/R-induced cells’ apoptosis [Citation8]. Another m6A catalyzing enzyme methyltransferase-like 14 (METTL14) is upregulated in vitro IRI models using AC16 cardiomyocytes and neonatal rat ventricular cardiomyocytes, which catalyzes the m6A modification of lncRNA KCNQ1OT1 in doxorubicin-induced cardiac injury [Citation9]. Overall, the evidence indicates that m6A plays a critical role in cardiovascular disease.
Fat mass and obesity-associated protein (FTO) are a m6A demethylase uninstalling the m6A modification on RNA [Citation10,Citation11]. In existing research, the functions of FTO have been partially explored in other pathophysiological process, however, the roles and mechanism of FTO in IRI are still unclear. Here, our study constructed the myocardial IRI mice and H/R cardiomyocytes, and found that FTO down-regulated in the pathophysiology, which participate in the apoptosis of cardiomyocytes. The purpose of this study was to elucidate the role of FTO in myocardial IRI and myocardial ischemia-reperfusion injury, thereby investigating its mechanism affecting myocardial ischemia-reperfusion injury.
2. Materials and methods
2.1. Experimental animals myocardial ischemia-reperfusion injury models
Male C57BL/6 mice (25–27 g) were provided by the Vital River Laboratory Animal Technology (Beijing, China). Mice were fed a standard diet for 1 week of adaptive feeding. Mice were randomly divided into two groups: the myocardial IRI group (n = 6) and Sham group (n = 6). The myocardial IRI mice models were performed as previously described [Citation12]. In brief, after anesthesia (50 mg/kg pentobarbital sodium, intraperitoneal injection), the left thorax was cut to expose the heart, and the left anterior descending (LAD) coronary artery was ligated by 7/0 sterile suture. Myocardial ischemia was induced by 30 min of LAD coronary artery ligation and following 2 h of reperfusion. Sham group mice underwent the same surgical procedure without LAD coronary artery ligation. All animal experiments were approved by the Ethics Committee of The Affiliated Hospital of Guangdong Medical University (No. GZMH2019078).
2.2. Cardiomyocyte isolation and hypoxia/reoxygenation (H/R) injury administration
Cardiomyocytes of primary neonatal ventricular myocytes were acquired from mice's hearts as described previously [Citation13]. Mice hearts were surgically collected, and the tissue were pruned and enzymatically digested with a trypsin enzyme (Beyotime Institute of Biotechnology, Beijing, China). After digestion, the cardiomyocytes were collected and centrifuged (1500 rpm, 5 min) for purifying. A monolayer of cardiomyocytes was cultured with a DMEM/high glucose medium (Hyclone, Logan, UT, USA) containing 10% FBS, 1% penicillin-streptomycin in 37°C incubator containing 5% CO2. To mimic hypoxia/reoxygenation (H/R), cardiomyocytes were exposed to hypoxia condition (1% O2, 5% CO2, 94% Nitrogen) for 12 hours, and then followed by reoxygenation condition (5% CO2, 21% O2, 74% Nitrogen) for diverse time of duration (6, 12, or 24 hours).
2.3. Cellular transfection
FTO overexpression plasmids (FTO OE) and FTO knockdown (shRNA targeting FTO, sh-FTO), as well as controls, were transfected into cardiomyocytes as described previously [Citation13]. The transfection plasmid used in this study was constructed by GenePharma Co., Ltd. (Shanghai, China). Refer to the transfection reagent instructions for the transfection procedure.
2.4. Quantitative real-time polymerase chain reaction (qRT-PCR)
According to the protocol of PrimeScript RT reagent kit (TaKaRa), the total RNA of cardiomyocytes was extracted by TRIzol (Invitrogen). Concentration of RNA was detected by a spectrometer. Reverse transcription was performed using the Transcriptor First Strand cDNA Synthesis Kit (Cat# 04897030001, Roche, USA). qRT-PCR was performed according to the instructions of SYBR Green PCR kit (TaKaRa, Dalian, China) on Applied Biosystems 7500. β-actin acted as the internal control. Relative gene expression was calculated using the 2−ΔΔCt method. The primers were listed in Table S1.
2.5. Western blotting
Cardiomyocytes were lysed by RIPA lysis buffer after different treatments with protease inhibitor (Sigma-Aldrich, St. Louis, Missouri, USA) condition. Cellular proteins were dissolved and collected for protein concentration quantification using the BCA (bicinchoninic acid) protein detection kit (Pierce, Rockford, IL, USA). Protein samples (50 μg) were added to SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis, 10%) and then separated by gel electrophoresis. Then, protein was transferred to PVDF (polyvinylidene fluoride) membrane (Millipore, Billerica, MA, USA) for incubation with primary antibodies (anti-FTO, 1:1000, ABE552 Sigma-Aldrich; anti-Yap1, 1:1000, ab205270, Abcam) at 4°C overnight. After primary antibodies incubation, members were incubated with horseradish peroxidase conjugated secondary antibody (anti-β-actin, 1:2000, Cell Signaling Technology) at room temperature. Blot images of bands were enhanced with chemiluminescence (ECL) reagents (Thermo, Waltham, MA, USA) and then captured using densitometry (Quantity One software; Bio-Rad).
2.6. Annexin V-FITC staining and flow cytometry
Cell apoptosis and cycle were analyzed by flow cytometry as previously described [Citation14]. In brief, transfected cells were harvested and discriminated into viable cells, dead cells, early apoptotic cells, and apoptotic cells. The apoptosis was measured using FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, San Jose, CA, USA) following the manufacturer’s protocol. For the cycle analysis, cells were washed in PBS and fixed in 70% ethanol for 2 h at 4°C. DNA staining was performed using propidium iodide (PI). Cell cycle profiles were generated using flow cytometry with Modifit software (BD Biosciences).
2.7. Enzyme-linked immunosorbent assay (ELISA)
Cardiomyocytes were transfected as previously indicated and culture condition. The culture supernatants were harvested and the concentrations of IL-1β, TNF-α, and IL-6 were detected using ELISA kits (Abcam, Cambridge, UK) following the manufacturer’s recommendation.
2.8. Actinomycin D assays
Cardiomyocytes were seeded in six-well plates (1 × 105 cells per well). After 24 hours, cardiomyocytes were added with 2 μg/ml Actinomycin D (Act D, Sigma) and collected at indicated time (3 h, 6 h). The RNA stability was exhibited by remaining level was analyzed using qRT-PCR, which was normalized to the mock treatment group (0 h).
2.9. Methylated RNA immunoprecipitation quantitative polymerase chain reaction (MeRIP-qPCR)
Total RNA was extracted from cardiomyocytes using Trizol (Thermo Fisher). MeRIP buffer containing RNA (100 μg) was incubated with anti-m6A antibody (ab151230) or IgG (1 μl) coated on protein A/G beads (10 mM Tris-HCl, 150 mM NaCl, pH 7.5, 0.1% NP-40). Finally, m6A bound RNA was extracted and measured by qRT-PCR.
2.10. RIP-qPCR
The RNA immunoprecipitation (RIP)-PCR was carried using the EZ-Magna RIP Kit (Millipore, Merck Millipore Darmstadt, Germany) according to the manufacturer’s protocol. Cardiomyocytes were lysed in complete RIP lysis buffer. Anti-m6A antibody or anti-FTO antibody were incubated with protein A/G agarose beads conjugated with antibodies or control IgG for 2 h at 4°C. The purified RNA fragments from m6A MeRIP were used for library construction. RNAs were extracted and subjected to qRT-PCR using primers normalizing to input.
2.11. Quantification of the m6A modification
Total RNA was isolated from cardiomyocytes using TRIzol (Invitrogen) according to manufacturer’s instructions. The quality of RNA was analyzed using a NanoDrop (). The global m6A enrichment in mRNA was measured by the EpiQuik m6A Methylation Quantification Kit (Epigentek, P-9005-48, colorimetric) based on manufacturer’s protocol. Each sample RNA (200 ng) was coated in assay wells. M6A antibody (ab195352, 1:1000) in solution was added to the assay wells separately at a suitable dilution concentration. The m6A levels are colorimetrically quantified at a wavelength of 450 nm absorbance.
2.12. Bioinformatic analyses
The SRAMP dataset (https://www.ncbi.nlm.nih.gov/geo/) was applied to predict potential m6A modification sites of YAP mRNA.
2.13. Statistical analysis
GraphPad Prism version 7.0 (La Jolla, CA, USA) and SPSS version 19.0 (IBM) were employed for statistical analysis, and the data were shown as mean ± SD. Differences between groups were identified by Student’s t-test. p less than 0.05 was considered as statistical significance.
3. Results
Results discovered that FTO was down-expressed in myocardial IRI mice and hypoxia/reoxygenation (H/R)-induced cardiomyocytes. Functionally, FTO overexpression attenuated the H/R-induced apoptosis and inflammation of cardiomyocytes. Mechanistically, methylated RNA immunoprecipitation quantitative polymerase chain reaction (MeRIP-qPCR) assay and RIP assay revealed that Yap1 mRNA acted as the target of FTO in cardiomyocytes. Moreover, FTO uninstalled the methylation of Yap1 mRNA, and enforced the stability of Yap1 mRNA.
3.1. FTO was down-regulated in myocardial IRI mice and H/R-induced cardiomyocytes
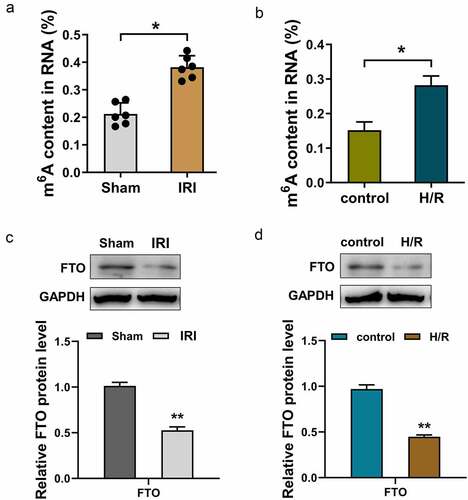
To investigate the function of FTO in myocardial IRI, we first analyzed the m6A modification level in myocardial IRI mice and H/R-induced cardiomyocyte cells. Results illustrated that m6A modification levels were up-regulated in myocardial IRI mice () and H/R-induced cardiomyocyte cells (), suggesting a high m6A modification status in pathophysiology. Moreover, the protein levels of FTO were detected in myocardial IRI mice and H/R-induced cardiomyocyte cells. Results illustrated that FTO protein levels were both down-regulated in myocardial IRI mice () and H/R-induced cardiomyocyte cells () as compared to control groups. In conclusion, these data found that m6A modification was up-regulated and FTO was down-regulated in myocardial IRI mice and H/R-induced cardiomyocyte cells. The data suggested that m6A demethylase FTO participated in myocardial IRI pathophysiology.
Figure 1. FTO was down-regulated in myocardial IRI mice and H/R-induced cardiomyocytes. (a) m6A RNA methylation colorimetric quantification analysis detected the m6A content in total RNA of myocardial IRI mice and control (Sham) mice. (b) m6A RNA methylation colorimetric quantification analysis detected the m6A content in total RNA of H/R and control cardiomyocytes. (c) Western blot analysis detected the FTO protein level in myocardial IRI mice and control (Sham) mice. (d) Western blot analysis detected the FTO protein level in H/R-induced and control cardiomyocytes. *p < 0.05, **p < 0.01.
3.2. FTO mitigated the apoptosis and inflammatory reaction induced by H/R
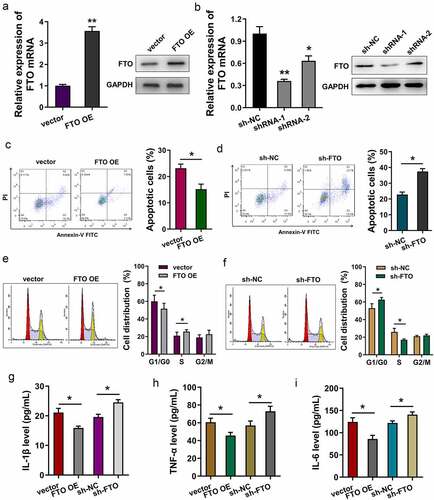
To investigate the functions of FTO in H/R-induced cardiomyocytes, gain and loss-of-function assays were performed. Transfection efficiency was detected using RT-PCR and Western blot assay, showing that FTO mRNA and protein levels were effectively up-regulated or down-regulated upon overexpression or knockdown (). Apoptosis analysis using flow cytometry reflected that FTO overexpression reduced the apoptotic cells (), and the knockdown of FTO increased the apoptosis of cardiomyocytes (). Cell cycle analysis found that FTO overexpression promoted the cycle progression of cardiomyocytes (), and FTO knockdown induced the cycle arrest at G0/G1 phase (). Regarding inflammatory reaction, inflammatory factors were detected upon FTO overexpression and knockdown. Results indicated that FTO overexpression reduced the secretion level of IL-1β (), TNF-α () and IL-6 (). Besides, FTO knockdown up-regulated them. Overall, these findings revealed that FTO mitigated apoptosis and inflammatory reaction induced by H/R.
Figure 2. FTO mitigated the apoptosis and inflammatory reaction induced by H/R. (a, b) RT-PCR and Western blot assay were performed to detected the FTO mRNA and protein levels in H/R-induced cardiomyocytes transfected with FTO overexpression plasmids and shRNA-FTO. (c, d) Apoptosis analysis using flow cytometry revealed the apoptotic rate of H/R-induced cardiomyocytes transfected with FTO overexpression plasmids and shRNA-FTO. (e, f) Cell cycle analysis using flow cytometry detected the cellular distribution at G0/G1, S, G2/M phase. (g) IL-1β, (h) TNF-α, and (i) IL-6 were detected with ELISA. *p < 0.05.
3.3. YAP1 mRNA acted as the target of FTO, and FTO uninstalled the m6A modification of YAP1
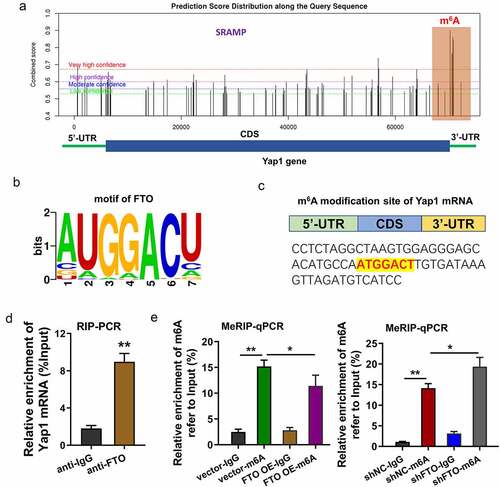
In the following research, we tried to investigate the downstream of FTO through which FTO exerted its functions in myocardial IRI. Previous research indicated that Yes-associated protein (YAP), also known as YAP1, participated in the progression of myocardial IRI and hypoxia/reoxygenation-induced cardiomyocytes [Citation15,Citation16]. Thus, our research tried to investigate the interaction within FTO and YAP1. An online predictive tool (SRAMP, http://www.cuilab.cn/sramp) revealed that there are several potential m6A modification sites in the 3’-UTR with very high probability (). The binding sites or motif of FTO were AUGGACU (). Moreover, the m6A modification sites of Yap1 mRNA were identified in the genomic sequence (). RNA immunoprecipitation (RIP)-PCR analysis revealed that the Yap1 mRNA was enriched by the anti-FTO antibody (). MeRIP-PCR assay indicated that FTO overexpression reduced the m6A modification level, while FTO knockdown increased the m6A modification level in H/R-induced cardiomyocytes (). Overall, the data indicated that YAP1 mRNA acted as the target of FTO, and FTO uninstalled the m6A modification of YAP1.
Figure 3. YAP1 mRNA acted as the target of FTO, and FTO uninstalled the m6A modification of YAP1. (a) Online predictive tool (SRAMP, http://www.cuilab.cn/sramp) revealed the potential m6A modification sites in the 3’-UTR with very high probability. (b) The binding sites or motif of FTO were AUGGACU. (c) The m6A modification sites of Yap1 mRNA were identified in the genomic sequence (ATGGACT). (d) RNA immunoprecipitation (RIP)-PCR analysis revealed the Yap1 mRNA level enriched by the anti-FTO antibody. (e) MeRIP-PCR assay indicated the m6A modification level of H/R-induced cardiomyocytes upon FTO overexpression or FTO knockdown. *p < 0.05.
3.4. FTO enhanced the stability of YAP1 mRNA through uninstalling the m6A modification of YAP1 mRNA
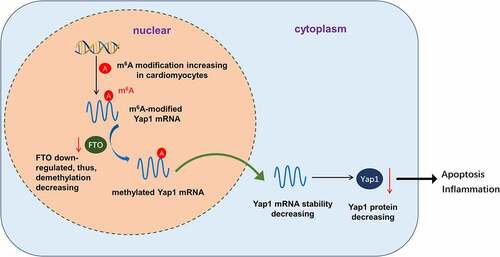
In the H/R-induced cardiomyocytes, the YAP1 mRNA level is down-regulated as compared to the control cells (). RNA immunoprecipitation (RIP)-PCR assay found that FTO overexpression reduced the YAP1 mRNA enrichment precipitated by anti-m6A antibody, besides, FTO knockdown up-regulated the YAP1 mRNA enrichment precipitated by anti-m6A antibody (). The data indicate that FTO uninstalled the m6A modification of YAP1 mRNA. RNA stability analysis found that FTO overexpression increased the YAP1 mRNA expression in H/R-induced cardiomyocytes treated with Actinomycin D, while FTO knockdown reduced the YAP1 mRNA expression (). Moreover, the Yap1 protein levels were detected using Western blotting assay, showing that FTO overexpression promoted the Yap1 protein and FTO knockdown inhibited the Yap1 protein (). Overall, these data provided the thought that FTO enhanced the stability of YAP1 mRNA by uninstalling the m6A modification of YAP1 mRNA ().
Figure 4. FTO enhanced the stability of YAP1 mRNA through uninstalling the m6A modification of YAP1 mRNA. (a) RT-PCR assay detected the YAP1 mRNA level in H/R-induced cardiomyocytes or control cells. (b) RNA immunoprecipitation (RIP) -PCR assay detected the YAP1 mRNA enrichment precipitated by anti-m6A antibody. Input acted as the positive control. (c) RNA stability analysis detected the YAP1 mRNA expression in H/R-induced cardiomyocytes treated with Actinomycin D (act D). Cardiomyocytes were transfected with FTO overexpression (FTO OE) and FTO knockdown (sh-FTO). (d) Western blotting assay indicated the Yap1 protein in FTO overexpression or FTO knockdown. *p < 0.05.
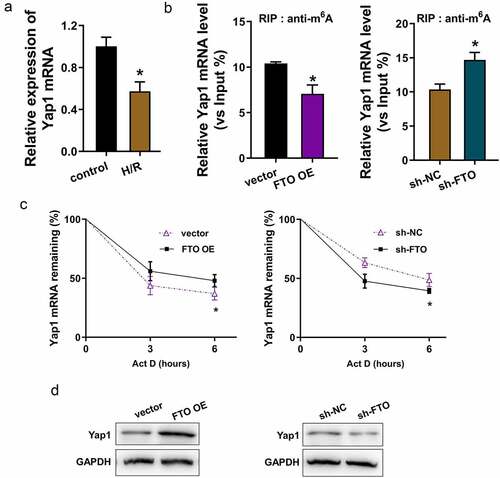
Figure 5. FTO enhanced the stability of YAP1 mRNA through uninstalling the m6A modification of YAP1 mRNA.
4. Discussion
N6-Methyladenosine (m6A) modification is a specific methylation of adenosine at the N6 position, which is widespread in messenger RNA (mRNA) and non-coding RNAs (ncRNAs) [Citation17,Citation18]. Hypoxia/reoxygenation (H/R)-induced myocardial cell injury is the main cause of acute myocardial infarction (AMI). Emerging evidence indicates that epigenetics are closely correlated with the pathogenesis of myocardial ischemia/reperfusion injury, suggesting that epigenetics act as a novel therapeutic target to prevent myocardial IRI. The purpose of the present study was to investigate the role of FTO in H/R-induced myocardial injury.
Emerging studies have revealed that m6A modification is widely distributed in pathophysiological processes, including myocardial fibrosis, myocardial infarction and so on. M6A could effectively influence the fate of RNA, including transcription splicing, RNA stability, cell differentiation, and drug resistance. FTO, a key component of demethylase, has been proven to regulate a variety of pathophysiological processes.
Here, the present research aims to investigate the function of FTO in myocardial IRI. First, the levels of FTO were detected in the heart tissues of myocardial IRI mice and H/R-induced isolated cardiomyocytes. Results showed that FTO was down-regulated in myocardial IRI mice and H/R-induced cardiomyocytes and the m6A modification level was up-regulated in the myocardial IRI mice and H/R-induced cardiomyocyte cells. Functional assay indicated that FTO mitigates the apoptosis and inflammatory reaction induced by H/R in vitro. However, there is a limitation that neonatal cardiomyocytes are different from adult cardiomyocytes in response to H/R. In vivo experiments were performed on adult mice, while in vitro experiments were performed on neonatal cardiomyocytes. These data indicate that FTO might function as a beneficial factor for myocardial IRI.
More evidence is emerging of the role of m6A in myocardial IRI. For instance, m6A methyltransferase METTL3 downregulates in mice I/R myocardial tissues and H/R cardiomyocytes, and METTL3 suppresses the H/R-induced cell apoptosis by upregulating miR-25-3p and activating the PI3K/Akt pathway in H/R-treated cardiomyocytes [Citation8]. Previous reports found an FTO-induced m6A station in the heart. For example, FTO is downregulated in heart tissues of heart failure mice, and the total level of m6A is increased. FTO overexpression upregulates the Mhrt and reduces m6A modification in the H/R-treated myocardial cells [Citation19].
Here, we performed methylated m6A RNA immunoprecipitation PCR to detect the m6A enrichment in myocardial IRI and validate m6A in individual transcripts. We found that YAP1 mRNA acted as the target of FTO, and FTO uninstalled the m6A modification of YAP1. Previous research indicated that Yes-associated protein (YAP), also known as YAP1, participated in the progression of myocardial IRI and hypoxia/reoxygenation-induced cardiomyocytes. In the present research, FTO knockdown up-regulated the YAP1 mRNA enrichment precipitated by anti-m6A antibody. Thus, FTO uninstalled the m6A modification of YAP1 mRNA. RNA stability analysis found that FTO overexpression increased the YAP1 mRNA expression in H/R-induced cardiomyocytes treated with Act D. Overall, the findings provide a thought that FTO enhanced the stability of YAP1 mRNA through uninstalling the m6A modification of YAP1 mRNA.
5. Conclusion
Overall, we demonstrated that m6A demethylase FTO can regulate the apoptosis and inflammation of H/R-induced cardiomyocytes in myocardial IRI. Moreover, FTO can uninstall the m6A modification of YAP1 mRNA to increase YAP1 mRNA stability and expression (). These novel findings may be a new direction to explore the therapeutic strategy for myocardial IRI.
Supplemental Material
Download MS Word (16.9 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
No research data was shared.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Carbone F, Bonaventura A, Montecucco F. Neutrophil-related oxidants drive heart and brain remodeling after ischemia/reperfusion injury. Front Physiol. 2019;10:1587.
- Chong J, Bulluck H, Fw Ho A, et al. Chronic remote ischemic conditioning for cardiovascular protection. Conditioning medicine. 2019;2(4):164–169.
- Edupuganti MM, Ganga V. Acute myocardial infarction in pregnancy: current diagnosis and management approaches. Indian Heart J. 2019;71(5):367–374.
- Iannattone PA, Zhao X, VanHouten J, et al. Artificial intelligence for diagnosis of acute coronary syndromes: a meta-analysis of machine learning approaches. Can J Cardiol. 2020;36(4):577–583.
- Guo J, Zheng J, Zhang H, et al. RNA m6A methylation regulators in ovarian cancer. Cancer Cell Int. 2021;21(1):609.
- Issah MA, Wu D, Zhang F, et al. Epigenetic modifications in acute myeloid leukemia: the emerging role of circular RNAs (Review). Int J Oncol. 2021;59(6). DOI:10.3892/ijo.2021.5287.
- Zhang Z, Wei W, Wang H, et al. N6-methyladenosine-sculpted regulatory landscape of noncoding RNA. Front Oncol. 2021;11:743990.
- Zhao X, Yang L, Qin L. Methyltransferase-like 3 (METTL3) attenuates cardiomyocyte apoptosis with myocardial ischemia-reperfusion (I/R) injury through miR-25-3p and miR-873-5p. Cell Biol Int. 2021. DOI:10.1002/cbin.11706
- Zhuang S, Ma Y, Zeng Y, et al. METTL14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol Toxicol. 2021. DOI:10.1007/s10565-021-09660-7.
- Zheng QK, Ma C, Ullah I, et al. Roles of N6-methyladenosine demethylase FTO in malignant tumors progression. Onco Targets Ther. 2021;14:4837–4846.
- Zhou LL, Xu H, Huang Y, et al. Targeting the RNA demethylase FTO for cancer therapy. RSC chemical biology. 2021;2(5):1352–1369.
- Zhou YY, Wang SQ, Zhu WZ, et al. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol. 2000;279(1):H429–436.
- Mahmoud AI, Kocabas F, Muralidhar SA, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497(7448):249–253.
- Li Y, Ren S, Xia J, et al. EIF4A3-Induced circ-BNIP3 aggravated hypoxia-induced injury of H9c2 cells by targeting miR-27a-3p/BNIP3. Mol Ther Nucleic Acids. 2020;19:533–545.
- Cai WF, Wang L, Liu GS, et al. Manipulating the Hippo-Yap signal cascade in stem cells for heart regeneration. Ann Palliat Med. 2016;5(2):125–134.
- Flinn MA, Link BA, O’Meara CC. Upstream regulation of the Hippo-Yap pathway in cardiomyocyte regeneration. Semin Cell Dev Biol. 2020;100:11–19.
- Chen C, Guo Y, Guo Y, et al. m6A modification in non-coding RNA: the role in cancer drug resistance. Front Oncol. 2021;11:746789.
- Wang X, Ma R, Zhang X, et al. Crosstalk between N6-methyladenosine modification and circular RNAs: current understanding and future directions. Mol Cancer. 2021;20(1):121.
- Shen W, Li H, Su H, et al. FTO overexpression inhibits apoptosis of hypoxia/reoxygenation-treated myocardial cells by regulating m6A modification of Mhrt. Mol Cell Biochem. 2021;476(5):2171–2179.