
ABSTRACT
Our study aimed to explore the effect of C-type lectin-like receptor 2 (CLEC2) expression level on oxidized low-density lipoprotein (ox-LDL)-induced macrophage damage and the regulatory mechanism of macrophage foaming. Foam cells were derived from RAW264.7 by ox-LDL, and the cell viability was detected by cell counting kit-8 (CCK-8) assay. Enzyme-linked immunosorbent assay (ELISA) was applied to detect the levels of inflammatory cytokines tumor necrosis factor (TNF-α), Interleukin-6 (IL-6), and Interleulin-1β (IL-1β). Small interfering CLEC2 (si-CLEC2) was synthesized and transfected into RAW264.7, and the apoptosis rate was analyzed by flow cytometry. Western blotting was employed to detect the protein expressions of Janus kinase 1 (JAK1), Signal transducers and activators of transcription-1 (STAT1), phosphorylation-JAK1 (p-JAK1), phosphorylation-STAT1 (p-STAT1), CLEC2, and the apoptosis-related proteins. The levels of total cholesterol (TC) and free cholesterol (FC) were measured using colorimetric kits. Results showed that ox-LDL could activate the JAK1/STAT1 pathway of macrophages and up-regulate the expression of CLEC2. CLEC2 knockdown could reduce macrophage inflammation and lipid accumulation. Inactivating JAK1/STAT1 pathway with JAK1 inhibitor can significantly reduce the phosphorylation of STAT1 and alleviate the ox-LDL-induced damage in macrophages by regulating the expression of CLEC2. In conclusion, targeting JAK1/STAT1 to inhibit CLEC2 can attenuate ox-LDL-induced macrophage damage. This study enriched the pathogenesis of atherosclerosis and provided the possible treatment targets.
Graphical abstract
Introduction
Atherosclerosis (AS) is the pathological basis of many diseases, which seriously threatens human health [Citation1]. Inflammatory induced by macrophages is the mean cause. Macrophages deposit cholesterol by up-taking ox-LDL and then form foam cells, that is the pathophysiological basis of AS and plays an important role in regulating AS severity [Citation2,Citation3]. Therefore, studying the regulatory mechanism of macrophage foaming is of great clinical and scientific significance, which may provide new targets for the development of AS targeted drugs.
Macrophages are essential for the development of AS, and macrophages are the largest number of inflammatory cells in plaques [Citation4–6]. M1 macrophages secrete pro-inflammatory cytokines to aggravate the inflammatory response of AS. Thus, inhibiting the M1 polarization of macrophages can significantly slow down the progression of AS [Citation7]. CLEC2 is a surface receptor that mediates platelet aggregation and activation [Citation8]. Recent studies have found that CLEC2 is one of the key points in the inflammatory response. For example, the pro-inflammatory response and tissue damage of Clec2d−/− mice are reduced under the hepatotoxic injury, and the survival rate is higher [Citation9]. CLEC2 aggravates the mice lung inflammation by interacting with podoplanin, a ligand on inflammatory alveolar macrophages [Citation10]. Based on the importance of macrophages on inflammatory, we focused on the regulation of CLEC2 in macrophages. Or rather, regulating CLEC2 in macrophages may be a vital function in the progress of AS [Citation11,Citation12]. The expression of signal transducers and activators of transcription-1 (STAT1) in ox-LDL-induced macrophages was up-regulated, indicating that STAT1 activation promoted M1 type polarization of macrophages and aggravated atherosclerotic inflammation [Citation13,Citation14]. Dengue virus activated platelets through CLEC2 to release extracellular vesicles (EV) to promote the release of pro-inflammatory cytokines while blocking STAT1 can effectively attenuate the inflammatory response induced by dengue virus [Citation15]. These results suggest that STAT1 and CLEC2 are closely related to inflammation. Therefore, this study will focus the study on the role of CLEC2 and STAT1 signaling pathways in ox-LDL-induced foam macrophage inflammation.
Methods
Cell culture and treatment
Murine RAW264.7 cells were obtained from the American Type Culture Collection (USA), and cultured in Dulbecco’s modified eagle medium (DMEM) medium (Sigma-Aldrich, USA) with 10% FBS (Gibco, USA) in a 37°C incubator containing 5% CO2. Cells were cultured with ox-LDL (Solarbio, China) for 24 h, and then foam cells were successfully formed. For the inhibitor treatment, 1 μM baricitinib or 1% DMSO was added to the cells for 2 h to inhibit JAK1.
Cell counting kit-8 (CCK-8) assay
According to CCK-8 kit’s (Beyotime, China) instruction, RAW264.7 macrophages with a density of 1 × 104cells/ml were seeded into a 96-well plate (100 μl/well). Gradient concentrations of ox-LDL (0, 25, 50, 100 μg/ml) were added for 24 h. After the treatment, 10 μl CCK8 was added to the wells and the cells were incubated for 3 h. Afterward, the optical density (OD) value was determined at 450 nm.
Synthesis and transfection of small interfering RNA (siRNA)
siCLEC2 and the control siRNA were provided by Sangon Biotech (Shanghai, China). 2 × 104 cells/well were inoculated into 6-well plates. The cells were transfected with 50 nM siCLEC2 using Lipofectamine 2000 in the next day, and siRNA control was also transfected as the siControl group [Citation16]. The cells were then divided into Control, ox-LDL, ox-LDL+siControl and ox-LDL+siCLEC2 groups. After being transfected for 48 h, 100 μg/ml ox-LDL was treated for another 24 h, then the cells were collected to detect.
Enzyme-linked immunosorbent assay (ELISA)
The grouping of cells is the same as above. The ELISA kit (MSK, China) was applied to detect inflammatory cytokines (inflammatory cytokines tumor necrosis factor (TNF-α), Interleukin-6 (IL-6), and Interleukin-1β (IL-1β)) in the samples. According to the manufacture’s protocol, 40 μl sample diluent and 10 μl sample were added to each well, and standard control and blank control were set accordingly. 100 μl horseradish peroxidase (HRP)-labeled antibody, samples, and conjugates were added to each well, and then incubated for 1 h at 25°C. The plate was washed and then incubated at 37°C for 15 min with 50 μl substrates A and B successively. After adding 50 μl termination buffer, the OD value was determined at 450 nm. Finally, based on the standard curve linear regression equation we calculate the sample concentration.
Western blotting
Total proteins from each group were extracted and detected with Western blotting as previously [Citation17]. The antibodies were used as following: anti-CLCE2 (1:1000), anti-p-STAT1 (1:1000), anti-p-JAK1 (1:1000), anti- GAPDH (1:1000).
Flow cytometry assay
The cells were inoculated into 6-well plates. The adherent cells were digested with trypsin. The suspended cells were collected and the concentration was adjusted to 3 × 106/ml. The cell apoptosis was then detected with Annexin V- Fluoresceine Isothiocyanate (FITC) apoptosis detection kit (Beyotime, China). Specifically, 5 μl Annexin V-FITC, and 5 μl propidium iodide were added, respectively; then, the cells were staining for 15 min, and the flow cytometry was applied to measure the apoptosis ratio within 1 h.
Oil red O staining
Oil red O staining was performed according to the reference [Citation18]. After fixing and staining, decolorization was performed with 60% isopropyl alcohol, and the staining results were observed under a microscope, and the results were measured by Image Pro Plus.
Detection of total cholesterol and free cholesterol
The content of total cholesterol (TC) and free cholesterol (FC) in the cells were determined according to the instructions of the TC and FC colorimetry kits (Applygen, China), respectively [Citation19]. The lysis buffer was added at a ratio of 0.1 ml per 1 × 106 cells for 10 min. The lysate was then quantified with a BCA protein assay kit. 10 μl standard and samples were added, respectively, into 96-well plates and then treated with 190 μl working solution for 20 min at 37°C. The OD value was determined at 550 nm. The detection procedure of FC in cells was the same as TC’s.
Plasmid construction and cell transfection
CLEC2 plasmids and control plasmids were synthesized by Sangon Biotech. The cells were grouped as follows in 6-well plate: Control, ox-LDL, ox-LDL+DMSO, ox-LDL+Baricitinib and ox-LDL+Baricitinib+CLEC2 group. The next day, Lipofectamine 2000 and plasmids were mixed and added to the cells and mixed gently. After being transfected for 48 h, the cells were induced with 100 μg/ml ox-LDL for another 24 h.
Statistical analysis
The experiments were carried out 3 times repeatedly, and statistical analysis was conducted using Statistical Product and Service Solutions 22.0. The data were expressed as mean ± standard deviation. Differences were evaluated by t-test or one-way analysis of variance, P < 0.05 was regarded as statistical significance.
Results
Macrophages deposit cholesterol to form foam cells is the pathophysiological basis of AS and has a decisive role in AS development. Here, we study the effect of CLEC2 expression level on ox-LDL induced macrophage damage and the possible regulatory mechanism of macrophage foaming. Results indicate that ox-LDL could induce macrophage damage by activating the JAK1/STAT1 pathway and up-regulate the expression of CLEC2. And the JAK1 inhibitor could alleviate the damage.
Ox-LDL activated the JAK1/STAT1 pathway and up-regulated CLEC2 expression level in macrophages
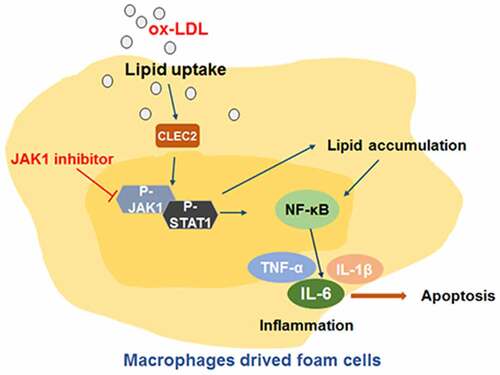
The cell viability was inhibited after treatment with gradient concentrations of ox-LDL, and the effect was concentration-dependent (). In addition, the levels of the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α had a significant increase when compared with the control group (). After treating macrophages with ox-LDL, the JAK1/STAT1 pathway was activated, the phosphorylation of JAK1 and STAT1 was promoted, and CLEC2 was upregulated (). These results show that the JAK1/STAT1 pathway and CLEC2 may be involved in the ox-LDL-induced macrophage damage.
Figure 1. The effect ox-LDL on the RAW264.7 macrophages. (a) CCK-8 assay was used to measure the cell viability. (b) ELISA assay was applied to detect the levels of IL-1β, IL-6, and TNF-α. (c) related-proteins expression were detected by Western blotting. *P < 0.05, **P < 0.1, compared with 0 μg/mL.
CLEC2 knockdown inhibited ox-LDL-induced macrophage apoptosis and up-regulated cell viability
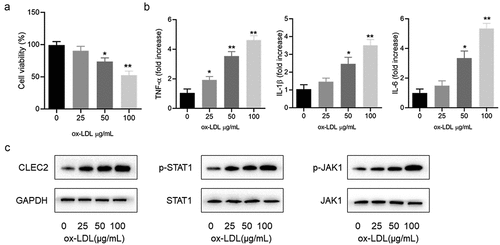
To decrease the expression of CLEC2, we synthesized siCLEC2 and transfected it into RAW264.7 cells. CLEC2 was significantly down-regulated in the cells transfected with siCLEC2#2 (). CCK-8 results showed that downregulation of CLEC2 improved the cell viability of ox-LDL-induced macrophages () but reduced the apoptosis rate (). In addition, CLEC2 inhibition in ox-LDL-treated macrophages upregulated the BCL2 expression, but decreased BAX expression (). As a result, it was displayed that CLEC2 knockdown could inhibit ox-LDL-induced apoptosis, thus up-regulate the viability of macrophages.
Figure 2. CLEC2 knockdown reversed the cell activity and apoptosis in ox-LDL-induced macrophages. (a) The expression level of CLEC2 was observed by Western blotting (transfected for 48 h). (b) Cell viability was detected using CCK-8 assay. (c) Apoptosis was detected by flow cytometry. (d) Western blotting was applied to detect the protein expression levels. **P < 0.01 compared with control; #P < 0.05 compared with ox-LDL+siControl.
CLEC2 knockdown inhibited ox-LDL-induced inflammatory response and lipid accumulation in macrophages
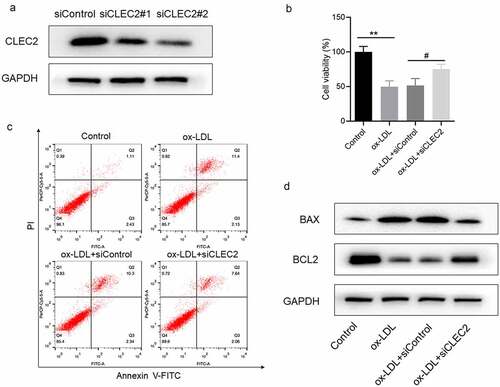
Further study showed that the levels of IL-1β, IL-6 and TNF-α were decreased in macrophages transfected with siCLEC2 (). Oil red O staining suggested that CLEC2 knockdown significantly inhibited the accumulation of intracellular lipids, as the levels of intracellular TC and FC decreased ()). In addition, the protein expression levels of HMGB1 and NF-κB decreased (). All evidence suggests that CLEC2 knockdown may suppress lipid accumulation and ox-LDL-induced inflammatory response in macrophages.
Figure 3. CLEC2 knockdown inhibited ox-LDL-induced inflammatory response and lipid accumulation in macrophages. (a) The levels of inflammatory cytokines were detected by ELISA. (b) Lipid accumulation was detected by oil red O staining, the area was measured by Image Pro Plus. (c) The contents of TC and FC were measured using colorimetric kits. (d) Western blotting was applied to detect the levels of protein expression. **P < 0.01 compared with control; #P < 0.05 compared with ox-LDL+siControl.
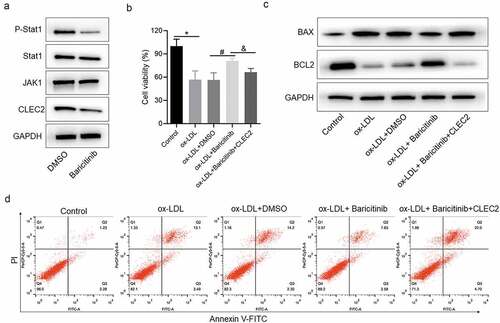
JAK1/STAT1 pathway affected the apoptosis and cell viability of ox-LDL-induced macrophage by regulating CLEC2
To verify the regulation mechanism between JAK1/STAT1 pathway and CLEC2, the JAK1 inhibitor baricitinib was used in the cells. The expression levels of CLEC2 and p-STAT1 were decreased after Baricitinib treatment (). Next, the cell viability detection indicated that inhibiting JAK1/STAT1 pathway with baricitinib could increase the cell viability when compared with ox-DL + DMSO treatment group, and overexpressed CLEC2 reversed this effect consequently (). In addition, the apoptosis rate was decreased in the baricitinib treatment group, but increased when CLEC2 was overexpressed (). For the expression of apoptosis-related proteins, we found that baricitinib treatment promoted BCL2 expression but reduced BAX expression, while CLEC2 overexpression promoted BAX expression but inhibited BCL2 expression (). The above findings indicate that the JAK1/STAT1 pathway affects the apoptosis and cell viability of ox-LDL-induced macrophages by regulating CLEC2.
Figure 4. JAK1/STAT1 signaling regulated the expression of CLEC2 and affected the cell viability and apoptosis. (a) The expression of JAK1/STAT1 signaling was detected with Western blotting. (b) The cell viability was detected with CCK-8 assay. (c) Western blotting was applied to detect the expression of proteins. (d) Apoptosis was detected with flow cytometry. *P < 0.05 compared with control, or ox-LDL+siControl; #P < 0.05 compared with ox-LDL+ DMSO; &P < 0.05 compared with ox-LDL+Baricitinib.
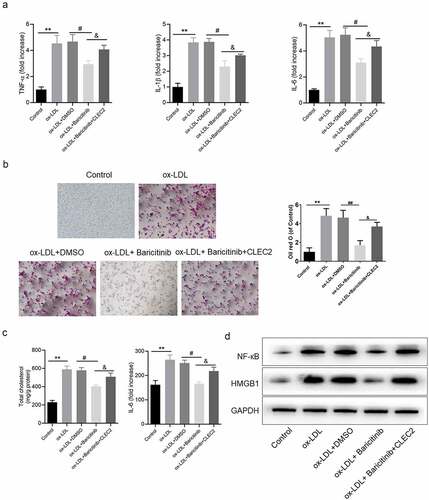
JAK1/STAT1 pathway affected the inflammatory response and lipid accumulation by regulating CLEC2 in ox-LDL-induced macrophage injury
Furthermore, inflammatory response with the effect of JAK1/STAT1 pathway was studied. In the ox-LDL-induced macrophage model, the levels of pro-inflammatory cytokines were decreased after baricitinib treatment, but increased after overexpression of CLEC2 (). Meanwhile, inhibiting JAK1 with baricitinib could significantly inhibit the accumulation of intracellular lipids and reduce the levels of TC and FC, but this phenomenon was reversed by the overexpression of CLEC2 ()). Protein expressions of HMGB1 and NF-κB demonstrated the same trend (). Consequently, it was indicated that JAK1/STAT1 pathway affected the inflammatory response and lipid accumulation by regulating CLEC2 in ox-LDL-induced macrophage injury.
Figure 5. JAK1/STAT1 pathway affected the inflammatory response and lipid accumulation. (a) The levels of inflammatory cytokines were detected by ELISA. (b) Lipid accumulation was detected by oil red O staining, and the area was measured by Image Pro Plus. (c) The contents of TC and FC were measured by colorimetric kits. (d) The protein expression levels were measured by Western blotting. **P < 0.01 compared with control; #P < 0.05 compared with ox-LDL+ DMSO; &P < 0.05 compared with ox-LDL+Baricitinib.
Discussion
Inflammation and ox-LDL have been proven to be important factors that cause AS [Citation20]. ox-LDL is formed by the oxidative modification of LDL, and it promotes the secretion of various chemokines and helps monocytes adhere to the endothelium [Citation21]. Besides, it activates monocytes to transform into macrophages, allowing them to absorb lipids through scavenger receptors (SRs) to form into foam cells [Citation22], which is crucial in the early development of AS [Citation23]. Meanwhile, studies have shown that inflammation stimulates the macrophage to release inflammatory cytokines and accelerates the formation of atherosclerotic plaque [Citation24]. Since ox-LDL is a key factor causing AS, targeting the ox-LDL-induced inflammatory pathway is a proven and effective method for treatment of AS. In this study, RAW264.7 cells, as a type of macrophage, were selected, and ox-LDL treatment was used to induce foam cells formation. Results demonstrated that ox-LDL could up-regulate the secretion of inflammation factors and down-regulate the viability of RAW264.7 cells. In addition, the protein expressions of p-JAK1, p-STAT1, and CLEC2 were increased, indicating that the JAK1/STAT1 pathway was activated by ox-LDL. As a result, it was suggested that the JAK1/STAT1 pathway and CLEC2 may be involved in the ox-LDL-induced macrophage damage.
CLEC2 is abundantly expressed in vascular endothelial cells [Citation25]. Studies have proved that CLEC2 can bind to the tumor cell surface protein podoplanin to activate platelets, thus causing platelet aggregation [Citation26]. However, no study has been conducted to clarify the effect of CLEC2 on the occurrence and development of AS. Macrophage foaming is the main pathogenesis in the inflammation of AS. Therefore, we studied the role of CLEC2 in ox-LDL induced macrophage foam. NF-κB is involved in regulating the inflammatory response [Citation27]. Activating NF-κB can help regulate the expression of IL-6, IL-1β and TNF-α, and promote the amplification of inflammatory cascade signals, thus regulating the occurrence and development of AS [Citation28,Citation29]. In order to explore whether and how CLEC2 mediates the up-regulation of NF-κB and related pro-inflammatory cytokines in RAW264.7, we transfected siCLEC2 into RAW264. Seven cells to knockdown CLEC2 expression. The results showed that the apoptosis rate of macrophages decreased, while cell viability was obviously improved by the down-regulation of CLEC2. At the same time, CLEC2 knockdown inhibited the ox-LDL-induced inflammatory response regulated by the activation of NF-κB, reflecting on the reduced levels of IL-1β, IL-6, TNF-α. Combined with the decrease in TC and FC, these results suggest that CLEC2 down-regulation can inhibit macrophage inflammatory response and lipid accumulation in ox-LDL-induced RAW264.7 cells.
Phosphorylation of JAK1 and Tyk2 could activate STAT1 and STAT2, which forms a signal transducer and activator of transcription 1 complex with interferon regulatory factor 9 to activate downstream genes [Citation30]. The JAK1/STAT1 signaling pathway has a great influence on the inflammatory response. The LYN proto-oncogene, protein phosphatase regulator (SHP)-1, and SHP-2 have been found to be involved in the activation of JAK1/STAT1 pathway in inflammatory responses [Citation31,Citation32]. In the present study, the JAK1 inhibitor baricitinib was used to inhibit the activity of JAK1/STAT1 pathway in RAW264.7 cells to help confirm whether JAK1/STAT1 pathway is involved in the differentiation of macrophages by regulating CLEC2. The results indicated that the prote in expressions of p-STAT1 and CLEC2 were positively correlated, and the overexpressed CLEC2 could reverse the effect of baricitinib. All the above findings revealed that JAK1/STAT1 pathway affects ox-LDL-induced macrophage damage by regulating CLEC2.
Conclusions
In conclusion, ox-LDL was used to induce RAW264.7 cells to form foam cells, and the new regulation mechanism of ox-LDL in the process of macrophage damage was investigated in this study. Our study found that CLEC2 and phosphorylation of STAT1 were increased in macrophages treated with ox-LDL.The results confirm that ox-LDL can activate the JAK1/STAT1 pathway to up-regulate CLEC2 expression and aggravate macrophage damage. Inhibiting STAT1 phosphorylation could significantly decrease the protein expression of CLEC2 and attenuate the damage of macrophages treated with ox-LDL. However, these results have not been validated in vivo. This study may enrich the regulatory mechanism of macrophages in the development of AS and provide a new target for the prevention and treatment of AS.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Liu Y, Li C, Yin H, et al. NLRP3 Inflammasome: a potential alternative therapy target for atherosclerosis. Evid Based Complement Alternat Med. 2020;2020:1561342.
- Boucher P, Matz RL, Terrand J. Atherosclerosis: gone with the Wnt? Atherosclerosis. 2020;301:15–22.
- Guerrini V, Gennaro ML. Foam cells: one size doesn’t fit all. Trends Immunol. 2019;40(12):1163–1179.
- Groh L, Keating ST, Joosten LAB, et al. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. 2018;40(2):203–214.
- Sharma M, Schlegel MP, Afonso MS, et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res. 2020;127(3):335–353.
- Kim H, Kumar S, Kang DW, et al. Affinity-driven design of cargo-switching nanoparticles to leverage a cholesterol-rich microenvironment for atherosclerosis therapy. ACS Nano. 2020;14(6):6519–6531.
- Wang Q, Chen K, Zhang F, et al. TRPA1 regulates macrophages phenotype plasticity and atherosclerosis progression. Atherosclerosis. 2020;301:44–53.
- Chauhan A, Sheriff L, Hussain MT, et al. The platelet receptor CLEC-2 blocks neutrophil mediated hepatic recovery in Acetaminophen induced acute liver failure. Nat Commun. 2020;11(1):1939.
- Lai JJ, Cruz FM, Rock KL. Immune sensing of cell death through recognition of histone sequences by C-Type lectin-receptor-2d causes inflammation and tissue injury. Immunity. 2020;52(1):123–135 e6.
- Lax S, Rayes J, Wichaiyo S, et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol. 2017;313(6):L1016–L1029.
- Chen Q, Lv J, Yang W, et al. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics. 2019;9(22):6424–6442.
- Ivashkiv LB. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18(9):545–558.
- Vinue A, Navarro J, Herrero-Cervera A, et al. The GLP-1 analogue lixisenatide decreases atherosclerosis in insulin-resistant mice by modulating macrophage phenotype. Diabetologia. 2017;60(9):1801–1812.
- Jian D, Dai B, Hu X, et al. ox-LDL increases microRNA-29a transcription through upregulating YY1 and STAT1 in macrophages. Cell Biol Int. 2017;41(9):1001–1011.
- Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. 2019;10(1):2402.
- Haider M, Al-Rashed F, Albaqsumi Z, et al. Candida albicans induces foaming and inflammation in macrophages through FABP4: its implication for atherosclerosis. Biomedicines. 2021;9(11):1567.
- Shao W, Xu J, Xu C, et al. Early-life perfluorooctanoic acid exposure induces obesity in male offspring and the intervention role of chlorogenic acid. Environ Pollut. 2021;272:115974.
- Lin X, Hu H, Liu Y, et al. Allicin induces the upregulation of ABCA1 expression via PPARγ/LXRα signaling in THP-1 macrophage-derived foam cells. Int J Mol Med. 2017;39(6):1452–1460.
- Schwarz A, Bonaterra GA, Schwarzbach H, et al. Oxidized LDL-induced JAB1 influences NF-κB independent inflammatory signaling in human macrophages during foam cell formation. J Biomed Sci. 2017;24(1):12: 12.
- Jialal I, Devaraj S. Low-density lipoprotein oxidation, antioxidants, and atherosclerosis: a clinical biochemistry perspective. Clin Chem. 1996;42(4):498–506.
- Ahotupa M. Oxidized lipoprotein lipids and atherosclerosis. Free Radic Res. 2017;51(4):439–447.
- Makowski MR, Henningsson M, Spuentrup E, et al. Characterization of coronary atherosclerosis by magnetic resonance imaging. Circulation. 2013;128(11):1244–1255.
- Yu XH, Fu YC, Zhang DW, et al. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245–252.
- Farahi L, Sinha SK, Lusis AJ. Roles of macrophages in atherogenesis. Front Pharmacol. 2021;12:785220.
- Colonna M, Samaridis J, Angman L. Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur J Immunol. 2000;30(2):697–704.
- Kato Y, Kaneko MK, Kunita A, et al. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. 2008;99(1):54–61.
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336(15):1066–1071.
- Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–234.
- Wu Y, Zhang W, Liu W, et al. The double-faced metabolic and inflammatory effects of standard drug therapy in patients after percutaneous treatment with drug-eluting stent. Atherosclerosis. 2011;215(1):170–175.
- Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662.
- Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320(26):1731–1735.
- Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–529.