
ABSTRACT
As a chronic brain disease, epilepsy affects ~50 million people worldwide. The traditional antiepileptic drugs (AEDs) are widely applied but showing various problems. Although the new AEDs have partially solved the problems of traditional AEDs, the current clinical application of traditional AEDs are not completely replaced by new drugs, particularly due to the large individual differences in drug plasma concentrations and narrow therapeutic windows among patients. Therefore, it is still clinically important to continue to treat patients using traditional AEDs with individualized therapeutic plans. To date, our understanding of the molecular and genetic mechanisms regulating plasma concentrations of AEDs has advanced rapidly, expanding the knowledge on the effects of genetic polymorphisms of genes encoding drug-metabolizing enzymes on the plasma concentrations of AEDs. It is increasingly imperative to summarize and conceptualize the clinical significance of recent studies on individualized therapeutic regimens. In this review, we extensively summarize the critical effects of genetic polymorphisms of genes encoding drug-metabolizing enzymes on the plasma concentrations of several commonly used AEDs as well as the clinical significance of testing genotypes related to drug metabolism on individualized drug dosage. Our review provides solid experimental evidence and clinical guidance for the therapeutic applications of these AEDs.
Graphical Abstract
1. Introduction
Epilepsy is a group of chronic brain diseases in humans characterized by transient dysfunctions in the central nervous system caused by abnormal discharge of brain neurons, generally happening unexpectedly and repeatedly. Approximately 50 million people are affected worldwide and nearly 80% of patients can completely control their seizures and restore their normal quality of life through medication [Citation1]. The antiepileptic drugs (AEDs) function by directly inhibiting the excessive discharge of neurons in the lesion and acting on the normal brain tissue around the lesion to prevent the spread of abnormal discharge of brain neurons [Citation2]. The AEDs function following several known mechanisms [Citation3]. First, the AEDs block the sodium channels. By acting on voltage-dependent sodium channels, the frequency of continuous action potentials in the neurons is reduced. This effect is further enhanced during depolarization. It is now well-known that repeated depolarization is an important mechanism underlying seizures of epilepsy. Second, the AEDs enhance the bioactivities of γ-aminobutyric acid (GABA) in various ways to ultimately inhibit the high-frequency repetitive action potentials. Third, the AEDs antagonize excitatory amino acids, which, together with their receptors, are involved in the occurrence of epilepsy. It has been well demonstrated in a variety of animal models of epilepsy that the seizures of epilepsy are inhibited by reducing the activities of excitatory transmitters (i.e., glutamate) in different ways.
The old generation AEDs, e.g., carbamazepine (CBZ), phenytoin (PHT), valproic acid (VPA), and phenobarbital (PB), have been widely used in clinical settings, even though they have shown serious problems, including the nonlinear pharmacokinetics, large individual differences, narrow treatment ranges, severe adverse reactions, and drug interactions. The new generation AEDs that have been introduced in the past 10 years, such as levetiracetam (LEV), lamotrigine (LTG), and oxcarbazepine (OXC), have shown fewer adverse reactions, higher bioavailability, and less drug interactions, partially solving the problems of the traditional AEDs. However, the current clinical applications of traditional AEDs cannot be completely replaced by the new drugs, particularly due to the large individual differences in plasma concentrations of AEDs and narrow therapeutic windows, which are the important measurements in clinical treatment of epilepsy.
In recent years, significant progress has been made to explore the genetic mechanisms affecting the plasma concentrations of AEDs [Citation3]. The relevant genes have been extensively investigated and reviewed in various ethnic groups worldwide [Citation4–12]. Furthermore, studies have showne that the genetic polymorphisms of these relevant genes also play important roles in the diagnosis of epilepsy. For example, Li et al. [Citation13] have investigated the functions of TUG1 in restoring hippocampal neuronal activities and preventing neuronal cell death. Their results show that silence of TUG1 increases the viability of hippocampal neurons and inhibits their apoptosis. The diagnostic capability of the elevated levels of TUG1 measured in children with temporal lobe epilepsy (TLE) has been further explored, revealing that TUG1 shows high sensitivity and specificity in children with TLE, strongly suggesting the diagnostic potential of TUG1 in children with TLE. Ahmed et al. [Citation14] have investigated the genetic polymorphisms of CYP2C19 in the healthy individuals of six distinct ethnic populations of Pakistan. The results show that the minor allele is present at a frequency of 15.06% and 8.14% for CYP2C19*2 and CYP2C19*3, respectively, suggesting that a significant portion of the Pakistani population and its certain ethnic groups contain high frequencies of polymorphisms with reduced functions. However, a comprehensive review of studies in this area is still lacking in Chinese populations. In this review, we first summarize the main factors affecting the metabolism of six types of AEDs (i.e., three traditional AEDs including VPA, CBZ, and PHT and three new AEDs including LEV, OXC, and LTG) currently commonly used in treating patients of epilepsy, including their general metabolic characteristics. We further analyze the effects of the genetic polymorphisms of genes encoding the metabolic enzymes on the plasma concentrations of AEDs and the clinical significance of the drug metabolism-related genotype testing on individualized drug dosage. We strongly believe that this comprehensive review on most of AEDs currently popularly used in clinical practice provides strong evidence and clinical guidance in the therapeutic applications of these AEDs.
2. Pharmacokinetic characteristics of traditional antiepileptic drugs
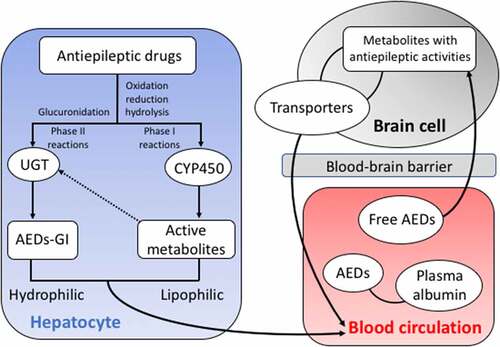
The metabolism of traditional AEDs in the liver includes phase I and phase II reactions (). With a large amount of exogenous substances undergoing biological transformations in the body, the liver is the main location for the phase I metabolic reactions of the biological transformation. During the phase I reaction, the drugs undergo oxidation, reduction, and hydrolysis catalyzed by the monooxygenase system with cytochrome P450 (CYP450) as the core to change the molecular structure, to increase the polarity of the drugs and the water solubility, ultimately to change the activities and functions of the drugs. The phase I metabolic reactions are the rate-limiting steps for the elimination of drugs from the body, causing either detoxification or poisoning effects [Citation15]. A portion of the active metabolities derived in phase I reactions enters the phase II reactions. During the phase II reaction, the drug and its metabolites combined with endogenous substances are excreted from the body, showing a detoxification effect. However, some active metabolites generated simultaneously during the phase II reactions probably cause the liver damage [Citation16]. The uridine diphosphate glucuronidase (UGT) is the main metabolic enzyme catalyzing the binding reaction during phase II reaction [Citation17]. Following the phase I and II reactions, the AEDs are absorbed into the blood and partially combined with plasma albumin. Then, the AEDs penetrate the blood-brain barrier and are eventually distributed in the brain, while the drug transporters (i.e., the functional membrane proteins located on the cell membrane) mediate the drugs to enter the cells by taking up the substrate to the target site to exert its effect and to efflux the drugs out of the cells thus reducing the plasma concentrations and drug efficacy.
Figure 1. Schematic pathways of antiepileptic drug (AED) metabolizing enzymes in both liver and brain cells and the blood circulation system. The arrowed dashed line indicates the participation in phase II reactions of a portion of the active metabolites derived in phase I reactions. AEDs-GI represents the glucuronidation products of AEDs.
We here review the studies on the relationships between the plasma concentrations of these traditional AEDs (i.e., VPA, CBZ, and PHT) and the genetic polymorphisms of genes encoding enzymes involved in the metabolism of these AEDs.
3. Valproic acid
3.1. Metabolic characteristics of valproic acid
As a type of broad-spectrum AEDs, the valproic acid (VPA) has been widely used in clinical applications. VPA shows antiepileptic effects by inhibiting the sodium ion channels and possibly the T-type calcium ion channels, inhibiting the metabolic enzymes GABA to cause the accumulation of GABA in brain, and inhibiting excessive discharge of neurons at the lesion and simultaneously preventing the spread of abnormal discharge of brain neurons [Citation18]. The effective plasma concentrations of VPA range from 50 to 100 ug/ml.
The pharmacokinetics of VPA conforms to the one-compartment model and the first-order kinetic process. The oral absorption of VPA is rapidly distributed in the body. Generally, the highest plasma concentration is obtained in 0.5–2 hours after the medicine is taken with an empty stomach or 2–4 hours after taken with food. The effective threshold of VPA varies among different patients with 50–100 ug/ml for 50% of patients, 25–50 ug/ml for 20% of patients, while 10% of patients have a range of either 100–150 ug/ml, 10–25 ug/ml, or less than 10 ug/ml [Citation19]. It is generally believed that the toxic reactions would be caused by the plasma concentrations over 100 ug/ml, while clinical improvement of symptoms is observed with the drug plasma concentrations of 50–60 ug/ml.
After the VPA is absorbed into the blood, it is mainly combined with plasma albumin with a binding rate of 84%-94%, while the concentration of free VPA is generally low. Different patients show different plasma albumin binding rates with VPA, probably due to the fatty acids and applications of other medicines with high binding capability with the plasma albumin. VPA can easily penetrate the blood-brain barrier and is evenly distributed in the brain. VPA can also pass through the placental barrier to cause teratogenic effects on the fetus, such as the spina bifida [Citation20]. VPA shows a slightly higher rate of teratogeny than those of other tranditional AEDs, e.g., CBZ and PHT.
3.2. Effect of cytochrome P450 on the metabolism of valproic acid
As a group of monooxygenases, the cytochrome P450 (CYP450) enzyme system contains enzymes that mainly exist in the microsomes of liver cells and are located on the endoplasmic reticulum membranes. These enzymes are a group of structurally and functionally related isozymes involved in the metabolism of many endogenous and exogenous substances (i.e., the detoxification of exogenous substances and activation of pre-carcinogens) and encoded by genes in the CYP superfamily containing more than 10 families of over 100 genes [Citation21,Citation22]. There are only three groups of genes (i.e., CYP1, CYP2, and CYP3) involved in regulating the metabolism of most drugs and foreign substances with CYP2 as the largest family containing 15 subfamilies (CYP2A to 2Q), metabolizing 40% of clinical drugs currently prescribed. It is estimated that about 56% of drugs are mainly metabolized by CYP450 enzymes, with CYP3A4 involved in metabolism of more than 50% of human drugs, followed by CYP2D6 (30%), CYP2C9 and CYP2C19 (12%), CYP1A2 (4%), CYP2E1 (2%), and CYP2A6 (2%) [Citation23,Citation24].
We here review the recent advancement in our understanding of the functions of these CYP450 enzymes in the metabolism of VPA. Studies have shown that four enzymes (i.e., CYP2A6, CYP2B6, CYP2C9, and CYP2C19) are the main metabolizing enzymes of VPA with their metabolic activities closely related to the metabolism of VPA [Citation25]. The wild types of these genes show significantly stronger metabolizing capability on VPA than the mutants. Patients carrying the mutant CYP genes show decreased capability of removing VPA, causing the accumulation of VPA in the body to induce adverse reactions, such as decreased carnitine, increased blood ammonia, and abnormal blood index [Citation26]. Furthermore, studies have also confirmed that the hepatotoxic metabolites of VPA are mainly produced by the CYP pathway [Citation27,Citation28]. Therefore, the genotypes encoding the CYP450 enzymes play important regulatory roles in maintaining the level of VPA in patients.
3.2.1. CYP2A6
The enzymatic activity of CYP2A6 regulates the metabolic rate of sodium valproate in vivo. The CYP2A6 gene contains the alleles of wild-type CYP2A6*1 and mutant CYP2A6*4 [Citation29]. The generation of CYP2A6*4 is caused by the deletion of CYP2A6 gene. Oscarson et al. have shown that the activity of CYP2A6 with *4 allele is reduced or even lost completely [Citation30]. Furthermore, Several studies have reported that the allele frequency of CYP2A6*4 is 13.3–15.1% in the Chinese populations [Citation30–32]. Sun et al. have reported that the average standard plasma concentration of sodium valproate in patients with CYP2A6*4 allele is significantly higher than that of the patients without CYP2A6*4 allele, suggesting that the polymorphism of CYP2A6 gene affects the plasma concentration of sodium valproate [Citation31]. The patients with CYP2A6*4/*4 genotype generate inactivated enzyme due to the complete deletion of the CYP2A6 gene [Citation30], while genotype CYP2A6*1/*4 generates enzyme with reduced activity due to having one copy of wild type CYP2A6 allele, indicating that the plasma concentration of sodium valproate in patients with *4 allele is significantly higher than that of patients without *4 allele [Citation30]. This study also showed that when given regular doses, the plasma concentration of the medicine in patients with CYP2A6*4 allele was higher than that of the patients without CYP2A6*4. Therefore, it is highly recommended that patients with CYP2A6*4 allele should be clinically treated with reduced dosage of sodium valproate and monitored closely for the occurrence of adverse reactions.
3.2.2. CYP2B6
CYP2B6 is the main member of the CYP2B subfamily and is the only homologous protein with enzymatic activity in this subfamily of humans. The CYP2B6 gene has been mapped to the location 19q12 ~ 19q13.2 on chromosome 19, containing 9 exons and 6 alleles with 9 mutation sites identified [Citation32–34]. As one of the 6 alleles, CYP2B6*6 contains two mutation sites, i.e., 516 G > T on exon 4 and 718A>G on exon 5, with the mutation rates of 21% and 28%, respectively, in the Chinese Han population [Citation35,Citation36]. Using the gene point mutation detection approach, Hideto et al. [Citation37] reported that at the Lys262Arg site, amino acid substitutions were observed in three enzymes (i.e., encoded by CYP26B*4, CYP26B*6, and CYP2B6*7), showing higher Vmax and Vmax/Km values than those of the non-mutant type. These results indicate that the catalytic activity of the CYP2B6 enzyme coded by the mutated CYP2B6 genotype has been significantly altered [Citation37], which is probably the main molecular mechanism generating the variations in the metabolic functions of each genotype of CYP2B6 on the heterogeneous compounds [Citation38]. Tan et al. have reported that the average plasma concentration of VPA in patients with CYP2B6*6 allele is higher than that of wild-type patients (i.e., homozygous for CYP2B6*1), while the metabolism of VPA slows down in the group with mutations of CYP2B6 gene [Citation39]. Therefore, it is speculated that the genetic polymorphism of CYP2B6 affects the plasma concentration of VPA, while the metabolism of VPA is regulated by CYP2B6 gene. Furthermore, studies have shown that patients with CYP2B6*1/*6 genotype show higher average drug plasma concentrations than patients with CYP2B6*1/*1 genotype, suggesting that patients with mutant genes may lose the enzymatic activity due to the lack of the gene, decrease the VPA metabolism, and keep the drug plasma concentration relatively high [Citation39]. These studies indicate that in the clinical application of VPA, in order to avoid the VPA poisoning caused by excessive drug concentration, patients with mutant CYP2B6 gene are initially treated with a small dose to achieve effective drug plasma concentration in order to control the symptoms [Citation37–39]. This strategy could reduce the waste of medicinal resources and, more importantly, prevent the adverse reactions of medicines and possible serious consequences. Therefore, it is recommended that when treating patients with mutant CYP2B6 genes, clinicians should follow the principle of starting with a small dose, while closely observing symptoms and monitoring changes in drug plasma concentration in patients.
3.2.3. CYP2C9 and CYP2C19
CYP2C is the main component of CYP450, accounting for 20% of the entire family of CYP450. Both in vitro and in vivo studies have shown that both CYP2C9 and CYP2C19 are the key metabolic enzymes for the biological transformation of VPA [Citation40]. CYP2C9 is responsible for most (~75-80%) of VPA terminal desaturation and hydroxylation reactions and closely related to the generation of the hepatotoxic products of VPA [Citation41]. CYP2C9*3 is a common polymorphic site of CYP2C9 allele in Chinese Han population. The A1075C site of CYP2C9*3 is the main mutation site in the Chinese population with the allele frequency of ~3.3% [Citation42]. The two common polymorphic sites of CYP2C19 allele are CYP2C19*2 (G681A; rs4244285) and CYP2C19*3 (rs4986893, c.636 G > A). Studies have shown that CYP2C19*2 causes the splicing mutation inactivation of the transcription protein, while CYP2C19*3 forms a terminator to disrupt the activity of the transcription proteins, causing decreased enzymatic activity to metabolize VPA, ultimately increasing the drug plasma concentration and causing side effects related to plasma concentration [Citation43].
Among the known polymorphic sites of CYP2C9 gene, the CYP2C9*3 mutation is commonly detected in the Chinese population, while the mutation of CYP2C9*2 is rarely reported [Citation44]. Studies have shown that the metabolites of VPA include 4-Ene-VPA, 4-OH-VPA, and 5-OH-VPA, with 4-Ene-VPA confirmed to show liver toxicity [Citation45]. In the CYP2C9 heterozygous mutant, the production of 4-Ene-VPA is 29% less than that of the wild homozygotes, while the mutant homozygotes generate 61% less of 4-Ene-VPA compared to wild homozygotes, indicating that patients with wild-type CYP2C9*1 are more likely to show liver toxicity than the carriers of CYP2C9*3.
Studies have shown that the plasma concentration of VPA in AA genotype at CYP2C19*2 locus is significantly higher than that of the GG genotype after the medicine is taken for 2, 4, and 8 hours [Citation46], while the plasma concentration of VPA in AA genotype at CYP2C19*3 locus is significantly higher than that of the GG genotype 8 hours after the medicine is taken [Citation47]. No significant difference is detected in the plasma concentration of VPA among other genotypes. The allele frequencies of CYP2C19*2 mutant (AA type) and CYP2C19*3 mutant (GG type) in Chinese population are 37.16% and 14.19%, respectively. These studies demonstrate that the genetic polymorphism of CYP2C19*2 gene is closely related to the plasma concentration of VPA in patients with epilepsy, ultimately affecting the therapeutic effect of VPA. These results are consistent with those reported previously [Citation48,Citation49]. Therefore, it is recommended that when VPA is used in the treatment of epilepsy, the dosage should be increased for patients with CYP2C19*3 GG genotype showing fast metabolism, while the dosage be appropriately reduced for patients with CYP2C19*3 AA genotype.
Using a combined analysis based on the mutant alleles of both CYP2C9 and CYP2C19 genes, Tan et al. categorized patients of epilepsy into three groups, including (1) extensive metabolizers (EM) with the wild-type genotypes of the homozygous combination of CYP2C9*1*1 and CYP2C19*1*1, (2) intermediate metabolizers (IM) with the heterozygous combinations of two out three genotypes (CYP2C9*1*3, CYP2C19*1*2, or CYP2C19*1*3), and (3) poor metabolizers (PM) with genotypes of mutant homozygous combination of either CYP2C9*3*3, CYP2C9*1*3, or CYP2C19*2*2 [Citation50]. The frequencies of the three groups of patients are 47.5%, 25%, and 27.5%, respectively. The number of mutant genes is positively correlated with the standardized plasma concentration, while the plasma concentration of PM patients is significantly higher than that of EM patients with equal dose of VPA taken.
These studies conclude that both CYP2C9 and CYP2C19 are involved in the metabolism of VPA in humans with their genetic polymorphisms significantly affecting the metabolic process of VPA [Citation45–50]. Therefore, in clinical treatments, it is substantially important to detect the genotype of patients in order to establish precise formulation of individualized drug dosages and to effectively avoid drug waste and adverse reactions, exhibiting significant clinical importance for individualized drug application.
3.2.4. CYP2D6
The gene encoding CYP2D6 is located in region 1 (i.e., band 3 and subband 1) on the long arm of chromosome 22 in humans, playing important roles in genetic pharmacology. Studies have shown that CYP2D6 is clinically involved in the metabolism of more than 25% of commonly used drugs, such as isoquinoline (adrenergic blocking drug), spapartin and propafenone (antiarrhythmic drugs), and amitriptyline (antidepressants) [Citation51]. In recent years, the distribution of CYP2D6 genetic polymorphisms in different human populations and its relationship with the effects of clinical therapeutic drugs have been widely explored [Citation52,Citation53]. The frequency of CYP2D6*10 allele is relatively high in Chinese population (50–70%), affecting the safety and effectiveness of substrate drugs in clinical applications.
By comparing the plasma concentration of sodium valproate and standardized drug plasma concentration in patients with epilepsy of different genotypes of CYP2D6*10, studies have reported that the drug plasma concentrations of three genotypes of CYP2D6*10 are not significantly different from the standardized drug plasma concentration [Citation54]. Specifically, patients of epilepsy with different genotypes of CYP2D6*10 not reaching the minimum effective plasma concentration and those exceeding the minimum toxic concentration are compared to investigate their genetic polymorphisms. The results show that there is no significant difference between the number of CYP2D6*10 genotypes not reaching the minimum effective concentration and the number of cases exceeding the minimum toxic concentration, indicating that the genetic polymorphism of CYP2D6*10 is not involved in the regulation of the plasma concentration of sodium valproate in patients with epilepsy.
3.3. Effect of uridine diphosphate glucuronidase on the plasma concentration of valproic acid
The uridine diphosphate glucuronidase (UGT) is widely distributed in many organs and tissues in humans such as liver, kidney, heart, brain, and skin, with the liver showing the highest activity [Citation55,Citation56]. UGT is mainly expressed in the liver with a wide range of substrates, catalyzing the metabolic reactions of both endogenous substances (i.e., steroid hormones, bile acid, and tretinoin) and exogenous substances such as drugs (i.e., CBZ, valproate, non-steroidal anti-inflammatory drugs, morphine, zidovudine, chloramphenicol, and farnesol) [Citation57]. As an important enzyme participating in the phase II metabolic reaction in the human body, UGT increases the water solubility of its metabolic substrates through glucuronidation, ultimately promoting the excretion of these substrates. Due to its polymorphic nature, the UGT gene encodes enzymes with varied metabolic efficiency and metabolic rates on their substrates [Citation58]. In humans, the genes encoding UGT are mainly divided into two families, UGT1 containing subfamily UGT1A and UGT2 containing subfamilies UGT2A and UGT2B [Citation59].
Studies have shown that compared with the effect of CYP450 genetic polymorphism on the plasma concentration of VPA, the UGT genetic polymorphism shows larger impact (generally by >30% or up to 50% for some sites) on the plasma concentration of VPA than that of CYP450 [Citation57–60]. In humans, a total of 19 active UGT subtypes have been identified with 7 subtypes (i.e., UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, and UGT2B7) involved in the process of VPA glucuronidation, while UGT1A8 and UGT1A10 are expressed in extrahepatic tissues but rarely expressed inside the liver, showing trivial effect on the metabolism of VPA.
3.3.1. UGT1A3
UGT1A3 is mainly expressed in the liver as well as the biliary and gastrointestinal tracts. The UGT1A3 gene shows a high mutation rate with many mutation sites identified. To date, a total of 31 single nucleotide polymorphisms (SNPs) have been identified in the promoter region and the first exon of UGT1A3 gene, caused mainly by alkaline substitutions [Citation61].
Studies have shown that mutations are identified in four (i.e., T31C, G81A, A477G, and A17G) out 7 loci of UGT1A3, while no mutation is revealed in other three loci (i.e., C133T, A808G, and G342A) [Citation61]. The standardized plasma concentration of sodium valproate in children with heterozygous mutations is significantly lower than that of the wild type homozygotes with only the T31C. With the high allele frequency (36.67%) of T31C, it is concluded that the T31C genetic polymorphism impacts greatly on the plasma concentration of sodium valproate in children with epilepsy, suggesting that children with heterozygous mutations should be treated with increased dosage of sodium valproate to reach the effective range of plasma concentration [Citation61]. These conclusions are consistent with the results reported by Cho et al. [Citation62] showing that the UGT1A3 gene can affect the activity of its own transcription enzymes. The inconsistency of these results may be caused by the small sample size of the study and the difference in the characteristics of drug metabolism between adults and children. To date, studies on the effects of UGT1A3 on AEDs worldwide are still lacking and further studies are needed to investigate the roles that UGT1A3 plays in the alteration of drug plasma concentration.
3.3.2. UGT1A6
UGT1A6 is distributed in the liver, gallbladder, colon, stomach, and brain. This enzyme is composed of 531 amino acids and exhibits catalytic activity in the form of either dimers or tetramers. To date, the studies on the effect of UGT1A6 on VPA glucuronidation are intensive and relatively highly advanced. Studies have shown that the UGT1A6 gene contains a total of 28 known SNPs with 3 closely related but not completely linked sites, i.e., T19G (S7A), A541 G (T181A), and A552C (R184S), showing the highest mutation frequency and being extensively studied [Citation61].
Studies by Kang et al. on the genetic polymorphisms of UGT1A6 A541 G and A552C genes show that the mutation rates of A541 G and A552C are 33.7% and 31.0% [Citation63], respectively, which are slightly higher than 22.0% and 24.7%, respectively, as reported by Xing et al. [Citation64]. Furthermore, Kang et al. have revealed no significant difference in the sodium valproate plasma concentration in patients with UGTlA6 A541 G genotype [Citation63]. The same results are also reported by Wang et al. [Citation65]. Jin et al. have documented that the A541 G mutation rate in patients with epilepsy in Chinese Han populations is 28.9% [Citation66]. Results of statistical analysis show that the plasma concentration of VPA in patients with heterozygous UGT1A6 mutant alleles is different from that of the wild-type patients. Most wild-type patients are normal or slow metabolizers with the measured drug plasma concentration consistent with the effective drug plasma concentration, while most mutant patients are fast metabolizers with the measured drug plasma concentration relatively lower than the effective drug plasma concentration. These results are consistent with those reported in previous studies [Citation67]. Based on the statistical analysis of the mutation of UGT1A6 A522C, it is concluded that the gene mutation frequency of UGT1A6 A522C is 26.25% and the plasma concentration of VPA is significantly higher in the carriers of mutated gene, while the plasma concentration in patients with the homozygous mutation is higher than that of the heterozygous patients [Citation68]. No statistical difference is revealed between the heterozygous and homozygous patients due to the small sample size of the homozygous carriers. These results are consistent with those reported by Kang et al. [Citation63]. Furthermore, studies by Mei et al. show that mutations at rs6759892 (T19G) and rs1105879 (A552C) cause the increase of enzymatic activity, leading to decreased plasma concentration of VPA [Citation69].
In summary, results based on various studies on each of the mutation sites of UGT1A6 gene are generally consistent, suggesting that mutations of UGT1A6 gene lead to enhanced enzymatic activity, accelerated VPA metabolism, and reduced drug plasma concentration [Citation63–69]. Therefore, it is recommended that the dosage of medicines for patients with mutations at these common mutation sites of UGT1A6 could be appropriately increased in order to quickly reach the therapeutic window and to optimize the therapeutic effect of the medicines.
3.3.3. UGT1A9
UGT1A9 is mainly expressed in the liver (with relatively high expression level) and kidney, playing an important role in the metabolism of drugs and foreign substances. To date, a total of 21 alleles have been identified in the UGT1A9 gene, including 11 located in the promoter, 4 in the first exon, and 6 in the first intron [Citation61]. Guo et al. have reported that the 4 gene mutations of UGT1A9 located in the first exon cause changes in metabolic capacity [Citation70]. However, these mutations are rare in various ethnic groups with a frequency of less than 1%, showing trivial significance in population studies of these mutations. Similarly, Lin et al. have reported that based on the results of statistical analysis on multiple mutation sites of UGT1A9, no mutations are detected in rs72551330 (T98C) in selected populations [Citation71]. These results are consistent with the findings by Guo et al. [Citation70], i.e., the UGT1A9 mutations, including rs13418420 (T1819C), rs2741045 (C441T), rs2741049 (CI399T), and rs6731242 (T1888G), showing no significant effects on the plasma concentration of VPA.
3.3.4. UGT2B7
UGT2B7 is mainly expressed in the liver and is the most important glucuronyl transferase with a wide range of substrates and the strongest activity in VPA metabolism. The UGT2B7 gene shows a high degree of polymorphism with more than 30 mutations reported in the promoter, intron, and exon regions [Citation61]. The results of a genetic study of UGT2B7 based on 239 Norwegian patients by Holthel et al. [Citation72] show that linked mutations are revealed in the UGT2B7 promoter region at the sites 1246, 1239, 840, 268, 211, 161, and 102, while the genetic polymorphisms at the sites 268 and 102 affect the clearance efficacy of VPA.
Results of a statistical analysis by Zou et al. show that the mutation rate of UGT2B7 A268G in the Chinese Han population is 69.95% [Citation73], which is similar to the mutation rate of 70.0% reported by Zhang et al. [Citation74]. Both of these studies conclude that the genotypes of the patients are associated with the VPA plasma concentration. In particular, the plasma concentration in patients of wild-type homozygous is significantly higher than those of the other genotypes, while the difference in the average plasma concentration among patients of different genotypes is statistically significant. The mutation of the bases from A to G at this site leads to an increase in the ability of patients to metabolize VPA and a decrease in drug plasma concentration. Because the metabolic capacity of wild-type patients is relatively weak, these patients may be treated with reduced dosage of medicine or adjusted medication program.
Zou et al. have reported that the mutation frequency of G211T locus in the Chinese Han population is 22.58% [Citation73], which is slightly higher than 15.8% as previously reported [Citation75]. No significant difference is revealed in the plasma concentration of VPA among patients with different genotypes at this locus. The same results are also reported by Sun et al. [Citation57]. Studies on the UGT2B7 C802T locus by Zhang et al. show that the mutation rate of the selected population is 63.73% [Citation76], which is comparable to the 68.5% reported by Chu et al. [Citation77]. The plasma concentrations of patients with both CT and TT genotypes are significantly lower than that of CC genotype, indicating that the mutation at C802T site leads to the enhanced enzymatic activity and increased VPA metabolism. These conclusions are supported by the results reported previously [Citation78]. Therefore, it is recommended that the patients of epilepsy with mutant UGT2B7 C802T gene be given a relatively high dose of VPA for the initial treatment. Then, after reaching a steady state, the plasma concentration is monitored to achieve an effective plasma concentration. This therapeutic strategy is suggested to avoid insufficient drug concentration to affect its efficacy.
In the UGT family, the relationship between UGT1A6 and UGT2B7 genetic polymorphisms and their effects on the VPA plasma concentration of patients with epilepsy is relatively clear, while the data on other genetic subtypes are lacking and the possible linked mutations in other genotypes cannot be ruled out [Citation79]. In order to study the overall effects of the genotypes on the plasma concentration of VPA, it is necessary to investigate not only the SNPs with prominent impact, but also the comprehensive analysis of multi-site polymorphisms [Citation80]. As an important metabolic enzyme involved in phase II reaction, the genetic polymorphism of UGT inevitably plays an important role in the clinical drug application, providing imperative guidance for precise therapy of patients.
3.4. Effect of transporters on the plasma concentration of valproic acid
Drug transporters are a group of functional membrane proteins located on the cell membranes. There are many types of transporters involved in drug transmembrane transport in the human body. Based on the direction of substrate transmembrane transport, these transporters are divided into two major types, one type involved in drug absorption and the other in drug efflux [Citation81]. The transporters mediating the drug to enter the cells take up the substrate to the target site to exert its effect. These transporters contain solute carriers (SLCs), mainly including L-type amino transporters (LATs), peptide transporters (PEPTs), sodium dependent secondary active transporters (SGLTs), sodium-independent facilitated diffusion transporters (GLUTs), monocarboxylate transporters (MCTs), organic anion transporters (OATs), and organic cation transporters (OCTs). Transporters mediating drug efflux mainly include P-glycoprotein (P-gp), multi-drug resistance associated proteins (MRPs), breast cancer resistance protein (BCRP), and bile salt export pump (BSEP) [Citation82]. These transporters belong to the ATP-binding cassette (ABC) family, using the energy of hydrolyzing ATP to transport drugs and other endogenous substances.
3.4.1. ABCC2
In the clinical application of sodium valproate, more than half of the patients show resistance to AEDs, not achieving the expected effects of AEDs in clinical treatment [Citation83]. To date, among the studies on the resistance mechanism of sodium valproate, it is generally believed that reduced sensitivity of the target site and excessive transporters may be the main mechanisms causing the resistance [Citation84]. As one of the members of the ABC family, the ATP-binding cassette-2 (ABCC2) shows high genetic polymorphism. The mutations in ABCC2 enhance its functions and in turn affect the drugs to enter the cytoplasm in brain tissue, reducing the plasma concentration of the drugs and ultimately affecting the therapeutic efficacy [Citation85]. The ABCC2 gene is located at position 24 of human chromosome 10 with a low content in normal brain tissue and mainly plays an important role in drug absorption and excretion [Citation86].
Studies by Liu et al. based on ABCC2 rs3740066 (C3972T) and rs717620 (C24T) sites show that the mutation rates of C3972T and C24T sites in the selected population are 28.84% and 25.92%, respectively [Citation87]. The C3972T genetic polymorphism is closely related to the efficacy and adverse reactions of neurological drugs. Although the mutation at C3972T site is synonymous, it still shows impact on the function of ABCC2, causing blocked drug absorption and excretion and affecting drug plasma concentration and therapeutic effects [Citation88]. This study also shows that the standardized drug plasma concentration of patients with CC genotype at C3972T locus is higher than those of CT and TT genotypes, suggesting that mutations at C3972T locus affect the function of ABCC2, causing the changes in transporter<apos;>s function, ultimately leading to the blocked absorption and excretion of sodium valproate and reduced plasma concentration. Furthermore, this study demonstrates that the clinical efficacy of carriers of the T allele at C3972T locus is lower than that of non-carriers, indicating that the C→T mutation at this site may affect the therapeutic effect in patients. The C24T locus is located in the 5’ untranslated region of the ABCC2 gene. Studies have shown that C24T polymorphism affects gene transcription activity, causing the individual variations in the function of ABCC2 [Citation89]. These studies also show that the standardized plasma concentration and plasma concentration in patients of C24T CC genotype are higher than those of CT and TT genotypes, suggesting that the SNPs of C24T may reduce the transporter activity and the plasma concentration of sodium valproate.
Studies by Chen et al. on the genotypes of rs2273697 (G1249A) and rs3470066 (C3972T) sites of ABCC2 gene show that the mutation rates of the G1249A and C3972T sites in the selected population are 9.37% and 24.16% [Citation90], respectively, which are comparable to those reported by Liu et al. [Citation87]. However, Chen et al. [Citation90] have revealed no correlation between ABCC2 C3972T site and either the plasma concentration or efficacy of VPA. These inconsistencies may be due to either the lack of appropriate statistical analysis of the possible synergetic effects between the sites or the biased statistical results due to insufficient sample size. The ABCC2 rs2273697 (G1249A) is a missense mutation with the conversion of the tyrosine to isoleucine at position 417. Chen et al. have reported that the plasma concentration of VPA in patients with genotype AA at ABCC2 G1249A site is significantly higher than those of carriers of genotypes GA and GG, probably due to the decreased efflux transport activity of ABCC2 [Citation90].
In summary, patients with ABCC2 mutation sites show lower VPA plasma concentration than those without mutation sites, and the drug efficacy in the carriers of ABCC2 gene mutations may be affected. Therefore, in the clinical application of VPA, patients with ABCC2 mutant genotypes could be treated with appropriately increased dosage to ensure the therapeutic effect.
3.4.2. ABCB1
The ATP-binding cassette subfamily B member 1 gene (ABCB1), also known as the multi-drug resistance gene (MDR1), is expressed in human brain in the form of P-glycoprotein (P-gp), which is a transporter functioning as an ATP-dependent drug efflux pump used for AEDs such as sodium valproate [Citation91]. These pumps reduce the level of therapeutic drugs in the cells by using the energy released by decomposing ATP to actively transport a large number of AEDs outside the brain capillary endothelial cells, ultimately developing drug resistance [Citation92]. Human P-gp is encoded by the ABCB1 (MDR1) and ABCB4 (MDR2) genes, while the P-gp encoded by MDR2 is not involved in drug delivery. The ABCB1 genetic polymorphism affects the expression and/or function of P-gp, causing the substantial variations in the multi-drug resistance among individuals [Citation93]. The G2677A/T mutation of the ABCB1 gene causes amino acid changes related to the function of transporting P-gp and the changes of protein expression, while the C3435T and C1236T mutations located in the coding region are also related to changes in protein expression without altering the amino acids [Citation94].
Results of the statistical analysis on the genotype of the ABCB1 gene and the VPA plasma concentration by Ding et al [Citation84]. show that the mutation rates of C1236T, C3435T, G2677T, and G2677A in the selected population are 55.24%, 35.31%, 47.20%, and 8.39%, respectively. Based on their efficacy, the patients of epilepsy in Chinese Han population treated with VPA monotherapy are grouped into (1) drug-resistance group (treated with regular AEDs either reaching the standardized plasma concentration or the maximum dosage commonly used but the frequency of seizures not reduced by 50%) and (2) the effective control group with the frequency of seizures reduced by more than 50%. Results of statistical analysis of the genotype distribution of each site of ABCB1 in both groups show that there is a significant linkage disequilibrium among the genetic polymorphisms of C1236T, G2677T, and C3435T [Citation95]. The VPA resistance group contains more haplotype CGC but less haplotype TTT than the effective control group, suggesting the association between haplotype CGC and the VPA resistance as well as the association between haplotype TTT and effective control of drugs. Furthermore, the incidence of CC type at C3435T locus in the drug-resistance group is higher than that of the effective control group, while the incidence of C1236T CT type is lower than that of the effective control group. No significant difference in the distribution of G2677T/A genotype is revealed between the two groups, suggesting that C3435T CC genotype may be more susceptible to drug resistance, while the application of VPA to patients of epilepsy with C3435T CT and C1236T CT genotypes may be more effective to control seizures than to patients with other genotypes.
4. Carbamazepine
4.1. Metabolic characteristics of carbamazepine
As a type of traditional clinical AED, carbamazepine (CBZ) shows optimal effect on psychomotor seizures. CBZ reaches a peak plasma concentration of 8–12 μg/mL in 4–5 hours after oral administration, while it takes 8–55 hours to reach a stabilized plasma concentration demonstrated by rapid distribution throughout the entire body with the plasma protein binding rate of ~76% and the bioavailability of 58–85%. By enhancing the inactivation efficiency of sodium channels, the CBZ restricts the divergence of high-frequency action potentials in the postsynaptic neurons, blocks the divergence of presynaptic sodium channels and action potentials, and blocks the release of neurotransmitters, ultimately regulating the nerve excitability and generating anti-convulsant and antiepileptic effects. Furthermore, CBZ has also been used in a wide range of clinical applications, including anti-peripheral neuralgia and anti-arrhythmia [Citation70]. However, this medicine should be used with caution because adverse reactions are frequently revealed in digestive, hematopoietic, and endocrine systems [Citation58,Citation96]. Moreover, there are still problems such as large individual differences in plasma concentration and drug resistance in the current clinical application of CBZ. Therefore, it is important to develop individualized treatment plans based on the detection of CBZ plasma concentration in patients.
4.2. Effect of cytochrome P450 on the plasma concentration of carbamazepine
Studies have shown that CBZ metabolism is mainly catalyzed by CYP3A4, CYP3A5, CYP2C8, and CYP1A2, with both CYP3A4 and CYP3A5 playing the main roles [Citation97Citation98]. CBZ is a highly efficient inducer of CYP3A4, CYP3A5, and other phase I and phase II metabolic enzymes in the liver, increasing the efficiency of substrate metabolism to reduce the plasma concentration of itself and other drugs metabolized by CYP3A4 and CYP3A5 [Citation99].
4.2.1. CYP3A4
CYP3A4 is the metabolic enzyme accounting for the highest content in the CYP450 family, participating in metabolism in vivo of 45–60% of currently available therapeutic drugs such as CBZ, cyclosporine, and lidocaine [Citation100]. The activity of CYP3A4 is affected by age, gender, and diet of the patients, and particularly, genetic components as the dominant factor causing individual differences [Citation101]. The CYP3A4 gene is located on chromosome 7, with a length of ~27.2 kb, containing a total of 13 exons and 12 introns. To date, the CYP3A4*1 G (rs2242480) mutation changing from G to A at position 82,266 is located in intron 10 and identified as having relatively high mutation rate of 22.1–29.9% in Chinese population [Citation74]. As another mutation site in intron 10 of CYP3A4 gene, rs4646440 is revealed to have a mutation rate of ~25% in Chinese population [Citation102]. The CYP3A4 mutations increase the activity of CYP3A4 enzyme and accelerate drug clearance. Results of the statistical analysis by Wang et al. [Citation103] on loci rs2242480 and rs4646440 of CYP3A4 reveal no correlation between the genotypes of these two sites and the CBZ plasma concentration. These results are consistent with those reported by Yun et al. [Citation104], while Wang et al. [Citation105] have report that the adjusted concentration and standardized concentration in patients of genotype GG at CYP3A4*1 G locus are significantly higher than those of genotypes AG and AA, though no significant difference is found between genotypes AG and AA. These results indicate that patients with wild type CYP3A4*1 G exhibit low metabolism, supporting that CYP3A4 mutations can lead to increased enzymatic activity. However, these studies do not reveal the correlation between the CYP3A4 genetic polymorphism and the efficacy of CBZ [Citation103–105], probably due to the trivial changes in the plasma concentration of CBZ caused by the CYP3A4 polymorphism not making significant change in the drug efficacy [Citation105]. Alternatively, after offsetting the effects of other genes, CYP3A4*1G is not sufficient to be a sole factor affecting the efficacy of CZB.
4.2.2. CYP3A5
CYP3A5 is located on human chromosome 7 q21.1–22.1 with a length of ~31.8 kb, containing a total of 13 exons and encoding 502 amino acids. The expression and activity of CYP3A5 vary considerably among individuals in different ethnic populations. High expression of CYP3A5 is revealed in ~10-20% of adults with CYP3A5 accounting for more than 50% of the total CYP3A enzyme content in liver [Citation106]. Jounaidi et al. have discovered the first SNP of CYP3A5, i.e., the substitution from cytosine (C) to adenine (A) at position 1280 of the 11th exon of CYP3A5, resulting in a mutation at position 398 with tryptophan changed to asparagine and the creation of a Tsp509 restriction site named CYP3A5*2 [Citation107]. The CYP3A5 A6986G is a single-base mutation of intron 3 from adenine (A) to guanine (G) (CYP3A5*3), producing several splice variant mRNAs to shift the stop codon forward and resulting in the absence of CYP3A5. Studies have shown that the CYP3A5*3/*3 genotype (GG genotype) is not expressed to generate CYP3A5 [Citation106], while the CYP3A5*3 is the most common type of SNP with a mutation rate of 71–76% in the Chinese population [Citation108].
Ren et al. have reported that the mutation rate of the CYP3A5 A6986G locus in the selected population is 79.17% [Citation109], which is comparable to those reported previously [Citation68]. Ren et al. further recognize the genotypes AA and AG as the CYP3A5 expression group and genotype GG as the CYP3A5 non-expression group [Citation109]. Results show that the dosage-corrected plasma concentration and the standardized plasma concentration of the CYP3A5 non-expression group are significantly increased by 27.9% and 32.2% in comparison to those of the CYP3A5 expression group, respectively. These results are similar to those reported by Park et al. [Citation110], i.e., the plasma concentration of CBZ in the CYP3A5 non-expression group is 31% higher than that of the CYP3A5 expression group, which show a 29% higher oral clearance efficacy. These results are consistent with those reported by Liu et al. [Citation111], indicating that patients with heterozygous genes have weakened metabolism, and their corrected drug plasma concentration is significantly higher than that of patients with wild type homozygous genes. Furthermore, no significant difference in efficacy and adverse reactions are identified between these two groups of patients with different genotypes of CYP3A5. However, using a population pharmacokinetic model to study the effect of CYP3A5 genotype and CBZ clearance, Seo et al. [Citation112] have reported conflicting results, i.e., the CYP3A5 non-expression group shows increased CBZ oral clearance efficacy by 8% than that of the CYP3A5 expression group. The inconsistency of these results may be due to either the use of combination of drugs in some patients, while the interaction between drugs cannot be completely ruled out, or the un-corrected variations in the metabolic rate caused by CBZ self-induction.
4.3. Effect of uridine diphosphate glucuronidase on plasma concentration of carbamazepine
In the UGT family, the UGT2B is encoded by a unique gene independent of the UGT1 family and located on chromosome 4q13 with UGT2B4, UGT2B7, UGT2B15, and UGT2B28 showing the genetic polymorphisms [Citation104]. Because UGT2B7 is the only member metabolizing CBZ in UGT family, we here review the studies focusing on the effects of UGT2B7 on the plasma concentration of CBZ.
As a member of the UGT2B family, the UGT2B7 gene has a full length of 16 kb, containing 6 exons and encoding 529 amino acids. Studies have shown that both CBZ and its main effective metabolites (i.e., 10,11-epoxidized carbamazepine) are metabolized by UGT2B7. The C802T is the identification location of the coding region of UGT2B7 gene and is confirmed to affect the stabilized plasma concentration of CBZ. Zhang et al. [Citation113] have investigated UGT2B7 using a statistical analysis and reported that the mutation rate of C802T site in the selected population is 29%, which is similar to those reported in Asian populations [Citation114]. Results of Zhang et al. suggest that the standard drug plasma concentrations in the group of UGT2B7*2 genotypes are not statistically significant [Citation113]. However, an increasing trend of the standardized drug plasma concentration is observed in the patients in the order of genotypes of mutant, heterozygote, and wild type. Qu et al. have report that UGT2B7*2 genotype can affect the stabilized plasma concentration of CBZ [Citation115]. To date, the relationship between UGT2B7 genotype and CBZ metabolism is not fully verified. The establishment of an improved statistical model with the exclusion of other factors involved is still needed in order to further investigate the relationships between UGT2B7 genotype and CBZ metabolism.
4.4. Effect of transporters on the plasma concentration of carbamazepine
The drug transporters involved in the metabolism of CBZ are classified into two groups, including the families of ATP-binding cassette (ABC) transporters and solute transporters (solute carriers or SLCs). The ABC transporter family contains a total of ~50 members numbered starting with ABC, e.g., ABCB1 (MDR1), ABCC2 (MRP2), and ABCG2 (BCRP) [Citation104].
4.4.1. ABCB1
Multi-drug resistance protein 1 (MDR1) belongs to the P-gp family containing polymorphic members [Citation91]. The P-gp is encoded by the ABCB1 gene located on chromosome 7q21 and is a transmembrane P-glycoprotein (P170) with a molecular weight of 170 KD composed of 1,280 amino acid residues. The P-gp contains two homologous halves each containing 6 hydrophobic transmembrane domains and one ATP binding site [Citation116]. The P-gp binds not only to drugs but also to ATP, which provides energy to pump intracellular drugs out of the cell, ultimately decreasing the drug concentration in the cell to make the cells resistant to drugs. Studies have shown that CBZ is a substrate of P-gp and the overexpression of P-gp may be one of the mechanisms regulating the resistance to AEDs [Citation117].
Yun et al. [Citation104] have selected three most common SNPs of ABCB1 gene, i.e., C1236T (rs1128503), G2677T/A (rs2032582), and C3435T (rs1045642), to investigate the relationships between the genetic polymorphisms of these three loci and the plasma concentrations of CBZ as well as its two metabolites (i.e., CBZE and CBZD). The results demonstrate that patients with the ABCB1 3435CT mutation genotype and the 3435 TT homozygous mutation genotype show lower plasma concentrations than that in patients with the 3435CC wild genotype. However, other studies show that the C3435T genetic polymorphism is not correlated with the CBZ plasma concentration [Citation118,Citation119]. These conflicting experimental results may be caused by: (1) the activity of P-gp and the distribution of ABCB1 polymorphisms are different in different populations, (2) other transporters such as ABCC2 also play important roles during the CBZ transport process, (3) CBZ may induce the expression of P-gp in both the intestine or brain tissues, thereby reducing drug absorption, while the expression level of P-gp may also be affected by the long-term use of CBZ [Citation120], and (4) the stage of treatment or the time to collect blood samples vary in different studies. Furthermore, studies have reported that patients carrying ABCB1 3435CC wild type show higher CBZE and CBZD plasma concentrations than that of patients with 3435CT heterozygous mutations, suggesting that CBZE may be a substrate of P-gp [Citation121].
4.4.2. ABCC2
The drug transporter ABCC2 (MRP2) is encoded by the ABCC2 gene and is overexpressed in the endothelial cells of the blood-brain barrier, liver, intestine, kidney, placenta, and lung. However, the expression level of ABCC2 is relatively low in normal brain tissue, whereas the expression level of ABCC2 in the brain tissue of patients with epilepsy is relatively increased [Citation122]. In Korean populations, the 1249 G > A mutation decreases the ability of ABCC2 to efflux and increase the intracellular concentration of CBZ, causing severe side effects [Citation123]. Several possible reasons could cause these varied experimental results. First, it is uncertain whether CBZ is a substrate of ABCC2. Second, these studies may have used different standards to determine the controlled epilepsy, subsequently affecting the interpretation of clinical data [Citation124]. Third, there are potentially genetic differences between different ethnic groups of populations. Lastly, other polymorphic sites or epigenetic factors of ABCC2 may affect the metabolism and efficacy of CBZ.
5. Phenytoin
5.1. Metabolic characteristics of phenytoin
Phenytoin (PHT), also known as dalantine, is a type of sodium salt of diphenylhydantoin and is the first-line drug for the treatment of severe seizures of epilepsy. In addition to its antiepileptic and anti-arrhythmic properties, PHT has shown wide clinical applications. The novel pharmacological effects of PHT have been discovered in recent years, such as treating acute cerebral infarction and atherosclerosis, burns, and hiccups, promoting wound healing, achieving satisfying therapeutic effects, and showing broad application prospects [Citation125]. However, due to its saturated metabolism and non-linear dynamics characteristics as well as the narrow treatment window and large individual variations, the application of PHT is likely to cause many types of adverse reactions in the central nervous system and hematopoietic system [Citation126,Citation127].
PHT is highly lipid-soluble and is transported to multiple tissues and organs of the body after joining plasma proteins. It can quickly pass through the blood-brain barrier [Citation128], inhibit the passive influx of Na+ outside the cell to stabilize the cell membrane and increase its excitement threshold, and prevent the spread of abnormal brain potential activities to surrounding normal tissues, thereby inhibiting seizures of epilepsy [Citation129]. PHT is eliminated through liver metabolism, while the metabolites show no drug activity and are mainly excreted in urine with a small amount (<5%) excreted in its original form. The effective therapeutic functions of PHT require a certain degree of saturation of liver metabolic enzymes (i.e., hydroxylase). PHT exhibits the first-order kinetics at low dosage but follows the zero-order kinetics once the enzyme becomes saturated. Within the treatment range, the ability of liver to metabolize PHT reaches the highest level with the increase of drug plasma concentration, while further increase of a trivial dosage may cause an enormous change in the concentration of PHT [Citation130].
5.2. Effect of cytochrome P450 on the metabolism of PHT
In general, the phenotypes of CYP450 enzymes are categorized into four types, i.e., poor metabolizers (PMs), intermediate metabolizers (IMs), extensive metabolizers (EMs), and ultrarapid metabolizers (UMs). The effects of genetic polymorphism of CYP450 on drug metabolism, efficacy, and toxicity have been extensively studied. For example, studies have shown that under the same conditions, poor drug metabolizers often carry related mutant genes, causing the drug to accumulate in their bodies, showing either over-estimated efficacy or prone to drug poisoning with the regular dosage, while the fast metabolizers often suffer the excessive drug resistance showing low efficacy [Citation131]. We here review the studies on the effects of CYP2C9 and CYP2C19 on the plasma concentration of PHT in the treatment of epilepsy.
Studies have shown that among the ten known alleles of CYP2C9, besides the wild-type CYP2C9*1, other two most common alleles (i.e., CYP2C9*2 and CYP2C9*3) encode enzymes with reduced activities of 70% and 3–5%, respectively, in comparison to that of the wild type [Citation132]. The carriers of the variant genes account for 40% among Caucasians and only 5% in Asians with the CYP2C9*3 as the most frequent mutation in Asian populations [Citation132]. The studies of enzymatic kinetics in vitro have shown that the intrinsic clearance rate of PHT metabolism by CYP2C9*3 decreases by 6–13.5%, in comparison to that of CYP2C9 wild-type (i.e., CYP2C9*1) [Citation133]. Clinical studies have also confirmed that CYP2C9*3 has shown the ability to significantly affect the clearance of PHT drugs [Citation134]. Among the two most common mutation sites (i.e., CYP2C9*2 and CYP2C9*3), the mutations in the active site resulting in a decrease in affinity with the substrate is considered as the mechanism causing the decrease in enzymatic activity by CYP2C9*3. Furthermore, the activity of CYPs may be affected by the electrons delivered by their coenzymes, i.e., the CYP450 oxidoreductase (POR). The CYP2C9*2 is a mutation encoding the outer surface proteins. The mechanism of CYP2C9*2 causing a decrease in the metabolic rate of CYP2C9 substrate may be associated with the decrease in POR binding ability [Citation135]. Studies by Ramasamy et al. [Citation136] show that the metabolism of PHT in heterozygous carriers of CYP2C9 gene (*1/*3) is significantly lower than that of wild-type homozygous carriers (*1/*1). The activity of the enzymes encoded by CYP2C9*3 is decreased to only 3–5% of that of the wild type. With the same dose, the plasma concentration of slow metabolizers is 34% higher than that of fast metabolizers [Citation137].
Besides the wild-type CYP2C19*1, the CYP2C19*2 and CYP2C19*3 are two most common alleles among the eight alleles of CYP2C19. In the Asian populations, 55.4% of Japanese patients of epilepsy carry the CYP2C19 variant with 13–23% poor metabolites in Asian populations and 2%-5% in white and black populations [Citation138,Citation139]. Although the CYP2C19 is only responsible for 10% of PHT metabolic clearance, studies in vivo have demonstrated that patients with CYP2C19*2 or CYP2C19*3 show significantly reduced ability to metabolize PHT [Citation140]. Furthermore, the differences in drug metabolism caused by genetic polymorphisms increase the steady-state plasma concentration of PHT to toxic level and cause toxic reactions [Citation141].
Results of statistical analysis on both CYP2C9 and CYP2C19 by Ren et al. [Citation142] show that the allele frequencies of CYP2C9*3, CYP2C19*2, and CYP2C19*3 are 7.30%, 33.50%, and 3.70%, respectively, in accordance with the distributions of these genetic sites in Chinese population [Citation143]. Based on the genetic polymorphisms of CYP2C9 and CYP2C19, patients are categorized into three groups, including the extensive metabolizers (EMs) containing the genotype CYP2C9*1/*1 combined with CYP2C19*1/*1, the intermediate metabolizers (IMs) with either the CYP2C9*1/*3, CYP2C19*1/*2, or CYP2C19*1/*3, and the poor metabolizers (PMs) containing either the CYP2C19*2/*2 or CYP2C19*1/*2 combined with CTP2C9*1/*3. The study of Ren et al. [Citation142] has demonstrated that the drug plasma concentrations of the extensive, intermediate, and poor metabolizer groups significantly increase sequentially, while the significant differences are revealed in patients not reaching the minimum effective concentration and those exceeding the minimum toxic concentration among the three groups of patients. Specifically, the drug plasma concentration in 95.5% of patients of extensive metabolizers is less than the minimum effective concentration, while as high as 23.1% of poor metabolizers reaching the minimum toxic concentration. Zhou et al. [Citation144] have conducted a statistical analysis on the CYP2C19 gene of children with epilepsy treated with PHT monotherapy. The patients are categorized into three groups based on the CYP2C19 genotypes detected, including the extensive metabolizers (EMs) carrying the wild homozygous genotype *1/*1, the intermediate metabolizers (IMs) carrying the point mutation genotypes of *1/*2 and *1/*3, and the poor metabolizers (PMs) with the double site mutation genotypes of *2/*2, *2/*3, and *3/*3. After each group of patients are given the same initial dose of PHT (5 mg/kg taken in 2–3 times), then different maintenance doses of PHT are administrated to different groups, i.e., the group of EMs are given 250 mg/m2 (taken in 2–3 times), while the groups of IMs and PMs are given sequentially reduced doses. After the patients have taken PHT for more than 5 half-lives and the PHT in the body reaches the steady-state plasma concentration [Citation145], the plasma concentration of PHT is evaluated. Results show that there is no statistically significant difference in either the steady-state plasma concentration of different metabolizers or the incidence of adverse reactions.
In summary, these results indicate that the examination of drug metabolism-related genes prior to the treatment of patients with epilepsy as well as the establishment of individualized dosage plan formulated based on the results of the genotype examination would greatly facilitate the accurate control of the drug plasma concentration within the treatment window and obtaining improved therapeutic effects and decreased adverse reactions in patients.
5.3. Effect of transporters on the metabolism of PHT
To date, it is commonly accepted that P-gp is associated with the formation of refractory epilepsy. This is because P-gp is not only the expression product of ABCB1 gene, but also part of the drug efflux transport system. By hydrolyzing ATP, the drug is pumped out of the cell, thereby the effective drug plasma concentration is not reached in the cells of affected tissues, ultimately affecting the therapeutic effect of the drug. Studies have shown that P-gp is closely related to refractory epilepsy by affecting the transport of a variety of AEDs [Citation146]. We here review the studies on the effects of ABCB1 on the plasma concentration of PHT.
Kimchi et al. [Citation147] have studied the SNPs of the ABCB1 gene and reported that the C3435T site affects DNA folding by changing codon selection and application, leading to changes in protein structure. Three SNP sites (i.e., 3435C>T, 1236C>T, and 2677 G > T) on ABCB1 gene subtly alter the structure of P-gp by affecting the speed and rhythm of translation of the codon substitution, thereby changing the affinity of either regulatory factors or substrates. Siddiqui et al. [Citation95] have investigated the relationship between the ABCB1 genetic polymorphism and AED resistance based on studies of 315 patients of epilepsy. Results show that the proportion of the CC genotype of ABCB1 C3435T in AED resistant patients is significantly higher (27.5%) than that of drug sensitive patients (15.7%), while the proportion of TT genotype is significantly lower (19.5%) than that of drug sensitive patients (29.6%). Furthermore, Zimprich et al. have reported that the haplotype composed of C1236T, C3435T, and G2677T/A is related to drug resistance in patients of epilepsy [Citation148], while Hung et al. have demonstrated that the genetic polymorphisms of ABCB1 genes G2677T and C3435T are related to AED resistance [Citation149]. However, several studies have revealed no association between the genetic polymorphism of these genes and the AED resistance [Citation150,Citation151]. For example, Jiang et al. have conducted statistical analyses on the C1236T, C3435T, and G2677T/A sites of the ABCB1 gene and revealed no statistical difference in the plasma concentration of PHT among the patients of the three SNP genotypes [Citation152]. Based on the criteria for defining epilepsy resistance proposed by Shahwan et al. [Citation153], who categorize the enrolled patients into either resistance group or control group and report that different alleles at the C1236T locus show statistically different distribution frequencies in the resistance and control groups. Specifically, the distribution frequency of the C allele in the resistance group (37.9%) is significantly higher than that in the control group (23.3%), while the distribution frequency of the T allele in the control group (76.6%) is significantly higher than that in the resistance group (62.0%). At the G2677T/A locus, the distribution frequency of the G allele in the resistant group (55.0%) is significantly higher than that in the control group (32.0%), while the distribution frequency of the T allele in the control group (53.8%) is significantly higher than that in the resistant group (29.6%). The distribution frequencies of C3435T alleles are not statistically significant. It is noted that studies on the ABCB1 genetic polymorphism, epilepsy, and epilepsy resistance have reached inconsistent results. Some studies support the hypothesis of the association between genetic factors (i.e., allele, genotype, or haplotype) and epilepsy as well as epilepsy resistance [Citation154,Citation155]. However, studies on patients of epilepsy in different regions and ethnic groups do not support the association hypothesis [Citation155,Citation156]. Studies with more statistical methods to conduct the meta-analyses have verified these inconsistent and contradictory conclusions, revealing no association between ABCB1 polymorphism and epilepsy as well as drug-resistant epilepsy [Citation156–158].
Currently, the relationship between the ABCB1 genetic polymorphism and PHT plasma concentration as well as the epilepsy resistance remains unknown. Further studies are necessary based on an improved model established to adjust factors such as geographic location, ethnic difference, and site interactions.
6. Pharmacokinetic characteristics of new antiepileptic drugs
In the past 10 years, new AEDs (e.g., OXC, LEV, and LTG) have been developed and widely used in clinical treatments. These new AEDs mostly show linear pharmacokinetic characteristics with the plasma concentration proportional to the dose as well as sound oral absorption and quickly reaching peak plasma concentration. Furthermore, due to their high bioavailability, the absorption of these AEDs are not affected by the type of food taken, while the plasma protein binding rate is low and the drug is generally widely distributed in the tissues [Citation159,Citation160].
The new AEDs are partially metabolized in the liver and are excreted from the urine through the kidneys either in the original form or as metabolites [Citation159–161]. Compared with the traditional AEDs, the new AEDs rarely interact with other drugs [Citation162]. To date, the new AEDs globally investigated and significantly affected by metabolic enzymes and their genetic polymorphisms include LTG, OXC, and LEV. We here review the studies on the relationship between the plasma concentration of these new AEDs and the genetic polymorphism of genes encoding enzymes involved in the metabolism of these new AEDs.
7. Lamotrigine
Lamotrigine (LTG) is one of the most commonly used new generation AEDs with a broad therapeutic spectrum. It is effective for some common types of seizures, such as simple partial seizures, complex partial seizures, partial seizures with secondary generalization, primarily generalized tonic-clonic seizures, and absence seizures. LTG is also used as a mood stabilizer for the treatment of bipolar disorder, particularly effective for patients in depressive phase of bipolar disorder without inducing mania [Citation163]. LTG mainly shows antiepileptic effects by blocking the voltage-sensitive sodium ion channels of the presynaptic membrane, thereby inhibiting the release of excitatory amino acids (i.e., glutamate and aspartate). Recent studies have suggested that LTG can also exert antiepileptic activity by blocking the voltage-sensitive calcium channels [Citation164]. LTG can be rapidly and completely absorbed by oral administration without evident first pass elimination. Its bioavailability is high (98%) with the plasma protein binding rate of 55%.
7.1. Metabolic characteristics of lamotrigine
LTG is mainly metabolized by UGT enzymes in the human body to convert into conjugated products of hydrophilic glucuronic acid and excreted by the kidneys, while the phase I metabolic enzyme CYP does not participate in the metabolism of LTG in vivo. LTG shows no evident hepatic enzyme induction, while its self-induction appears in the early stage of treatment with no distinct clinical significance [Citation77]. About 75–90% of the dose of LTG is excreted in the form of glucuronic acid conjugated products, consisting of mainly 2-N-glucuronide and 5-N-glucuronide accounting for ~10%, while less than 10% of the dose of LTG is excreted in its original form with the fecal excretion accounted for only 2%. The highly safe LTG shows a half-life of 15–30 hours and is the first choice of AED for women of childbearing ages.
Similar to other AEDs, LTG passes through the blood-brain barrier to reach the epileptic center to exert its antiepileptic activity. Studies have shown that the concentration of LTG in the cerebrospinal fluid is correlated with the concentration of LTG in the circulating blood [Citation165]. Therefore, the concentration of LTG in plasma is a strong indication of the efficacy of LTG. Some patients with epilepsy take large doses of medicine without achieving satisfactory therapeutic effects, while other patients show severe adverse reactions with small doses. Therefore, it is clinically important to monitor the plasma concentration of LTG in order to achieve satisfactory therapeutic effects and to avoid severe adverse reactions in patients of epilepsy.
7.2. Effect of uridine diphosphate glucuronidase on the metabolism of lamotrigine
The enzymes UGT1 and UGT2 play important roles in the metabolism of endogenous and exogenous substances in the human body. These enzymes use UDP-glucuronic acid groups as the glycosyl donors showing high activity in catalyzing the phase II binding reactions. The compounds glycosylated by UGT1 and UGT2 enzymes show increased hydrophilicity and are easily excreted from the human body. The function of UGT3 enzyme is currently unclear. It has been reported that UGT8A1 enzyme uses UDP-galactosyl as a glycosyl donor to catalyze the synthesis of cell membrane components, while enzyme UGT1A4 is the main isoenzyme involved in the acidification reaction of LTG [Citation166]. Studies have shown that enzymes UGT2B7 [Citation167], UGT1A3 [Citation168], and UGT1A1 [Citation169] participate in the acidification reaction of LTG with both UGT2B7 and UGT1A3 showing lower catalytic activities, while UGT1A4 showing 10 times higher catalytic activities than that of UGT1A3 [Citation167,Citation168].
7.2.1. UGT1A4
Enzyme UGT1A4 is mainly expressed in the human liver, bile duct, small intestine, and colon, mainly involved in the metabolism of drugs containing primary and secondary amine structures. It has been reported that the mutation frequencies of UGT1A4 C457T, G419A, G163A, and C471T in Chinese population are 0.190, 0.191, 0.194, and 0.998, respectively [Citation170]. Ehmer et al. have experimentally confirmed for the first time the association between UGT1A4 genetic polymorphism and the enzymatic functions [Citation171]. Specifically, it is revealed that the mutation of exon 1 of UGT1A4 gene affects the function of the enzyme. Two mutation sites are identified with one located at the 70th base causing the change from proline to threonine at position 24 (P24T), and the other at the 142nd base causing the change from leucine to valine at position 48 (L48V). These two mutations also affect the enzymatic activity of UGT, i.e., UGT1A4 gene mutations P24T and L48V cause the decrease of the UGT enzymatic activity [Citation171].
Yang et al. have investigated the relationship between UGT1A4 T142G locus and the plasma concentration of LTG to show that the distribution frequencies of UGT1A4 142 T > G alleles T and G are 87.74% and 12.26%, respectively [Citation172], which are similar to that of the Japanese population (12.9–16.5% for G allele) as previously reported [Citation173,Citation174]. Furthermore, the standardized plasma concentration of LTG in patients with TT genotype at UGT1A4 T142G locus is significantly higher than that of patients with TG and GG genotypes, and the clinical efficacy of patients with TT genotype is significantly higher than that of patients with TG and GG genotypes. Gulcebi et al. have reported the similar results, i.e., UGT1A4 T142G site mutation increases the catalytic activity of UGT enzyme, resulting in a decreased plasma concentration of LTG, e.g., the plasma concentration of LTG in T142G mutation carriers is 52% lower than that of wild-type patients [Citation175]. These results are consistent with those reported by He et al. [Citation176], further revealing that in the combined medication, the plasma concentration of VPA is related to the plasma concentration of LTG, i.e., VPA increases the plasma concentration of LTG by 1.0%. However, the results of Liu et al. [Citation177] have revealed no association between the UGT1A4 T142G polymorphism and the LTG plasma concentration with the LTG used alone, whereas when used in combination with VPA, the LTG plasma concentration of mutant patients is higher than that of wild type, similar to the results of the aforementioned studies. These results indicate that patients with UGT1A4 T142G allele G show accelerated metabolism of LTG and reduced plasma concentration in the body. Therefore, it is recommended that these patients be treated with appropriately increased dosage of LTG under the clinical treatment, whereas the wild-type (TT) patients with relatively weak metabolic capacity should be treated with appropriately reduced dosage.
7.2.2. UGT2B7
The subtype enzymes of UGT287 are also involved in the LTG metabolism. To date, studies have explored the effects of genetic polymorphism of UGT287-C161T and UGT287 G211T/A on LTG plasma concentration in the populations of epilepsy in Caucasian, Japan, Thailand, and China [Citation178–181], confirming that the SNPs are associated with LTG concentration in Caucasian, Japanese, and Thai populations but not in Chinese population of epilepsy. Therefore, it is concluded that the effects of SNPs on drug plasma concentration vary among different ethnic populations, ultimately causing different drug efficacy in different populations and individuals. Milosheka et al. have conducted a population pharmacokinetic analysis based on 100 patients treated with LTG [Citation182]. Results show that the UGT2B7 C161T and A372G genotypes are associated with the clearance rate of LTG, which is significantly lower in carriers of −161 TT than that of carriers of −161CC, and significantly higher in carriers of −372 GG than that of −372AA carriers. However, studies by Zhou et al. and Liu et al. on Chinese patients of epilepsy have not revealed the significant effect on LTG plasma concentration by UGT2B7 C161T, G211T, A372G, A735G, and C802T SNPs [Citation177,Citation182,Citation183]. These results indicate that the sites of the UGT2B7 gene have not shown any significant effects on the metabolism of LTG in the Chinese population. Further studies are still needed to verify these conclusions.
7.3. Effect of transporters on the metabolism of lamotrigine
Previous studies have shown that genetic polymorphisms of drug-metabolizing transporters, such as the P-gp encoded by the MDR1/ABCB1 gene, the BCRP encoded by ABCG2 gene, and the multi-drug resistance-related protein-2 encoded by MDR2/ABCC2 genes, significantly affect the pharmacokinetics and bioavailability of AEDs. The overexpression of these transporters has been shown to affect the plasma concentration of LTG and cause different therapeutic effects [Citation184]. As a subfamily of influx transporters, organic cation transporters (OCTs) play important roles in the distribution and excretion of cationic drugs as well as non-charged compounds.
7.3.1. OCT1
The organic cation transporters (OCTs) are the subtypes of internal transporter proteins and are mainly involved in the transport process of organic cation proteins and uncharged complexes [Citation185–187]. Studies by Dickens et al. show that OCT1-mediated active transport plays an important role in maintaining the intracellular concentration of LTG, suggesting that LTG may serve as a substrate of OCT1 in cerebral vascular endothelial cells [Citation188]. Lu et al. have reported that the mutation rate of OCT1 A1222G (rs628031) site in the selected population is 67.65% [Citation189], which is similar to that reported previously [Citation190], showing a correlation between the A1222G locus genotype and the LTG plasma concentration but not associated with the efficacy of LTG. Furthermore, the plasma concentration in patients of GG genotype is 25.6% lower than those of AA and AG genotypes, suggesting that patients with GG genotype require a higher maintenance dose of LTG in order to achieve the recommended effective plasma concentration. However, no significant correlation is identified between the OCT1 C1022T site and the LTG plasma concentration as well as its efficacy. To date, studies on the effects of OCT gene on the plasma concentration of LTG are sparse [Citation188–190]. The explicit relationship between OCT and the plasma concentration and metabolism of LTG remains to be investigated by further studies.
7.3.2. ABCG2
The BCRP belongs to the ABC transporter superfamily and is encoded by the ABCG2 gene, distributed in a variety of tumor cells and normal human tissues, such as the brain, liver, and small intestine, playing important roles in transport and efficacy of various types of drugs, including LTG [Citation191]. Studies based on the ABCG2 knockout mice have shown that ABCG2 may be involved in the distribution of LTG in the brain [Citation192], while the genetic polymorphism of ABCG2 may affect the expression and function of ABCG2 protein, thereby affecting the pharmacokinetics of the drugs [Citation193].
Lu et al. have reported that the patients with ABCG2 C421A (rs2231142) AA show a higher plasma concentration of LTG compared with those of CC and CA genotypes [Citation189]. However, no evident association is revealed between ABCG2 G34A (rs2231137) genetic polymorphism and plasma concentration of LTG. Zhou et al. have also reported that the ABCG2 rs2231142 genetic polymorphism show a significant effect on LTG plasma concentration [Citation136]. Among the genotype distribution, AA, CA, and CC genotypes account for 10.7%, 44.3%, and 45%, respectively [Citation190]. Similar to the above results, patients with ABCG2 rs2231142 allele A may be treated with a relatively lower dosage of LTG in order to reach the maintenance plasma concentration required for the treatment. Studies have shown that the expression of allele A increases the risk of transporters being degraded by the proteasome, ultimately reducing the binding of transporters to cell membranes [Citation194]. It is noted that He et al. have reported that there is no significant correlation between the ABCG2 rs17731538 locus and the LTG plasma concentration and drug resistance in Chinese patients of epilepsy [Citation195].
7.3.3. ABCB1
The P-gp is involved in the transport of a variety of drugs, while the ABCB1 genetic polymorphism causes varied effects and concentrations of multiple drugs in different genotypes. Studies have shown that LTG is a substrate of P-gp, which participates in the transport of LTG through the blood-brain barrier and endogenous cells [Citation196], while the P-gp actively transports drugs out of the cell using the energy released from ATP hydrolysis, reducing the intracellular drug concentration. Studies have shown that the mutation of ABCB1 C1236T is associated with both the dose-corrected concentration of LTG and the maintenance dose of LTG [Citation197,Citation198]. To date, the most comprehensively studied ABCB1 gene sites are C1236T, G2677T/A, and C3435T with their genetic polymorphisms affecting the expression and functions of P-gp. The G2677T/A is a missense mutation encoding either serine or threonine mutated from alanine [Citation199,Citation200].
Lovrić et al. have investigated the correlation between the LTG plasma concentration and ABCB1 SNPs based on 222 Croatian patients [Citation198]. Results show that the LTG dose-corrected concentration of 1,236 CC gene carriers is significantly higher than those of CT and TT carriers. However, no significant association is revealed between the ABCB1 G2677T and C3435T genes and the LTG plasma concentration [Citation198]. Furthermore, Huang et al. have reported that when the ABCB1 2677 G (rs2032582) TT genotype and the C3435T (rs1045642) TT genotype coexist, the distribution volume of LTG in the body increases by 136.0%, while the plasma concentration may decrease [Citation201]. However, other studies based on Chinese population have not found the correlation between the ABCB1 genes and the LTG plasma concentration [Citation189,Citation195,Citation202].
In summary, there is no specific mutation site affecting the LTG plasma concentration by LTG metabolic enzyme and transporter genetic polymorphisms, probably due to the complexity of the LTG metabolism and transport process not affected by a single metabolic enzyme and transporter. The LTG plasma concentration is generally affected by many non-genetic factors such as age, drug combination, and estrogen level of the patients. Therefore, in order to explicitly clarify the effects of metabolic enzymes and transporter SNPs on the LTG plasma concentration, it is necessary to further pursue clinical studies with multiple centers and large sample size to correct the effects of age, combination of medication, and estrogen levels of the patients, to introduce the population pharmacokinetics to quantitatively describe the effect of genetic polymorphism on LTG metabolism, and ultimately to provide evidence for establishing individualized clinical administration of LTG.
8. Oxcarbazepine
As a type of new generation AED, the oxcarbazepine (OXC) was approved by the U.S. Food and Drug Administration in 2000 for the treatment of epilepsy [Citation203]. As the first-line drug choice for patients with newly diagnosed or untreated focal epilepsy, OXC could be used either alone or in supplementary treatment [Citation204,Citation205]. OXC is a 10-keto derivative of CBZ, while the pharmacokinetics of OXC and CBZ are different with the tolerability of OXC significantly higher than that of CBZ [Citation206].
In the actual clinical applications for the treatment of the same type of epileptic seizure, the dosage of OXC used by different individuals varies greatly. One of the reasons causing these variations is probably the gene mutation with one of its genetic basis being the genetic polymorphism of the genes encoding related enzymes and receptors, ultimately causing the individual differences in the optimal dose of OXC in different individuals with epilepsy [Citation207,Citation208]. Furthermore, the effective plasma concentration of OXC fluctuates in the relatively narrow range of 5–30 μg/ml. The understanding of pharmacogenomics of OXC will facilitate the clinical drug selection, drug dosage adjustment, and reduction of adverse drug reactions.
8.1. Metabolic characteristics of oxcarbazepine
As a derivative of CBZ, the OXC shares the similar chemical structure with CBZ and the pharmacological mechanism of blocking the voltage-sensitive sodium ion channels [Citation209]. As an inactive prodrug, OXC is quickly reduced by cytoplasmic enzymes in the liver to a pharmacologically active monohydroxy oxide, i.e., the 10,11-dihydro-10-hydroxy-carbamazepine (MHD) [Citation210]. The MHD contains two types of enantiomers, i.e., S-type and R-type, with the ratio of ~4:1 in the body. Animal experiments have shown that the antiepileptic activities of both S-type and R-type are the same but with inconsistent pharmacokinetics. Previous studies have shown that UGT2B15 is the metabolizing enzyme for S-type glucuronidation, while UGT2B7 and UGT1A9 are the metabolizing enzymes for the R-type glucuronidation [Citation211]. Studies have shown that R-type glucuronidation is not affected by patient<apos;>s genotype and gender, whereas the gender and UGT2B15 D85Y genotype are the main factors affecting the S-type glucuronidation [Citation212]. The metabolism of OXC generating its active products is non-enzyme-inducible, which is hardly affected by CYP450 metabolic enzymes, though with trivial enzymatic induction or inhibitory effects, inducing the activity of CYP3A4/5 [Citation213].
8.2. Effect of cytochrome P450 on the metabolism of oxcarbazepine
The effects of CYP450 enzymes on the metabolism of OXC have been determined using hepatocytes and HepaRG cells in vitro. Results show that as three main enzymes involved in the metabolism of OXC, CYP1A2, CYP2B6, and CYP3A4 are induced by OXC [Citation214]. Experiments in vitro have demonstrated that OXC can induce CYP1A2, CYP2B6, and CYP3A4, while 10-hydroxyoxcarbazepine induces CYP2B6 and CYP3A4 but not CYP1A2, as measured at mRNA level in the hepatocytes and HepaRG cells cultured for 72 hours [Citation215].
8.2.1. CYP3A4 and CYP3A5
CYP3A is an important subfamily of CYP450 and is the most abundant hepatic drug-metabolizing enzyme in the liver. As two important members of the CYP3A family, CYP3A4 and CYP3A5 share high homology and are abundant in the liver with ~150 drugs as their substrates, accounting for about 50% of all drugs. The activity of CYP3A4 is mainly affected by genetic factors. Studies of the effects of the CYP3A4 genetic polymorphism on its enzymatic activity have shown that CYP3A4*1G mutation increases enzymatic activity and accelerates drug metabolism [Citation105,Citation216]. Studies by Wang et al. have not revealed the positive correlation between the plasma concentration of OXC and the CYP3A4/3A5 genetic polymorphisms [Citation217], indicating that OXC can inhibit CYP3A4/3A5 enzymes, but in turn affecting the metabolism of other drugs, while OXC itself is not affected by these enzymes.
8.2.2. CYP2C19
Although the OXC is not metabolized by CYP450, previous studies have shown that both OXC and its metabolite MHD inhibit the CYP2C19 enzyme, indicating that it is possible for the OXC to bind with enzyme to affect its own metabolism [Citation218]. To date, the main CYP2C19 variants in the Chinese population are CYP2C19*2 and CYP2C19*3, while no significant difference is revealed in the inhibitory effects of AEDs on patients with CYP2C19*2 and CYP2C19*3 genotypes [Citation209]. A large number of studies have shown that there are three phenotypes of CYP2C19 enzyme, including the fast metabolizing type (636 GG), the medium metabolizing type (636 GA), and the slow metabolizing type (636AA) [Citation218]. Zhou et al. have investigated the relationships between the CUP2C19*3 locus genotype and the OXC plasma concentration [Citation210]. Results show that the frequencies of alleles G and A are 68% and 32%, respectively. Patients of different metabolizing types show significantly different effects on MHD plasma concentration. Specifically, the drug plasma concentrations of patients with slow and fast metabolizations are the highest and the lowest, respectively, indicating that the slow metabolization causes a decrease in CYP2C19 enzymatic activity, thereby increasing the plasma concentration of MHD. These results further suggest that the CYP2C19*3 genetic polymorphisms of different patients lead to significant differences in MHD plasma concentrations, causing reduced therapeutic effects or aggravated adverse reactions. Therefore, these problems should be monitored cautiously in clinical settings.
Zhou et al. have studied the relationships between the CYP2C19 genotype and the metabolism of OXC in children with epilepsy in Chinese population [Citation219]. Results show that the genotype frequencies of CYP2C19*2 locus *1/*1, *1/*2, and *2/*2 are 53.85%, 33.85%, and 12.31%, respectively, while the allele frequency of *2 is 29.23%, which is in accordance with the distribution of this gene locus in the Chinese population reported previously [Citation220]. Results of univariate analysis have demonstrated that the CYP2C19*2 locus genotypes show no significant effect on the MHD plasma concentration. However, as the number of mutant alleles increases in patients, the average drug plasma concentration shows an increasing trend. These results indicate that OXC is not metabolized by CYP450 enzymes. Therefore, the difference in enzymatic activity caused by CYP450 genetic polymorphism does not necessarily lead to the differences in the plasma concentration of OXC. Instead, as the metabolite of OXC, the MHD may be joined with CYP2C19 to affect its own metabolism, leading to the differences in therapeutic efficacy and possible risk of adverse reactions [Citation221]. Therefore, it is recommended that OXC should be prescribed cautiously in clinical treatment of patients with epilepsy.
8.3. Effect of uridine diphosphate glucuronidase on oxcarbazepine metabolism
OXC is rapidly and completely absorbed orally and is then quickly metabolized into the active metabolite MHD in the body by the aromatic ketone degrading enzymes. MHD is mainly metabolized by UGT to produce glucuronide and excreted through the kidneys. Studies have reported that the MHD plasma concentration in carriers of UGT1A9 or UGT2B7 mutant genes is significantly higher than that of non-mutant patients [Citation179,Citation222]. However, more comprehensive studies on the effects of UTG on the metabolism of OXC are currently lacking. Further investigations are necessary to clarify the effects of UTG on the metabolism of OXC.
8.3.1. UGT1A9
UGT1A9 is expressed widely in liver, kidney, stomach, small intestine, colon, testis, prostate, and ovary. Results of in vitro studies based on human liver microsomes show that the UGT1A9 intron polymorphic variant CI399T is associated with enhanced glucuronidation of substrates of UGT1A1 and UGT1A9 [Citation223]. Zhang et al. have explored the relationship between UGT1A9 genotype and OXC plasma concentration in Chinese patients of epilepsy [Citation224]. Results show that the mutation rate of UGT1A9 CI399T gene in selected children with epilepsy is 56.9%, which is higher than 43.5% reported by Lu et al. [Citation225] but similar to 55% reported by Guo et al. [Citation226]. No correlation is identified between the UGT1A9 CI399T genetic polymorphism and either the MHD plasma concentration or the clinical efficacy in children with epilepsy, probably due to the weakened expression of UGT gene in children [Citation227]. Lu et al. have categorized patients of epilepsy into seizure group and seizure-free group based on SNPs and therapeutic efficacy [Citation225]. Results show that there is a significant difference between the seizure group and the seizure-free group at the UGT1A9 CI399T site. In the seizure-free group, there are more CC genotypes than either CT or TT genotypes, while there is no statistical difference between CT and TT genotypes, suggesting that the mutant T allele is associated with the increase of UGT1A9 activity. These results are consistent with those reported previously [Citation223].
8.3.2. UGT2B7
A large amount (94%) of OXC is metabolized by UGT, which shows significant differences in the activity among different ethnic groups and individuals. As an important SNP of UGT, the UGT2B7 gene is located at 4q13 and is 16 kb in length containing a total of 6 exons and 5 introns, encoding 529 amino acids [Citation228]. Ma et al. have conducted a statistical analysis on the relationship between the genetic polymorphism of UGT2B7 T802C locus and the OXC plasma concentration in Chinese patients of epilepsy [Citation229]. Results show that the mutation rate of the UGT2B7 T802C locus in the selected population is 66%, which is consistent with that reported previously [Citation230]. Furthermore, there are significant differences in the maintenance dose of OXC in patients with three genotypes at UGT2B7 T802C locus in the order of CC > CT > TT. Patients with the C allele require a significantly higher maintenance dose of OXC than those without the mutant allele. These results are consistent with those reported by Ma et al. [Citation208]. Furthermore, results of the studies on the UGT2B7 rs7439366 locus by Shen et al. suggest that the genetic polymorphism of this locus may affect the sensitivity of individuals to OXC but show no significant effect on its concentration [Citation231]. Zhang et al. have reported that the mutation rate of UGT2B7 T802C site is 67.1% [Citation224], which is similar to 66% as reported previously but lower than 78% in Greek population [Citation232]. The mutation rate of UGT2B7 T802C gene is significantly higher in Japanese population (73.2%) than that in Caucasian population (55.1%), likely due to the ethnic and regional differences [Citation233]. However, no significant correlation is revealed between the UGT2B7 T802C locus and either the OXC plasma concentration or its therapeutic efficacy in children with epilepsy, probably due to the weakened expression of the UGT gene in children.
In summary, it is recommended that patients with UGT2B7 mutants be given a higher maintenance dose of OXC in order to obtain a satisfactory therapeutic effect. Used in clinical treatment of epilepsy, the dose of OXC can be individualized based on the UGT2B7 genotype. However, due to the weakened expression of UGT gene in children with epilepsy, the clinical treatment of drug therapy for children cannot be completely dependent on genetype testing but in combination with closely monitoring the drug plasma concentration to ensure that the individualized dosages are reasonably formulated to avoid adverse reactions.
8.4. Effect of transporters on the metabolism of oxcarbazepine
ABC transporters play important roles in the metabolism of OXC in vivo. Studies have shown that the ABC transporters play an important role in AED tolerance [Citation234]. ABCB1 is the first enzyme investigated in the ABC family, while OXC is the substrate of P-gp. ABCC2 is mainly distributed in the luminal (apical) membranes of both hepatocytes and the proximal tubule cells in the kidney with a small portion located in the apical membrane of polar cells such as the intestine, gallbladder epithelial cells, embryos, and endothelial cells of the blood-brain barrier. Studies have shown that the up-regulation of ABCC2 expression in brain tissue in patients with epilepsy may be related to the efficacy and distribution of the drug in the body [Citation235].
8.4.1. ABCB1
The P-gp is expressed by the ABCB subfamily member 1 transporter gene (ABCB1), also known as the multi-drug resistance gene 1 (ABCB1) [Citation236]. To date, the C3435T (rs1045642) locus in exon 26 is the most extensively studied ABCB1 SNP [Citation237]. Results of a systematic review and meta-analysis show that the ABCB1 C3435T genetic polymorphism, particularly the TT genotype, plays important roles in treatment of refractory epilepsy [Citation238]. Shen et al. have investigated the effects of ABCB1 gene mutations on the plasma concentration of OXC and the treatment of epilepsy [Citation231]. Results show that the ABCB1 C3435T genetic polymorphism is significantly associated with standardized OXC plasma concentration and its efficacy. Patients with the CC genotype show higher plasma concentration than those of CT and TT genotypes with patients of homozygous CC genotype at the ABCB1 C3435T locus showing the best therapeutic effect with OXC. Seo et al. have reported that the therapeutic efficacy of drugs varies in patients of epilepsy with different ABCB1 C3435T genotypes and there is no statistical difference in the plasma concentration dose ratio (C/D) of CBZ in the plasma of different genotype groups, suggesting that patients in the drug-resistant genotype group may develop drug resistance by restricting AEDs to pass through the blood-brain barrier to reach brain cells [Citation119].
In addition to the C3435T locus, Yue et al. have analyzed the relationship between the genetic polymorphisms of ABCB1 rs1002204 and rs10234411 loci and the OXC plasma concentration by comparing the OXC plasma concentration dose ratio (C/D) of different genotype groups [Citation239]. Results show that there is no significant difference in the C/D ratio among the three genotype groups, which is consistent with the hypothesis proposed previously, i.e., the overexpression of P-gp in the blood-brain barrier of patients caused by various factors does not affect the drug absorption of AEDs and distribution in plasma but restrict AEDs from entering brain cells, causing the difficulty for AEDs to reach effective concentrations in the brain [Citation240–243]. These results suggest that the TT genotype at rs1002204 and rs10234411 may be a genetic risk factor for patients prone to drug resistance. The TT genotype may affect the brain tissue drug concentration of the patient without changing the C/D ratio of OXC in the patient<apos;>s blood, whereas leading to the drug resistance. Therefore, in order to compensate for the insufficient drug concentration in brain tissue, patients with TT genotype should be given increased doses of drugs to achieve high drug plasma concentrations.
8.4.2. ABCC2
To date, studies have shown that some AEDs (e.g., CBZ and PHT) are the substrates of MRP2, which is involved in the transport of AEDs in the blood-brain barrier [Citation244]. Most studies on the effects of ABCC2 on AEDs focusing on drug resistance derive inconsistent conclusions, while studies on ABCC2 affecting drug transport in the body are sparse [Citation245]. Recent studies have shown that ABCC2 G1249A mutation decreases the transport of CBZ on the cell membrane, leading to increased adverse reactions in the central nervous system, while no correlation is identified between ABCC2 and either the plasma concentration or the dose of CBZ [Citation246]. OXC is a derivative of CBZ, while studies focusing on the effects of ABCC2 on the maintenance dose and plasma concentration of OXC are currently sparse. Ma et al. have reported that the mutation frequencies of ABCC2 C3972T and G1249A in Chinese populations are 12.0% and 10.5%, respectively [Citation229], slightly different from those reported in other ethnic groups (18.8% and 12.5%) [Citation246]. Furthermore, there are significant differences in the maintenance dose of OXC in patients with three different genotypes at ABCC2 G1249A in the order of AA > GA > GG. Moreover, patients carrying the A allele require a significantly higher maintenance dose of OXC than patients without the mutant allele. However, Shen et al. have not identified the significant impact of the G1249A locus genetic polymorphism on the therapeutic concentration and efficacy of OXC, probably due to either insufficient samples of patients or differences in the adjustment methods of multiple factors involved in the mebabolism of OXC [Citation231].
9. Levetiracetam
As one of the new generation AEDs, levetiracetam (LEV) was approved for clinical use in the 1990s, showing ideal pharmacokinetic characteristics. The LEV is not metabolized by either the liver or the cytochrome P450 enzyme system, not interacting with other AEDs but mainly excreted through kidneys [Citation247]. Several studies have demonstrated that addition of LEV is effective and safe in the treatment of intractable or partial epilepsy. The possible functional mechanisms of LEV include: (1) LEV specifically binds to the SV2A protein to inhibit the abnormal discharge of the epileptic loop, thereby blocking the occurrence of epilepsy [Citation248], (2) LEV inhibits the release of calcium by binding with Sri Lanka cinnamicine receptors and inositol triphosphate receptors [Citation249], (3) LEV strengthens the inhibitory effect of the GABA circuit by preventing the decrease of GABA receptors without altering the concentration of GABA in brain tissue [Citation250,Citation251], and (4) LEV is not a substrate of either P-gp or MRP but can enter brain tissue through the blood-brain barrier [Citation252,Citation253].
The ~50 transporters of the ABC superfamily in humans are categorized into 7 subtypes, named as ABCA to ABCG, transporting multiple substrates including drugs, lipids, and ions. Three out of these transporters, i.e., P-gp, ABCC1, and ABCG2, are mainly related to epilepsy drug resistance [Citation254]. P-gp is a protein in the ABC family, highly expressed on the surface of capillary endothelial cells and astrocytes of the blood-brain barrier in patients with refractory epilepsy. P-gp decomposes ATP to provide energy to pump AEDs into the blood, reducing the drug concentration in the epileptic center and failing to effectively inhibit the epileptic discharge of brain cells [Citation255]. Several commonly used AEDs (i.e., PHT, PB, and CBZ) are substrates of P-gp. The ability of P-gp to efflux AEDs is associated with dosage and decreases in the order of PHT > CBZ > LTG > PB > VPA [Citation256], LEV, and gabapentin (GBP). The ABCB1 gene encoding P-gp is located in C3435T of exon 26 of chromosome 7, and contains three polymorphic genotypes CC, CT, and TT, with the up-regulation of the CC gene leading to the overexpression of P-gp [Citation257], ultimately causing resistance to AEDs in patients with epilepsy.
Zheng et al. have studied the relationship between the therapeutic effect of adding LEV to the treatment of refractory epilepsy and the ABCB1 genetic polymorphism [Citation258]. Results show that after the testing subjects with refractory epilepsy expressing CC gene and generating P-gp are treated with addition of LEV, the seizure rate is reduced by more than 66% (i.e., 6 out 9 patients), while the seizure rate in the placebo group is decreased by ~14% (i.e., 1 out 7 patients), suggesting that LEV is not the substrate of P-gp and is capable of escaping the P-gp to enter the epileptogenic center through the blood-brain barrier. Gao et al. have demonstrated that the ABCB1 C3435T genetic polymorphism is not related to AED resistance [Citation259], which is consistent with the results reported previously [Citation150,Citation260]. It is speculated that the cause of AED resistance may be related to the genetic polymorphism of other genetic sites. Furthermore, several studies have shown that ABCB1 C3435T genetic polymorphism may be the determining factor of AED resistance [Citation95,Citation119,Citation261]. The variations in these experimental results may be caused by the limitation of sample size, inconsistent classifications of seizure types, and multi-site effects of refractory epilepsy.
In summary, it is commonly believed that the genetic polymorphism of ABCB1 is associated with the occurrence of refractory epilepsy. However, some studies have not identified this significant association, while a large number of studies show that the overexpression of P-gp is the cause of intractable epilepsy. As a drug for the treatment of refractory epilepsy, LEV has shown satisfactory therapeutic effects and safety with its plasma concentration and efficacy not affected by ABCB1 genetic polymorphism. This is the commonly accepted mechanism regulating the LEV passing through the blood-brain barrier and effectively acting on the epileptic center.
10. Summary and prospect
In recent years, genetic testing has gradually become an important support for medication in the clinical application of AEDs in the treatment of patients with epilepsy, providing therapeutic guidance for the selection of clinical AEDs. Based on the metabolic characteristics of both traditional and new AEDs, the detection of important sites of drug-metabolizing enzymes and transporter genes is important and helpful to effectively analyze and predict the response of different patients to different types of AEDs, ultimately to establish the individualized doses. For patients with epilepsy, the individualized drug delivery would facilitate the quick and accurate establishment of the treatment window, avoid drug waste, reduce economic pressure, choose the appropriate type and dosage of drugs with the least adverse reactions, and choose the types of AEDs that are not easily tolerated to maintain long-term curative effects. Therefore, the investigation and detection of genes encoding the drug-metabolizing enzymes are of significant importance to the clinical treatment of epilepsy. This subject needs to be further seriously studied and systematically summarized. In this review, we have summarized the recent advancement in our understanding of the therapeutic effects of genetic polymorphism on the plasma concentration of six types of AEDs currently commonly used in the clinical treatment of epilepsy (). Based on these studies, the clinical dosages are recommended in the individualized treatment plan of patients of epilepsy using both the traditional and newly developed AEDs.
Table 1. Summary of effects of genetic polymorphism on the plasma concentration of six types of antiepileptic drugs. Symbol ‘ – ’ indicates information not available. Symbols ‘↑’ and ‘↓’ indicate the increase and decrease of the drug plasma concentration, respectively
To date, the detection of drug-metabolizing genes has been widely used in the clinical treatment of epilepsy in many major hospitals in China. The patients’ DNA samples are routinely collected from the patients’ oral mucosal epithelial cells to detect the target genes, subsequently determining the selection of appropriate AEDs for the patients with low risk of adverse reactions and high level of bioavailability based on the phenotypes of different target genes. These results facilitate significantly the establishment of a unique genetic database of the Chinese population, ultimately developing the clinical guidance distinctive for the Chinese population based on the characteristics of genetic distribution. Furthermore, the detection and acquisition of a large amount of data provide strong theoretical and experimental evidence for the development and production of novel AEDs. It is reasonably expected that in the future, it is highly possible to provide detailed recommendations of the types, dosages, and frequency of administration of the personalized AEDs based on the convenient collections of the oral mucosal epithelial cells.
11. Conclusion
Due to its severe impact on human health, epilepsy is extensively investigated to explore its effective treatments worldwide. Although the traditional AEDs have shown serious side effects, the current treatment of epilepsy is still commonly based on the traditional AEDs. The clinical knowledge on the effects of genetic polymorphism of genes encoding drug-metabolizing enzymes on the plasma concentration of AEDs has been significantly expanded. It is increasingly imperative to summarize and conceptualize the clinical significance of recent studies on individualized therapeutic regimens. Our review summarizes solid experimental evidence to provide the clinical guidance for the applications of AEDs.
Highlights
Clinical significance of individualized therapeutic regimens of antiepileptic drugs
Effects of genetic polymorphism on the plasma concentration of antiepileptic drugs
Clinical significance of genotypes related to drug metabolism on drug dosage
Clinical guidance is provided for the therapeutic application of antiepileptic drug
Acknowledgments
We would like to thank Professor Hui Li for her technical support in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Chen X, Jin Y, Tang G. New edition of pharmacology. In: The. 15th ed. Beijing: People’s Medical Publishing House,; 2005. p. 211.
- Lu Z, Yu Q. Pharmacokinetics of oxcarbazepine and its stereoselsctivity. Chin J Clin Pharmacol. 2003;19:134.
- Jiao Z, Zhong MK, Shi XJ. Therapeutic drug monitoring of anti-epilepsy drugs. China Pharm. 2004;13:386–389.
- Jiao Z, Zhong MK, Shi XJ. Frequent distribution of ultrarapid metabolisers of debrisoquine in an Ethiopian population carrying duplicated and multiduplicated functional CYP2D6 alleles. J Pharmacol Exp Ther. 1996;278(1):441–444.
- Evans WE, Relling MV, Rahman A, et al. Genetic basis for low prevalence of deficient CYP2D6 oxidative drug metabolism in black Americans. J Clin Invest. 1993;91(5):2150–2154.
- Evans WE, Relling MV, Rahman A, et al. Genetic analysis of the interethnic difference between Chinese and Caucasians in the polymorphic metabolism of debrisoquine and codeine. Eur J Clin Pharmacol. 1991;40(6):553–556.
- Evans WE, Relling MV, Rahman A, et al. Genetic analysis of the Chinese cytochrome P4502D locus: characterisation of variant CYP 2D and genes present in subjects with diminished capacity for debrisoquine hydroxylation. Mol Pharmacol. 1994;46(3):452–459.
- Masimirembwa C, Hasler M, J A. Genetic polymorphism of drug metabolising enzymes in African populations: implications for the use of neuroleptics and antidepressants. Brain Res Bull. 1997;44(5):561–571.
- Masimirembwa C, Hasler M, J A. Genetic polymorphism of cytochrome P450 2D6 in a Zimbabwean population. Pharmacogenetics. 1993;3(6):275–280.
- Masimirembwa C, Hasler M, J A. Phenotype and genotype analysis of debrisoquine hydroxylase (CYP2D6) in a black Zimbabwean population: reduced enzyme activity and evaluation of metabolic correlation of CYP2D6 probe drugs. Eur J Clin Pharmacol. 1996;51(2):117–122.
- Neyshaburinezhad N, Ghasim H, Rouini M, et al. Frequency of important cyp450 enzyme gene polymorphisms in the iranian population in comparison with other major populations: a comprehensive review of the human data. J Pers Med. 2021;11(8):804.
- Yokota H, Takamura S, Furuya H, et al. Evidence for a new variant CYP2D6 allele CYP2D6J in a Japanese population associated with lower in vivo rates of sparteine metabolism. Pharmacogenetics. 1993;3(5):256–263.
- Li C, Zheng X, Liu P, et al. Clinical value of lncRNA TUG1 in temporal lobe epilepsy and its role in the proliferation of hippocampus neuron via sponging miR-199a-3p. Bioengineered. 2021;12(2):10666–10673.
- Ahmed S, Gul S, Akhlaq M, et al. Estimation of polymorphisms in the drug-metabolizing enzyme, cytochrome P450 2C19 gene in six major ethnicities of Pakistan. Bioengineered. 2021;12(1):4442–4451.
- Hou J, Sun E, Song J, et al. Relationship between hepatic drug-metabolizing enzymes CYP450 and traditional Chinese medicine-induced hepatotoxicity. Chin J Chin Mater Med. 2016;41(15):2774–2780.
- Liao N, Chen W. Progress in cytochrome P450 enzyme in toxicity of traditional Chinese medicines. Chin J Pharmacol Toxicol. 2012;26:402.
- Bai X, Jiang D, Wang Y. Gene polymorphism and drug metabolism of valproic acid. Chin J Drug Depend. 2007;16:241–244.
- Jiang MX. Pharmacology, the fourth edition. Beijing: People’s Publishing House; 1996. p. 99.
- Wang XE, Feng Y, Kang Y. Clinical significance of determination of blood concentration of antiepileptic drugs. Chin J Hospital Pharm. 1992;12:422–423.
- Wang W, Gong S, Li C, et al. Comparison of antiepileptic drugs on embryotoxicity in rats. Chin Pharm J. 1998;33:617–618.
- Lu T, Su L, Ji D, et al. Interaction between CYP450 enzymes and metabolism of traditional Chinese medicine as well as enzyme activity assay. Chin J Chin Mater Med. 2015;40(18):3524–3529.
- Zhao Y, Xu Z, Liu Y, et al. Research progress in genetic polymorphism of CYP450 enzymes related to drug metabolism. ClinMed J. 2017;15:1–6.
- Ariyoshi N, Takahashi Y, Miyamoto M, et al. Structural characterization of a new variant of the CYP2A6 gene (CYP2A6*1B) apparently diagnosed as heterozygotes of CYP2A6*1A and CYP2A6*4C. Pharmacogenetics. 2000;10(8):687–693.
- Soyama A, Saito Y, Hanioka N, et al. Single nucleotide polymorphisms and haplotypes of CYP1A2 in a Japanese population. Drug Metab Pharmacokinet. 2005;20(1):24–33.
- Zhang T, Xu S, Liu C, et al. Effects of CYP450 enzymes polymorphisms on plasma valproic acid concentration. Chin J Biochem Pharm. 2016;36:1–4.
- Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323–1338.
- Sadeque AJ, Fisher MB, Korzekwa KR, et al. Human CYP2C9 and CYP2A6 mediate formation of the hepatotoxin 4-ene-valproic acid. J Pharmacol Exp Ther. 1997;283(2):698–703.
- Ghodke-Puranik Y, Thorn CF, Lamba JK, et al. Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2013;23(4):236–241.
- Oscarson M, McLellan RA, Gullstén H, et al. Characterisation and PCR-based detection of a CYP2A6 gene deletion found at a high frequency in a Chinese population. FEBS Lett. 1999;448(1):105–110.
- Tao W. Study on the relationship between CYP2A6 and CYP2B6 gene polymorphisms and sodium valproate plasma concentration. In: Innovation driven and transformational development, Proceedings of the Eleventh Annual Conference of Qingdao, 2013 (Shandong), pp. 133–137.
- Sun Y, Tan L, Song J. Effect of CYP2A6 genetic polymorphisms on serum concentration of sodium valproate. Chin J Neurol. 2006;11:745–747.
- Zhong S, Feng S, Jiang Y, et al. Determination of pharmacokinetics of sodium valproate in children with epilepsy. Chin J Appl Clin Pediatr. 1995;10:163–164.
- Zhang J, Yu L, Qu Z, et al. Interaction between sodium valproate and phenytoin or carbamazepine. Chin J Hospital Pharm. 1995;15:539–540.
- Wang X, Wang L. Effects of sodium valproate on clonazepam in pharmacokinetics and its related mechanism. Chin J Hospital Pharm. 1998;18:197–199.
- Zhou H. Pharmacogenetics. Vol. 53. Beijing: Science Press; 2001. p. 87–99.
- Liang W. Biopharmaceuticals and pharmacokinetics. Beijing: People’s Medical Publishing House; 2000. p. 102.
- Fumiyoshi Y, Hideto H, Takayuki I, et al. Novel hierarchical classification and visualization method for multiobjective optimization of drug properties: application to structure-activity relationship analysis of cytochrome P450 metabolism. J Chem Inf Model. 2008;48(2):364–369.
- Hoffman SM, Fernandez-Salguero P, Gonzalez FJ, et al. Organization and evolution of the cytochrome P450 CYP2A-2B-2F subfamily gene cluster on human chromosome 19. J Mol Evol. 1995;41(6):894–900.
- Ou J, Tan L, Sun Y. Association study between valproic acid plasma concentration and CYP286 gene polymorphism. Chin J Clin Neurosci. 2007;4:378–381.
- Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. J CNS Drugs. 2002;16(10):695–714.
- Kiang TK, Ho PC, Anari MR, et al. Contribution of CYP2C9, CYP2A6, and CYP2B6 to valproic acid metabolism in hepatic microsomes from individuals with the CYP2C9*1/*1 genotype. Toxicol Sci. 2006;94(2):261–271.
- Yang JQ, Morin S, Verstuyft C, et al. Frequency of cytochrome P450 2C9 allelic variants in the Chinese and French populations. Fundam Clin Pharmacol. 2003;17(3):373–376.
- Imai J, Ieiri I, Mamiya K, et al. Polymorphism of the cytochrome P450 (CYP) 2C9 gene in Japanese epileptic patients: genetic analysis of the CYP2C9 locus. Pharmacogenetics. 2000;10(1):85–89.
- Wang SL, Huang J, Lai MD, et al. Detection of CYP2C9 polymorphism based on the polymerase chain reaction in Chinese. Pharmacogenetics. 1995;5(1):37–42.
- Gu X, Zhu M, Sheng C, et al. Population pharmacokinetics of unbound valproic acid in pediatric epilepsy patients in China: a protein binding model. Eur J Clin Pharmacol. 2021;77(7):999–1009.
- Zhu Z, Wang H, Cao X, et al. Study on correlation between CYP2C19 gene polymorphism with serum valproic acid concentration and individualized medication. Int J Lab Med. 2018;39:14–16.
- Cheung NY. Law treatment of acute period of stroke: clinical observation of 98 cases. Labor Invest. 2014;32:21.
- Han R, Li Y, Wu W. Effects of CYP2C19 genetic polymorphism on plasma concentration of sodium valproate in epileptic patients. Neural Injury Funct Reconstr. 2015;10:295–297.
- Feng Y, Zhou J, Liu K, et al. Distribution of CYP2C19 gene polymorphisms related to valproic acid metabolism in children with epilepsy and individualized treatment study. Chin J Pract Nerv Dis. 2015;18:49–52.
- Tan X, Zhang Y, Wang S, et al. Study on the relationship between CYP2C9 and CYP2C19 gene polymorphisms and valproic acid concentration in epilepsy patients. Pharm Clin Res. 2011;19:123–128.
- Han L, Liu J. CYP2D6 gene polymorphism and its influence on drug metabolism, China Pharmacy. The Journal of Craniofacial Surgery. 2015;26(4):1066–1068.
- Neafsey P, Ginsberg G, Hattis D, et al. Genetic Polymorphism in Cytochrome P450 2D6 (CYP2D6): population distribution of CYP2D6 activity. J Toxicol Environ Health B Crit Rev. 2009;12(5–6):334–361.
- De Gregori M, Allegri M, De Gregori S, et al. How and why to screen for CYP2D6 interindividual variability in patients under pharmacological treatments. Curr Drug Metab. 2010;11(3):276–282.
- Fang F, Fang Z, Zhang D, et al. Gene polymorphism of CYP2D6*10 in patients with epilepsy and the effect on serum concentration of sodium valproate. China Pharm. 2015;26:1066–1068.
- Ke J, Wang D, Zhu Y. Research progress on the relationship between UGT gene polymorphisms and antiepileptic drug metabolism. Stroke Nervous Dis. 2021;28:226–228+234.
- Chen Y, Fan Q. Research progress on the effect of gene polymorphisms on the pharmacokinetics of common antiepileptic drugs. Shandong Med J. 2021;61:112–115.
- Sun Y, Zhuo W, Lin H, et al. Effects of UGT2B7-C802T and G211T genetic polymorphism on metabolism of valproic acid in epilepsy patients. Chin Hosp Pharm J. 2015;35:216–219.
- Nakamura A, Nakajima M, Yamanaka H, et al. Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab Dispos. 2008;36(8):1461–1464.
- He B, Wang W, Zhao X, et al. Genetic polymorphisms of UGT-glucurono-syltransferase 2B7 in Chinese healthy Han population. Chin J Clin Pharmacol. 2014;30:6–8.
- Wen Z, Xiao J, Chen X. Research progress of valproic acid pharmacogenomics. Central South Pharm. 2016;14:526–531.
- Zhang M, Li R, Zhou Y. Effect of UGT gene polymorphism on the plasma concentration of valproate acid. Central South Pharm. 2019;17:586–589.
- Cho SK, Oh ES, Park K, et al. The UGT1A3*2 polymorphism affects atorvastatin lactonization and lipid-lowering effect in healthy volunteers. Pharmacogenet Genomics. 2012;22(8):598–605.
- Kang W, Chen J, Chai D, et al. Relationship between UGTlA6 A541G and A552C genetic polymorphism and plasma concentration of sodium valproate. Chin J Health Care Med. 2016;18:280–283.
- Xing Y, Yang L, Wang L, et al. Systematic screening for polymorphisms within the UGT1A6 gene in three Chinese populations and function prediction through structural modeling. J Pharmacogenomics. 2009;10(5):741–752.
- Wang Y, Gao L, Liu Y, et al. Effect of UGTIA6 A541G genetic polymorphism on the metabolism of valproic acid in Han epileptic children from Henan. Chin J Contemp Pediatr. 2010;12:429–432.
- Jin L, Yang L, Ma M. Effect of UGT1A6 genetic polymorphism on serum concentration of valproic acid in Han epileptic patients. China Pharm. 2013;16:802–804.
- Nagar S, Zalatoris JJ, Blanchard RL. Human UGT1A6 pharmacogenetics: identification of a novel SNP, characterization of allele frequencies and functional analysis of recombinant allozymes in human liver tissue and in cultured cells. Pharmacogenetics. 2004;14(8):487–499.
- Tan X, Zhang X, Qiu Z. Study on the correlation between UGT1A6 gene polymorphism and valproic acid concentration in epilepsy patients. Pharm Clin Res. 2014;22:299–301.
- Mei S, Feng W, Zhu L. Genetic polymorphisms and valproic acid plasma concentration in children with epilepsy on valproic acid. Seizure. 2017;51:22–26.
- Guo D, Pang L, Zhou H. Progress in the pharmacogenomics of UDP-glucuronosyltransferase. Chin J New Drugs. 2011;20:1188–1193.
- Lin X, Zhang Y, Lin P, et al. Effects of UGTlA6 and UGTlA9 gene polymorphisms on blood concentration of valproic acid in Han epileptic patients. China Pharm. 2017;28:1013–1017.
- Holthe M, Rakvåg TN, Klepstad P, et al. Sequence variations in the UDP-glucuronosyltransferase 2B7 (UGT2B7) gene: identification of 10 novel single nucleotide polymorphisms (SNPs) and analysis of their relevance to morphine glucuronidation in cancer patients. Pharmacogenomics J. 2003;3(1):17–26.
- Zou M Study on the correlation between UGT2B7 gene polymorphism and valproic acid blood concentration. MS Thesis, Central South University: Changsha, 2011.
- Zhang W, Chang YZ, Kan QC, et al. CYP3A4*1G genetic polymorphism influences CYP3A activity and response to fentanyl in Chinese gynecologic patients. Eur J Clin Pharmacol. 2010;66(1):61–66.
- Yi J, Xie H, Zhou H. Distribution of UGT1A9-2152T and UGT2B7-G211T mutations in Chinese Han population. Chin J Clin Pharmacol Ther. 2007;12:460–464.
- Zhang Q, Tang H, Lyu X, et al. Effect of udp-glucuronosyltransferase UGT2B7 genetic polymorphisms on serum valproate concentrations in children with epilepsy. J Pediatr Pharm. 2016;22:33–36.
- Chu XM, Zhang LF, Wang GJ, et al. Influence of UDP-glucuronosyltransferase polymorphisms on valproic acid pharmacokinetics in Chinese epilepsy patients. Eur J Clin Pharmacol. 2012;68(10):1395–1401.
- Zhang H, Zhang W, Li Y, et al. Correlations between UGT2B7*2 gene polymorphisms and plasma concentrations of carbamazepine and valproic acid in epilepsy patients. Brain Dev. 2018;40(2):100–106.
- Rowland A, Miners J, Mackenzie P. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol. 2013;45(6):1121–1132.
- Mori H, Takahashi K, Mizutani T. Interaction between valproic acid and carbapenem antibiotics. Drug Metab Rev. 2007;39(4):647–657.
- Jetter A, Kullak-Ublick G. Drugs and hepatic transporters: a review. Pharmacol Res. 2020;154:104234.
- Giacomini KM, Huang SM, Tweedie DJ, et al., International Transporter Consortium. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–236. DOI:10.1038/nrd3028.
- Yan J, Gao Z, Gong C, et al. Effects of quetiapine combined with sodium valproate on agitation and impulsive aggressive behavior in patients with first-episode schizophrenia. J Southeast Univ (Med Sci Edi). 2015;34:148–150.
- Ding Y, Yang F, Liu J. The effect of multidrug resistance gene haploid on the blood concentration of sodium valproate in Chinese patients with epilepsy. Chin J Clin Pharmacol Ther. 2010;15:1267–1272.
- Li J, Chen B. Research status and progress of inhibiting the function of P-glycoprotein transporter in reversing tumor multidrug resistance. J Southeast Univ (Med Sci Edi). 2018;37:359–363.
- Cuffe S, Azad A, Qiu X, et al. ABCC2 polymorphisms and survival in the princess margaret cohort study and the NCIC clinical trials group BR.24 trial of platinum-treated advanced stage non-small cell lung cancer patients. Cancer Epidemiol. 2016;41:50–56.
- Liu Y, Zhang X, Wu J. Correlation analysis between ABCC2 gene polymorphism with blood concentration of sodium valproate and efficacy in epileptic patients. Mod Med J. 2019;47:646–651.
- Di Francia R, Siesto RS, Valente D, et al. Current strategies to minimize toxicity of oxaliplatin: selection of pharmacogenomic panel tests. Anticancer Drugs. 2013;24(10):1069–1078.
- Zhang Z, Xu H. The value of genetic polymorphisms of mycophenolate mofetil in individualized therapy. J Clin Pediatr. 2018;38:259–261.
- Chen J, Wang X, Zhou L, et al. Influence of transporter genetic polymorphisms on valproic acid blood concentration and clinical efficacy of epilepsy patients. Chin J Clin Pharmacol. 2016;32:215–217.
- Schinkel A. The physiological function of drug-transporting P-glycoproteins. Semin Cancer Biol. 1997;8(3):161–170.
- Marzolini C, Paus E, Buclin T, et al. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75(1):13–33.
- Zhao Z, Sheps JA, Ling V, et al. Expression analysis of ABC transporters reveals differential functions of tandemly duplicated genes in Caenorhabditis elegans. J Mol Biol. 2004;344(2):409–417.
- Leschziner GD, Andrew T, Pirmohamed M, et al. ABCB1 genotype and PGP expression, function and therapeutic drug response: a critical review and recommendations for future research. Pharmacogenomics J. 2007;7(3):154–179.
- Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med. 2003;348(15):1442–1448.
- Makmor-Bakry M, Sills GJ, Hitiris N, et al. Genetic variants in microsomal epoxide hydrolase influence carbamazepine dosing. Clin Neuropharmacol. 2009;32(4):205–212.
- Huang W, Lin YS, McConn DJ, et al. Evidence of significant contribution from CYP3A5 to hepatic drug metabolism. Drug Metab Dispos. 2004;32(12):1434–1445.
- Ferraro TN, Buono RJ. The relationship between the pharmacology of antiepileptic drugs and human gene variation: an overview. Epilepsy Behav. 2005;7(1):18–36.
- Xi S, Teng J. Medications for epilepsy. Henan: Zhengzhou University Press; 2002. p. 229.
- Danielson PB. The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr Drug Metab. 2002;3(6):561–597.
- Ozdemir V, Kalow W, Tang BK, et al. Evaluation of the genetic component of variability in CYP3A4 activity: a repeated drug administration method. Pharmacogenetics. 2000;10(5):373–388.
- Chen CH, Wang SC, Tsou HH, et al. Genetic polymorphisms in CYP3A4 are associated with withdrawal symptoms and adverse reactions in methadone maintenance patients. Pharmacogenomics. 2011;12(10):1397–1406.
- Wang P, Yin T, Ma H. Effects of CYP3A4/5 and ABCB1 genetic polymorphisms on carbamazepine metabolism and transport in Chinese patients with epilepsy treated with carbamazepine in monotherapy and biotherapy. Epilepsy Res. 2015;117:52–57.
- Yun W Genetic analysis of individual differences in the metabolism of antiepileptic drug carbamazepine. MS Thesis, Jilin University: Changchun, 2015.
- Wang J Correlation between CYP3A5 subfamily gene polymorphisms and carbamazepine concentration and efficacy in Chinese Han patients with epilepsy. PhD Dissertation, Fudan University: Shanghai, 2012.
- Kuehl P, Zhang J, Lin Y, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27(4):383–391.
- Jounaïdi Y, Hyrailles V, Gervot L, et al. Detection of CYP3A5 allelic variant: a candidate for the polymorphic expression of the protein. Biochem Biophys Res Commun. 1996;221(2):466–470.
- Li D, Zhang GL, Lou YQ, et al. Genetic polymorphisms in MDR1 and CYP3A5 and MDR1 haplotype in mainland Chinese Han, Uygur and Kazakh ethnic groups. J Clin Pharm Ther. 2007;32(1):89–95.
- Ren J Study on the correlation between CYP3A5 gene polymorphism and carbamazepine plasma concentration. MS Thesis, Jilin University: Changchun, 2010.
- Park PW, Seo YH, Ahn JY, et al. Effect of CYP3A5*3 genotype on serum carbamazepine concentrations at steady-state in Korean epileptic patients. J Clin Pharm Ther. 2009;34(5):569–574.
- Liu J, Li Y, Zhang G, et al. Effects of CYP3A5 gene polymorphism on steady state serum concentrations and therapeutic efficacy of carbamazepine in chinese han epileptic patients. China Pharm. 2014;25:3433–3435.
- Seo T, Nakada N, Ueda N, et al. Effect of CYP3A5*3 on carbamazepine pharmacokinetics in Japanese patients with epilepsy. Clin Pharmacol Ther. 2006;79(5):509–510.
- Zhang W Study on the relationship between UGT2B7*2 gene polymorphisms and blood levels of carbamazepine and valproic acid in patients with epilepsy. PhD Dissertation, BaoTou Medical College, Inner Mongolia University of Science and Technology: Baotou, 2015.
- Qu J Effects of ABCB1, ABCC2, CYP3A5 and UGT2B7 gene polymorphisms on the efficacy of antiepileptic drugs. PhD Dissertation, Central South University: Changsha, 2012.
- Saito K, Moriya H, Sawaguchi T, et al. Haplotype analysis of UDP-glucuronocyltransferase 2B7 gene (UGT2B7) polymorphisms in healthy Japanese subjects. Clin Biochem. 2006;39(3):303–308.
- Mollazadeh S, Sahebkar A, Hadizadeh F, et al. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018;214:118–123.
- Kwan P, Wong V, Ng PW, et al. Gene-wide tagging study of association between ABCB1 polymorphisms and multidrug resistance in epilepsy in Han Chinese. J Pharmacogenomics. 2009;10(5):723–732.
- Nakajima Y, Saito Y, Shiseki K, et al. Haplotype structures of EPHX1 and their effects on the metabolism of carbamazepine-10,11-epoxide in Japanese epileptic patients. Eur J Clin Pharmacol. 2005;61(1):25–34.
- Seo T, Ishitsu T, Ueda N, et al. ABCB1 polymorphisms influence the response to antiepileptic drugs in Japanese epilepsy patients. Pharmacogenomics. 2006;7(4):551–561.
- Meng H, Guo G, Ren J, et al. Effects of ABCB1 polymorphisms on plasma carbamazepine concentrations and pharmacoresistance in Chinese patients with epilepsy. Epilepsy Behav. 2011;21(1):27–30.
- Zhang C, Zuo Z, Kwan P, et al. In vitro transport profile of carbamazepine, oxcarbazepine, eslicarbazepine acetate, and their active metabolites by human P-glycoprotein. Epilepsia. 2011;52(10):1894–1904.
- Dombrowski SM, Desai SY, Marroni M, et al. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42(12):1501–1506.
- Kim WJ, Lee JH, Yi J, et al. A nonsynonymous variation in MRP2/ABCC2 is associated with neurological adverse drug reactions of carbamazepine in patients with epilepsy. Pharmacogenet Genomics. 2010;20(4):249–256.
- Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ilae commission on therapeutic strategies. Epilepsia. 2010;51(6):1069–1077.
- Zhang L. Research progress of individualized medication of phenytoin sodium guided by gene. Strait Pharm J. 2018;30:6–9.
- Kang Y, Chen M, Lai S. Potentially unsafe herb-drug interactions between a commercial product of noni juice and phenytoin-a case report. Acta Neurol. 2015;24:43–46.
- Taguchi Y, Takashima S, Tanaka K. a case of phenytoin intoxication caused by interaction between phenytoin and capecitabine. Brain Nerve. 2015;67(2):213–217.
- Marchi M, Besana E, Raiteri M. Oxiracetam increases the release of endogenous glutamate from depolarized rat hippocampal slices. Eur J Pharmacol. 1990;185(2–3):247–249.
- Copani A, Genazzani AA, Aleppo G, et al. Nootropic drugs positively modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-sensitive glutamate receptors in neuronal cultures. J Neurochem. 1992;58(4):1199–1204.
- Pittaluga A, Pattarini R, Raiteri M. Putative cognition enhancers reverse kynuremc acid antagonism at hippocampal NMDA receptors. Eur J Pharmacol. 1995;272(2–3):203–209.
- Vander Weide J, Steijns LS, Van Weelden MJ, et al. The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogentics. 2001;11(4):287–291.
- Llerena A, Alvarez M, Dorado P, et al. Interethnic differences in the relevance of CYP2C9 genotype and environmental factors for diclofenac metabolism in Hispanics from Cuba and Spain. Pharmacogenomics J. 2014;14(3):229–234.
- Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45(6):525–538.
- Zhou Y, Ingelman-Sundberg M, Lauschke VM. Worldwide distribution of cytochrome P450 alleles: a meta-analysis of population-scale sequencing projects. Clin Pharmacol Ther. 2017;102(4):688–700.
- Kusama M, Maeda K, Chiba K, et al. Prediction of the effects of genetic polymorphism on the pharmacokinetics of CYP2C9 substrates from in vitro data. Pharm Res. 2009;26(4):822–835.
- Ramasamy K, Narayan SK, Shewade DG, et al. Influence of CYP2C9 genetic-polymorphism and undernourishment on plasma-free phenytoin concentrations in epileptic patients. Ther Drug Monit. 2010;32(6):762–766.
- Hennessy S, Leonard CE, Freeman CP, et al. CYP2C9, CYP2C19, and ABCB1 genotype and hospitalization for phenytoin toxicity. J Clin Pharmacol. 2009;49(12):1483–1487.
- Anderson GD. Pharmacogenetics and enzyme induction inhibition properties of antiepileptic drugs. Neurology. 2004;63(10):S3–S8.
- Xie HG, Prasad HC, Kim RB, et al. CYP2C9 allelic variants: ethnic distribution and functional significance. Adv Drug Deliv Rev. 2002;54(10):1257–1270.
- Johnson JA, Gong L, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011;90(4):625–629.
- Niinuma Y, Saito T, Takahashi M, et al. Functional characterization of 32 CYP2C9 allelic variants. Pharmacogenomics J. 2014;14(2):107–114.
- Ren X, Wang X, Xu X, et al. Influence of CYP2C19 and CYP2C9 gene polymorphisms on phenytoin concentrations in Chinese Han epileptic patients. Chin J Clin Pharmacol. 2011;27:249–251+264.
- Yang ZF, Cui HW, Hasi T, et al. Genetic polymorphisms of cytochrome P450 enzymes 2C9 and 2C19 in a healthy Mongolian population in China. Genet Mol Res. 2010;9(3):1844–1851.
- Zhou J, Dong F, Wu G, et al. Clinical effects of individualized administration of PHT in epileptic children guided by TDM combined with CYP2C19 genotype. Chin J Pract Med. 2019;16:98–101.
- Qi X, Huang Y, Wang Y, et al. Association of plasma sodium phenytoin concentration with CYP2C19 gene polymorphism. Chin J New Drugs. 2004;10:922–925.
- Oka E, Ohtsuka Y, Yoshinaga H, et al. Prevalence of childhood epilepsy and distribution of epileptic syndromes: a population-based survey in Okayama,Japan. Epilepsia. 2006;47(3):626–630.
- Kimchi-Sarfaty C, Marple AH, Shinar S, et al. Ethnicity-related polymorphisms and haplotypes in the human ABCB1 gene. Pharmacogenomics. 2007;8(1):29–39.
- Zimprich F, Sunder-Plassmann R, Stogmann E, et al. Association of an ABCB1 gene haplotype with pharmacoresistance in temporal lobe epilepsy. Neurology. 2004;63(6):1087–1089.
- Hung CC, Tai JJ, Lin CJ, et al. Complex haplotypic effects of the ABCB1 gene on epilepsy treatment response. Pharmacogenomics. 2005;6(4):411–417.
- Vahab SA, Sen S, Ravindran N, et al. Analysis of genotype and haplotype effects of ABCB1 (MDR1) polymorphisms in the risk of medically refractory epilepsy in an Indian population. Drug Metab Pharmacokinet. 2009;24(3):255–260.
- Szoeke C, Sills GJ, Kwan P, et al. Multidrug-resistant genotype (ABCB1) and seizure recurrence in newly treated epilepsy: data from international pharmacogenetic cohorts. Epilepsia. 2009;50(7):1689–1696.
- Jiang D, Huang C, Wang B, et al. Association study of ABCB1 genetic polymorphism and the effect of epilepsy with phenytoin treatment. Chin J Clinicians. 2015;9:2482–2486.
- Shahwan A, Murphy K, Doherty C, et al. The controversial association of ABCB1 polymorphisms in refractory epilepsy: an analysis of multiple SNPs in an Irish populatio. Epilepsy Res. 2007;73(2):192–198.
- Smolarz B, Skalski D, Rysz A, et al. Polymorphism of the multi-drug resistance 1 gene MDRl G2677T/A (rs2032582) and the risk of drug-resistant epilepsy in the Polish adult population. Acta Neurol Belg. 2017;117(4):849–855.
- Keangpraphun T, Towanabut S, Chinvarun Y, et al. Association of ABCB1 C3435T polymorphism with phenobarbital resistance in Thai patients with epilepsy. J Clin Pharm Ther. 2015;40(3):315–319.
- Dong L, Luo R, Tong Y, et al. Lack of association between ABCB1 gene polymorphisms and pharmacoresistant epilepsy: an analysis in a western Chinese pediatric population. Brain Res. 2011;1391:114–124.
- Nurmohamed L, Garcia-Bournissen F, Buono RJ, et al. Predisposition to epilepsy–does the ABCB1 gene play a role? Epilepsia. 2010;51(9):1882–1885.
- Li SX, Liu YY, Wang QB. ABCB1 gene C3435T poly-morphism and drug resistance in epilepsy:evidence based on 8604 subjects. Med Sci Monit. 2015;21:861–868.
- McLean MJ. Clinical pharmacokinetics of gabapentin. Neurology. 1994;44:17–22.
- Wu W, Heebner JB, Streeter AJ. Evaluation of the absorption, excretion, pharmacokinefics and metabolism of anticonvulsant topiramate in healthy men. Pharm Res. 1994;11:336.
- Perucca E, Johannessen SI. The ideal pharmacokinetic properties of an antiepileptic drug:how close does leve tiracetam come. Epileptic Disord. 2003;5:17–26.
- Gatti G, Bonomi I, Jannuzzi G, et al. The new antiepileptic drugs: pharmacological and clinical aspects. Curr Pharm Des. 2000;6(8):839–860.
- Stoffel W, Bosio A. Myelin glycolipids and their functions. Curr Opin Neurobiol. 1997;7(5):654–661.
- Ehmer U, Kalthoff S, Fakundiny B, et al. Gilbert Syndrome redefined: a complex genetic haplotype influences the regulation of glucuronidation. Hepatology. 2012;55(6):1912–1921.
- Castel-Branco M, Lebre V, Falcao A, et al. Relationship between plasma and brain levels and the anticonvulsant effect of lamotrigine in rats. Eur J Pharmacol. 2003;482(1–3):163–168.
- Iyer L, Hall D, Das S, et al. Phenotype-genotype correlation of in vitro SN-38(active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A4 promoter polymorphism. Clin Pharmacol Ther. 1999;65(5):576–582.
- Rougier P, Van Cutsem E, Bajetta E, et al. Randomised trial of irinotecan versus fluorouracil by continuous infusion after fluorouracil failure in patients with metastatic colorectal cancer. Lancet. 1998;352(9138):1407–1412.
- Kawato Y, Aonuma M, Hirota Y, et al. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991;51(16):4187–4191.
- Ando Y, Saka H, Ando M, et al. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 2000;60(24):6921–6926.
- Yang J, Cai L, Huang H, et al. Genetic variations and haplotype diversity of the UGT1 gene cluster in the Chinese population. Plos One. 2012;7(4):e33988.
- Ehmer U, Vogel A, Schutte JK, et al. Variation of hepatic glucuronidation: novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology. 2004;39(4):970–977.
- Yang L, Lin W, Zhang J, et al. The relationship between the distribution characteristics of UGT1A4 gene and its polymorphisms and the blood concentration of lamotrigine in patients with epilepsy in Han nationality in northern China. Chinese J Neurol. 2015;48:180–184.
- Trottier J, Verreault M, Grepper S, et al. Human UDP-glucuronosyltransferase (UGT)1A3 enzyme conjugates chenodeoxycholic acid in the liver. Hepatology. 2006;44(5):1158–1170.
- Itaaho K, Mackenzie PI, Miners JO, et al. The configuration of the 17-hydroxy group variably influences the glucuronidation of β-estradiol and epiestradiol by human UDP-glucuronosyltransferase. Drug Metab Dispos. 2008;36(11):2307–2315.
- Liston HL, Markowitz JS, De Vane L. Drug glucuronidation in clinical psychopharmacology. J Clin Psychopharmacol. 2001;21(5):500–515.
- He Y, He F, Mo X, et al. Quantitative estimation of blood concentration of lamotrigine in Chinese Han pediatric patients with epilepsy based on UGT1A4 142T>G polymorphism and blood concentration of valproic acid. China Pharm. 2017;28:2737–2742.
- Liu L, Zhao L, Wang Q, et al. Influence of valproic acid concentration and polymorphism of UGT1A4*3, UGT2B7 −161C > T and UGT2B7*2 on serum concentration of lamotrigine in Chinese epileptic children. Eur J Clin Pharmacol. 2015;71(11):1341–1347.
- Singkham N, Towanabut S, Lertkachatarn S, et al. Influence of the UGT2B7 −161C>T polymorphism on the population pharmacokinetics of lamotrigine in Thai patients. Eur J Clin Pharmacol. 2013;69(6):1285–1291.
- Blanca SM, Herranz JL, Leno C, et al. UGT2B7_−161C>T polymorphism is associated with lamotrigine concentration-to-dose ratio in a multivariate study. Ther Drug Monit. 2010;32(2):177–184.
- Inoue K, Yamamoto Y, Suzuki E, et al. Factors that influence me pharmacokinetics of lamotrigine in Japanese patients with epilepsy. Eur J Clin Pharmacol. 2016;72(5):555–562.
- Zhou Y, Wang X, Li H, et al. Polymorphisms o fABCG2, ABCB1 and HNF4alpha are associated with lamotrigine trough concentrations in epilepsy patients. Drug Metab Pharmacokinet. 2015;30(4):282–287.
- Milosheska D, Lorber B, Vovk T, et al. Pharmacokinetics of lamotrigine and its metabolite N-2-glucuronide: influence of polymorphism of UDP-glucuronosyltransferases and drug transporters. Br J Clin Pharmacol. 2016;82(2):399–411.
- Wang Q, Zhao L, Liang M, et al. Effects of UGT2B7 genetic polymorphisms on serum concentrations of valproic acid in Chinese children with epilepsy comedicated with lamotrigine. Ther Drug Monit. 2016;38(3):343–349.
- Miller DS, Calmon RE. Signaling pathways that regulate basal ABC transporter activity at the blood-brain barrier. Curr Pharm Des. 2014;20(10):1463–1471.
- Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24(7):1227–1251.
- Misaka S, Knop J, Singer K, et al. The nonmetabolized β-blocker nadolol is a substrate of OCT1, OCT2, MATE1, MATE2-K, and P-Glycoprotein, but Not of OATP1B1 and OATP1B3. Mol Pharm. 2016;13(2):512–519.
- Matthaei J, Kuron D, Faltraco F, et al. OCT1 mediates hepatic uptake of sumatriptan and loss-of-function OCT1 polymorphisms affect sumatriptan pharmacokinetics. Clin Pharmacol Ther. 2016;99(6):633–641.
- Dickens D, Owen A, Alfirevic A, et al. Lamotrigine is a substrate for OCT1 in brain endothelial cells. Biochem Pharmacol. 2012;83(6):805–814.
- Lu R Study on the correlation between drug gene polymorphism and lamotrigine plasma concentration in patients with epilepsy. PhD Dissertation, Zhejiang University: Hangzhou, 2016.
- Zhou Y, Ye W, Wang Y, et al. Genetic variants of OCT1 influence glycemic response to metformin in Han Chinese patients with type-2 diabetes mellitus in Shanghai. Int J Clin Exp Pathol. 2015;8(8):9533–9542.
- Yanase K, Tsukahara S, Mitsuhashi J, et al. Functional SNPs of the breast cancer resistance protein-therapeutic effects and inhibitor development. Cancer Lett. 2006;234(1):73–80.
- Romermann K, Helmer R, Loscher W. The antiepileptic drug lamotrigine is a substrate of mouse and human breast cancer resistance protein (ABCG2). Neuropharmacology. 2015;93:7–14.
- Urquhart BL, Ware JA, Tirona RG, et al. Breast cancer resistance protein (ABCG2) and drug disposition: intestinal expression, polymorphisms and sulfasalazine as an in vivo probe. Pharmacogenet Genomics. 2008;18(5):439–448.
- Furukawa T, Wakabayashi K, Tamura A, et al. Major SNP (Q141K) variant of human ABC transporter ABCG2 undergoes lysosomal and proteasomal degradations. Pharm Res. 2009;26(2):469–479.
- He Y Pharmacogenomics-based research on individualized medication of lamotrigine in the treatment of epilepsy patients in southern China. PhD Dissertation, Southern Medical University: Guangzhou, 2017.
- Rivas N, Buelga DS, Elger CE, et al. Population pharmacokinetics of lamotrigine with data from therapeutic drug monitoring in German and Spanish patients with epilepsy. Ther Drug Monit. 2008;30(4):483–489.
- Bruce CR, Brolin C, Turner N, et al. Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol Endocrinol Metab. 2007;292(4):E1231–1237.
- Lovrić M, Božina N, Hajnšek S, et al. Association between lamotrigine concentrations and ABCB1 polymorphisms in patients with epilepsy. Ther Drug Monit. 2012;34(5):518–525.
- Brzaković B, Vučićević K, Kovačević SV, et al. Pharmacokinetics of lamotrigine in paediatric and young adult epileptic patients–nonlinear mixed effects modelling approach. Eur J Clin Pharmacol. 2014;70(2):179–185.
- Wegner I, Wilhelm AJ, Sander JW, et al. The impact of age on lamotrigine and oxcarbazepine kinetics: a historical cohort study. Epilepsy Behav. 2013;29(1):217–221.
- Huang W Population pharmacokinetics study of lamotrigine in the treatment of epilepsy based on genetic polymorphisms of metabolic enzymes, transporters and receptors. PhD Dissertation, Guangzhou Medical University: Guangzhou, 2017.
- Chen Y, Xu S, Qiu F, et al. Influences of sodium channel gene and transporter gene polymorphisms on lamotrigine concentration. Chin J Clin Pharmacol. 2016;32:2069–2072.
- Gamble C, Williamson PR, Chadwick DW, et al. A meta-analysis of individual patient responses to lamotrigine or carbamazepine monotherapy. Neurology. 2006;66(9):1310–1317.
- Beydoun A. Safety and efficacy of oxcarbazepine: results of randomized, double-blind trials. Pharmacotherapy. 2000;20(8 Part 2):152–158.
- Zou X, Chen J, An D, et al. Efficacy of low to moderate doses of oxcarbazepine in adult patients with newly diagnosed partial epilepsy. Seizure. 2015;29:81–85.
- Schmidt D, Elger C. What is the evidence that oxcarbazepine and carbamazepine are distinctly different antiepileptic drugs? Epilepsy Behav. 2004;5(5):627–635.
- Franco V, Perucca E. The pharmacogenomics of epilepsy. Expert Rev Neurother. 2015;15(10):1161–1170.
- Ma CL, Wu XY, Jiao Z, et al. SCN1A, ABCC2 and UGT2B7 gene polymorphisms in association with individualized oxcarbazepine therapy. Pharmacogenomics. 2015;16(4):347–360.
- Ambrósio AF, Soares-Da-Silva P, Carvalho CM, et al. Mechanisms of action of carbamazepine and its derivatives, oxcarbazepine, BIA 2-093, and BIA 2-024. Neurochem Res. 2002;27(1/2):121–130.
- Wellington K, Goa KL. Oxcarbazepine: an update of its efficacy in the management of epilepsy. CNS Drugs. 2001;15(2):137–163.
- Court MH, Duan SX, Guillemette C, et al. Stereoselective conjugation of oxazepam by human UDP-Glucuronosyltransferases (UGTs): s-oxazepam is glucuronidated by UGT2B15, while R-oxazepam is glucuronidated by UGT2B7 and UGT1A9. Drug Metab Dispos. 2002;30(11):1257–1265.
- Court MH, Hao Q, Krishnaswamy S, et al. UDP-glucuronosyltransferase (UGT) 2B15 pharmacogenetics: UGT2B15 D85Y genotype and gender are major determinants of oxazepam glucuronidation by human liver. J Pharmacol Exp Ther. 2004;310(2):656–665.
- Högler W, Wudy SA, Luef G, et al. Oxcarbazepine accelerates cortisol elimination via cytochrome P450 3A4 induction. Arch Dis Child. 2010;95(12):1065.
- Andreasen AH, Brosen K, Damkier P. A comparative pharmacokinetic study in healthy volunteers of the effect of carbamazepine and oxcarbazepine on CYP3A4. Epilepsia. 2007;48(3):490–496.
- Sugiyama I, Murayama N, Kuroki A, et al. Evaluation of cytochrome P450 inductions by anti-epileptic drug oxcarbazepine, 10-hydroxyoxcarbazepine, and carbamazepine using human hepatocytes and HepaRG cells. Xenobiotica. 2016;46(9):765–774.
- Miura M, Satoh S, Kagaya H, et al. Impact of the CYP3A4*1G polymorphism and its combination with CYP3A5 genotypes on tacrolimus pharmacokinetics in renal transplant patients. Pharmacogenomics. 2011;12(7):977–984.
- Wang P Study on the correlation between CYP3A4/3A5, ABCB1 gene polymorphism and the plasma concentration of carbamazepine, oxcarbazepine and its main metabolites. PhD Dissertation, Central South University: Changsha, 2014.
- Neels HM, Sierens AC, Naelaerts K, et al. Therapeutic drug monitoring of old and newer anti-epileptic drugs. Clin Chem Lab Med. 2004;42(11):1228–1255.
- Zhou X, Ma L, Xiang Q, et al. Relationship between cytochromes P450 2C19* 2 genotype and the concentration of the active oxcarbazepine metabolite in pediatric patients with epilepsy. Chin J Clin Pharmacol. 2017;33:595–598.
- Xia D, Zuo J, Zhao D, et al. Genetic polymorphism analysis of cytochrome CYP3A4, CYP2C9, CYP2C19, CYP2D6 in Chinese Han and Mongolian population. China Pharm. 2012;47:2017–2022.
- Mu D. Preliminary theoretical analysis of pharmacokinetics and metabolic differences between oxcarbazepine and carbamazepine. Tianjin: Tianjin University; 2006.
- Wang P, Lin XQ, Cai WK, et al. Effect of UGT2B7 genotypes on plasma concentration of valproic acid: a meta-analysis. Eur J Clin Pharmacol. 2018;74(4):433–442.
- Girard H, Villeneuve L, Court MH, et al. The novel UGT1A9 intronic I399 polymorphism appears as a predictor of 7-ethyl-10-hydroxycamptothecin glucuronidation levels in the liver. Drug Metab Dispos. 2006;34(7):1220–1228.
- Zhang W Analysis of the efficacy and influencing factors of oxcarbazepine in children with epilepsy. PhD Dissertation, Fujian Medical University: Fuzhou, 2019.
- Lu Y, Fang Y, Wu X, et al. Effects of UGT1A9 genetic polymorphisms on monohydroxylated derivative of oxcarbazepine concentrations and oxcarbazepine monotherapeutic efficacy in Chinese patients with epilepsy. Eur J Clin Pharmacol. 2017;73(3):307–315.
- Guo D, Pang LF, Han Y, et al. Polymorphisms of UGT1A9 and UGT2B7 influence the pharmacokinetics of mycophenolic acid after a single oral dose in healthy Chinese volunteers. Eur J Clin Pharmacol. 2013;69(4):843–849.
- Neumann E, Mehboob H, Ramírez J, et al. Age-Dependent Hepatic UDP-Glucuronosyltransferase Gene Expression and Activity in Children. Front Pharmacol. 2016;7:437.
- He N, Zhao X, Zhang W. Research progress of relationship between the UGT2B7 genetic polymorphisms and the risk of cancers. Chin J Clin Pharmacol. 2015;31:1796–1798.
- Ma C Effect of gene polymorphism on the pharmacokinetics and pharmacodynamics of carbamazepine and its derivative oxcarbazepine. PhD Dissertation, Fudan University: Shanghai, 2013.
- Chung JY, Cho JY, Yu KS, et al. Pharmacokinetic and pharmacodynamic interaction of lorazepam and valproic acid in relation to UGT2B7 genetic polymorphism in healthy subjects. Clin Pharmacol Ther. 2008;83(4):595–600.
- Shen C, Zhang B, Liu Z, et al. Effects of ABCB1, ABCC2, UGT2B7 and HNF4α genetic polymorphisms on oxcarbazepine concentrations and therapeutic efficacy in patients with epilepsy. Seizure. 2017;51:102–106.
- Chatzistefanidis D, Lazaros L, Giaka K, et al. UGT1A6- and UGT2B7-related valproic acid pharmacogenomics according to age groups and total drug concentration levels. Pharmacogenomics. 2016;17(8):827–835.
- Bhasker CR, McKinnon W, Stone A, et al. Genetic polymorphism of UDP-glucuronosyltransferase 2B7 (UGT2B7) at amino acid 268: ethnic diversity of alleles and potential clinical significance. Pharmacogenetics. 2000;10(8):679–685.
- Lazarowski A, Czornyj L, Lubienieki F, et al. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia. 2007;48(Suppl.):140–149.
- Kubota H, Ishihara H, Langmann T. Distribution and functional activity of P-glyeoprotein and multi-drug resistance-associated proteins in human brain microvascular endothelial cells in hippocarnpal sclerosis. Epilepsy Res. 2006;68(3):213–228.
- Dallas S, Miller DS, Bendayan R. Multidrug resistanceassociated proteins: expression and function in the central nervous system. Pharmacol Rev. 2006;58(2):140–161.
- Tazzite A, Kassogue Y, Diakité B, et al. Association between ABCB1 C3435T polymorphism and breast cancer risk: a Moroccan case-control study and meta-analysis. BMC Genet. 2016;17(1):126.
- Chouchi M, Kaabachi W, Klaa H, et al. Relationship between ABCB1 3435TT genotype and antiepileptic drugs resistance in Epilepsy: updated systematic review and meta-analysis. BMC Neurol. 2017;17(1):32.
- Yue L The distribution of ABCB1 gene polymorphism in patients with epilepsy and its correlation with the efficacy of antiepileptic drugs. PhD Dissertation, Fudan University: Shanghai, 2010.
- Loscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005;76(1):22–76.
- Lee G, Dallas S, Hong M, et al. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev. 2001;53(1):569–596.
- Chwab M S, Ezchelbaum M, Fromm MF. Genetic polymorphisms of the human drug transporter. Annu Rev Pharmacol Toxicol. 2003;43(1):285–307.
- Fromm MF. The influence of MDR1 polymorphisms on P-glycoprotein expression and function in humans. Adv Drug Deliv Rev. 2002;54(10):1295–1310.
- Potschka H, Fedrowitz M, Loscher W. P-glycoprotein and multi-drug resistance-associated protein are involved in the regulation of extracellular levels of the major antiepileptic drug carbamazepine in the brain. Neuroreport. 2001;12(16):3557–3560.
- Haufroid V. Genetic polymorphisms of ATP-binding cassette transporters ABCB 1 and ABCC2 and their impact on drug disposition. Curr Drug Targets. 2011;12(5):631–646.
- Hung CC, Chang WL, Ho JL, et al. Association of polymorphisms in EPHX1, UGT2B7, ABCB1, ABCC2, SCN1A and SCN2A genes with carbamazepine therapy optimization. Pharmacogenomics. 2012;13(2):159–169.
- Ueda K, Taguchi Y, Morishima M. How does P-glycoprotein recognize its substrates? Semin Cancer Biol. 1997;45:424–429.
- Silvio B, Sanja H, Poljakovic Z. Comment letters. Epilepsia. 2006;47(2):449–452.
- Wang-Tilz Y, Tilz C, Wang B, et al. Influence of lamotrigine and topiramate on MDR1 expression in difficult-to-treat temporal lobe epilepsy. Epilepsia. 2006;47(2):233–239.
- Volk HA, Loscher W. Multidrug resistance in epilepsy: rats with drug-resistant seizures exhibitenhanced brain expression of P-glycoprotein compared with rats with drug-responsive seizures. Brain. 2005;128(6):1358–1368.
- Rizzi M, Cacoia S, Guiso G. Limbic seizures induce P-glycoprotein in rodent brain:functional implications for pharmacoresistance. J Neurosei. 2002;22(14):5833–5839.
- Lv Y, Yan Y, Wang X, et al. Antiepileptic drug-induced overexpression of P-glycoprotein-multidrug resistance gene in astrocytes in vitro. Chin J Neurol. 2001;34:42–44.
- Kwan P, Sills GJ, Butler E, et al. Regional expression of multidrug resistance genes in genetically epilepsy-prone rat brain after a single audiogenic seizure. Epilepsia. 2002;43(11):1318–1323.
- Wu X, Shen D. Intractable epilepsy. Chin J Neurol. 1998;31:4–6.
- Ling V. Multidrug resistance: molecular mechanisms and mlinical relevance. Cancer Chemother Pharmacol. 1997;40(7):3–8.
- Hung C, Chen C, Lin C, et al. Functional evaluation of polymorphisms in the human ABCB1 gene and the impact on clinical responses of antiepileptic drugs. Pharmacogenet Genomics. 2008;18(5):390–402.
- Hoffmeyer S, Burk O, von Richter O, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97(7):3473–3478.
- Zheng X Study on the relationship between the drug resistance of refractory epilepsy and the pharmacological mechanism of levetiracetam and the polymorphism of MDR1 C3435T. PhD Dissertation, Tianjin Medical University: Tianjin, 2009.
- Gao Q Study on population pharmacokinetics of levetiracetam and polymorphism of MDR1 C3435T in patients with epilepsy. PhD Dissertation, Fujian Medical University: Fuzhou, 2014.
- Tan N, Heron S, Scheffer I, et al. Failure to confirm association of a polymorphism in ABCB1 with multidrug-resistant epilepsy. Neurology. 2004;63(6):1090–1092.
- Kwan P, Baum L, Wong V, et al. Association between ABCB1 C3435T polymorphism and drug-resistant epilepsy in Han Chinese. Epilepsy Behav. 2007;11(1):112–117.