
ABSTRACT
Interstitial Cells of Cajal (ICC) plays a critical role in the peristaltic contractions of the gastrointestinal and urinary tract. The dysfunction and loss of ICC contributes to hypokinetic disease, such as gallstoneand ureteropelvic junction obstruction . In the present study, we identified the underlying driving molecular signals of oxidative stress and apoptosis in ICC. ICC was isolated from small intestine of Balb/c mice, and stimulated with tumor necrosis factor-alpha (TNF-α). MTT and flow cytometry were performed to assess cell viability, apoptosis, and the level of reactive oxygen species in ICC, respectively. The level of malondialdehyde, superoxide dismutase, and glutathione peroxidase in cells were measured to assess oxidative stress. The expression of inflammatory factors (interleukin, IL-1 and IL-6) and apoptosis-related proteins were detected by western blot. We observed that TNF-αinduced inflammation, oxidative stress and cell apoptosis in ICC. By using quantitative real-time PCR , we verified that the expression of long non-coding RNAMEG3 was elevated by TNF-α in ICC. Silencing MEG3 reversed inflammation, oxidative stress, and cell apoptosisin TNF-α-treated ICC. Subsequently, we confirmed that MEG3 sponged cytoprotective miR-21 to upregulate the expression of I-kappa-B-kinase beta (IKKB) and activate the nuclear factor kappa-B (NF-κB) pathway. Both miR-21 overexpression and IKKB knockdown reduced TNF-α-induced above symptoms in ICC. Taken together, we can conclude that MEG3 mediates inflammation, oxidative stress and apoptosis in TNF-α-treated ICC via the miR-21/IKKB-NF-κB axis. The study improves our understanding of the molecular mechanism of ICC reduction related diseases.
Graphical Abstract

Introduction
Interstitial Cells of Cajal (ICC) is a kind of pacemaker cells initially identified in the gastrointestinal tract, located in the myenteric region between nerve endings and smooth muscle cells [Citation1]. Afterward, ICC has also been discovered in urogenital tissues, including ureter, prostate, and junction of renal pelvis and ureter, etc [Citation2,Citation3]. ICC and longitudinal smooth muscle cells have the same origin, mesenchymal stem/progenitor cells, which can become ICC under the stimulation of stem cell factor (SCF) during embryonic development. ICC plays a critical role in the peristaltic contractions of the gastrointestinal and urinary tract by generating a rhythmic depolarization and transmitting it to the musculature [Citation1,Citation4]. Recently, studies have proved that the dysfunction and loss of ICC are involved in hypokinetic disease, such as colonic motor dysfunction, gallstone and ureteropelvic junction obstruction (UPJO) [Citation5–7]. Of note, inflammatory stimulation could induce oxidative stress, thus impairing the pacemaker function or even inducing cell apoptosis in ICC [Citation8]. However, little is known about the biological characteristics of ICC under inflammatory stimulation and its molecular regulatory mechanism.
Long non-coding RNAs (lncRNAs) are a class of RNAs, each of which contains more than 200 nucleotides and does not encode protein. LncRNAs are widely involved in the regulation of cell biological processes, including apoptosis, inflammation, and stress response. LncRNA MEG3 is generally expressed in many normal tissues, but previous studies have shown the expression of MEG3 is missing in multiple cancer [Citation9–11]. Plenty of basic experiments evidence MEG3 acts a tumor suppressor, which can inhibit the proliferation of tumor cells [Citation10,Citation12,Citation13]. Besides, MEG3 is reported to promote inflammation, apoptosis, and oxidative stress of non-cancer cell [Citation14–17]. LncRNA is well known to participate in the post-transcriptional modification of genes as a competitive endogenous RNA. LncRNAs sponge microRNAs (miRNAs), a class of small non-coding RNAs with a length of about 22 nucleotides, to release target mRNA, so as to upregulate the expression of effector genes. Similar to lncRNAs, miRNAs are also involved in the biological behaviors of cells, and their aberrant expression is often associated with diseases. MiR-21 has been identified as an apoptotic suppressor and oncogenic miRNA in various cancer types [Citation18]. Recent studies suggest that miR-21 can protect cell from oxidative stress and apoptosis [Citation19–21]. Interestingly, increasing evidences show that miR-21 is negatively regulated by MEG3 in different types of tissues [Citation22–24]. The interaction between MEG3 and miR-21 and their function in ICC have not been studied yet.
Nuclear factor kappa-B (NF-κB) is a key transcription factor in cells, which mediates cell proliferation, bacterial and viral infection, inflammation, and so on. In recent years, studies have pointed out that it also plays an important role in cellular oxidative stress and apoptosis [Citation25–28]. I-kappa-B-kinase beta (IKKB) is an upstream activator of the NF-κB pathway [Citation29]. IKKB is crucial for the rapid activation of NF-κB pathway through the pro-inflammatory signaling cascade, so inhibition of IKKB expression and NF- κB signaling is often used as a treatment strategy for diseases, such as inflammation and cancer. In the early stage of the study, we performed bioinformatics analysis and found that there was a binding site between miR-21 and IKKB.
Based on those evidences mentioned above, we hypothesized that the MEG3/miR-21/IKKB network and the NF-κB pathway drive oxidative stress and apoptosis in ICC under pathological conditions. Therefore, we isolated ICC from murine small intestine, stimulated it with tumor necrosis factor (TNF-α) and carried out a set of in vitro experiments to verify the possible molecular mechanism. The work aimed to provide a potential target for the treatment of ICC dysfunction and loss-related diseases.
Materials and methods
ICC isolation and culture
Balb/c mice were used for ICC preparation. The care and studies on animals were conducted according to the guidelines of The Ethics Committee of The First Affiliated Hospital of Xinjiang Medical University. Small intestine of mouse was used as the tissue source of ICC, due to the abundance of ICC [Citation30]. In brief, the small intestine (10 cm from ileocecum) in each mouse was removed and dissected longitudinally, and the mucosal layer was stripped. The muscle strips were repeatedly rinsed with Ca2+-free Hank’s solution (pH7.4, MIBio, Shanghai, China) for 3 ~ 4 times, and then cut into 1 ~ 2 mm3 tissue pieces. The tissues were incubated with 20 mL of type II collagenase (1.3 mg/mL, BD, NJ, USA) for 30 min at 37°C. The mixture was centrifugated at 1500 r/min for 3 min, resuspended with DMEM medium. Then, cells were removed large pieces through a 200-mesh sieve, added to Ficoll 400 gradient density liquid (Reagan Biotechnology, Beijing, China), centrifugated at 30 g for 10 min, and the cell precipitation at the junction of the liquid level was stored.
The cells were cultured with the smooth muscle growth medium (Sciencell, CA, USA) containing 10% FBS (Gibco, NY, USA), 2% antibiotics/antimycotics (Gibco) and 5 ng/mL of SCF (Sigma, MO, USA), in which cover slips coated with murine collagen (BD) was placed in advance. The medium was then plated in an incubator with 5% CO2 at 37°C. Finally, ICC could be identified using a microscopy after labeled with anti-c-Kit antibody.
TNF-α intervention
Cells were initially treated with different concentration of TNF-α (Sigma, 0, 10, 20, 30 ng/mL) to ensure the appropriate concentration for intervention, the concentration design refers to the previous study [Citation31]. In the next experiment, cells were stimulated by 20 ng/mL of TNF-α.
Cell transfection
Cells were transfected with siRNA-NC, siRNA-MEG3, pcDNA-NC, pcDNA-MEG3, miRNA-NC, miR-21 mimic, miR-21 inhibitor, or siRNA-IKKB by using the transfection kit (Introvigen, CA, USA) according to the instruction. The siRNA, pcDNA, mimic and inhibitor were designed and synthesized at GenePharma Co., Ltd. (Shanghai, China). miR-21 mimic: 5′-UAGCUUAUCAGACUGAUGUUGA-3′, miR-21 inhibitor: 5′-UAGCUUAUCAGACUGAUGUUGA-3′.
Luciferase reporter gene assay
The binding site of miR-21 and MEG3 has been reported in the previous studied [Citation32], and the binding site of miR-21 and IKKB was predicted by miRwalk (http://mirwalk.umm.uni-heidelberg.de/). Then, Dual-Lumi™ Luciferase Reporter Gene Assay Kit (Beyotime, Shanghai, China) was used to test the binding relationship [Citation33]. Briefly, the wild type or mutant untranslated region (UTR) of MEG3 or IKKB was cloned to the reporter vector, and then the vector was co-transfected to the HEK cells (Sciencell) with miR-21 mimic or miRNA-NC. Forty-eight hours later, the luciferase activity was detected at a microplate reader. The mutant sequence of MEG3: 5’-GGGGGCAUUGGGCAUAGUCACC-3’, and the mutant sequence of IKKB: 5’-AGCACCGTACCAGCATGAG-3’.
MTT assay
MTT assay was performed to detect the cell viability in vitro as previous reported [Citation34]. First, cells were seeded in 96-well plates, and cultured overnight. Second, different groups of cells were given corresponding treatment (with or without TNF-α stimulation). Twenty-four hours later, MTT reagent (Sigma, MO, USA, 10 μL/well) was added into cells, and incubated for 2 h. Finally, a microplate reader was used to measure the absorption value of the mixture at 450 nm.
Apoptosis assay
Annexin V-fluoresecin isothiocyanate (FITC)/PI double staining kit (Beijing Biosea Biotechnology, Beijing, China) and flow cytometry were performed to evaluated cell apoptosis. In brief, cells were seeded into a 6-well plate at a density of 1 × 105/well and cultured overnight. Then, different groups of cells were given corresponding treatment (with or without TNF-α stimulation). Cells were suspended with 200 μL of binding buffer (containing 5 μL PI and 10 μL Annexin V-FITC), which was incubated for 30 min in the dark. Finally, the cells were analyzed through flow cytometry (CytoFLEX, Beckman Coulter, CA. USA).
Measurement of oxidative stress reaction levels
The levels of MDA, SOD, and GSHPx in the supernatant were determined using the corresponding kit (Beyotime, Shanghai, China) and a microplate reader (ThermoFisher, Shanghai, China). The ROS levels were detected using the Reactive Oxygen Species Assay Kit (Beyotime, Shanghai, China), and a flow cytometry (CytoFLEX). All operations were carried out according to the manufacturer’s instructions.
Quantitative Real-time PCR (qRT-PCR)
The RNA level was measured by qRT-PCR as previously reported [Citation35]. Total RNA in cells was extracted by using Trizol reagent (Beyotime). When its purity was qualified, the RNA sample was used as a template for reverse transcription PCR using the Primer Script RT reagent kit (Takara, Beijing, China). The product of RT-PCR (cDNA) was used as a template for the following quantitative PCR (qPCR) using a qPCR kit (Taqman, Shanghai, China). The primers were listed as follows: MEG3: 5′-CTGCCCATCTACACCTCACG-3′(forward) and 5′-CTCTCCGCCGTCTGCGCTAGGGGCT-3′(reverse); IKKB: 5’-CAGCAAGGAGAACAGAGGT-3’ (forward) and 5’-ACTGTGTACTTCTGCTGCTC-3’(reverse); GAPDH: 5’ -CACTCACGGCAAATTCAACGGCA-3’(forward) and 5’ -GACTCCACGACATACTCAGCAC-3’(reverse); miR-21: 5’-GCGGCGGTAGCTTATCAGACTG-3’ (forward) and 5’ -ATCCAGTGCAGGGTCCGAGG-3’ (reverse); U6: 5’-GCGCGTCGTGAAGCGTTC-3’ (forward) and 5’ -GTGCAGGGTCCGAGGT-3’(reverse). The GAPDH and U6 were used as the internal reference genes. The relative expression was calculated through the formula ‘2−ΔΔct’.
Western blot
The protein expression was measured by western blot as previous reported [Citation36]. RIPA lysis buffer (CWBIO, Beijing) was used to release protein in cells. 20 μg of protein sample was subjected to electrophoresis, and transferred to polyvinylidene (PVDF) membrane. The membrane was blocked with skimmed milk. Next, they were incubated at 4°C overnight with the primary antibody (IL-1: # M421B, IL-6: #M621B, Bax: #PA5-11378, Bcl2: #PA5-20068, cleaved caspase3: #PA5-20068, IKKB: # MA5-16162, GAPDH: # 14–9523-82, p65: # PA1-186, H3: # MA5-15721, ThermoFisher, 1:1000 dilution). After being washed, the membrane was incubated with horseradish peroxidase-labeled secondary antibody (#31430, ThermoFisher, 1:5000 dilution). Finally, the membrane was photographed under a gel imaging analyzer (Bio-Rad, CA, USA).
Statistical analysis
Data was analyzed by using the Prism 7.0 software (GraphPad, CA, USA). All data was presented as the ‘average ± standard deviation’, and data from two groups was compared by using the student’s t-test. Differences were considered to be statistically significant when p value was less than 0.05.
Results
TNF-α induced oxidative stress, inflammation and apoptosis in ICC
TNF-α is usually used for construction of pathological model. In the present study, we also used TNF-α to induce oxidative and apoptosis in ICC. The viability of ICC in different concentration of TNF-α (0, 10, 20, 30 ng/mL) was assessed by MTT assay, and data showed that cell viability was reduced by TNF-α in a concentration-dependent manner (p < 0.05, ). According to the results, 20 ng/ml of TNF-α had a moderate effect on the survival rate of cells, so 20 ng/ml was selected as the intervention concentration of TNF-α in the following experiments. Flow cytometry was then performed to assess the apoptosis, and results confirmed that 20 ng/ml of TNF-α promoted the apoptosis in ICC (p < 0.05, ). Next, the oxidative stress reaction markers were detected through ELISA kit and the flow cytometry. We found that TNF-α elevated the level of MDA and ROS in ICC, and decreased the level of SOD and GSHPx (p < 0.05, ). In addition, we detected the expression of inflammation or apoptosis-related protein, and consistent with the above results, TNF-α increased the inflammation markers (IL-1 and IL-6) and the apoptogenic factor (Bax and cleaved caspase3), but reduced the anti-apoptotic protein Bcl2 in ICC (p < 0.05, ). These findings indicated that TNF-α induced oxidative stress, inflammation, and apoptosis in ICC, and it could be used as an inducer of ICC cell apoptosis in vitro.
Figure 1. TNF-α induced oxidative stress, inflammation and apoptosis in ICC. a:Cell viability in ICC treated with different concentrations of TNF-α(0, 10, 20, 30 ng/mL), ‘*’ means P < 0.05 compared with ICC treated with 0 ng/mL of TNF-α. ICC was treated with or without 20 ng/mL of TNF-α. b-c: Flow cytometry was performed to detect cell apoptosis rate (b) and ROS level (c). d: Oxidative stress reaction markers were detected by ELISA method with a microplate reader. e:Western blot was used to determine the expression of IL-1, IL-6, Bax, Bcl2 and cleaved caspase3.‘*’ means P < 0.05 compared with control group.
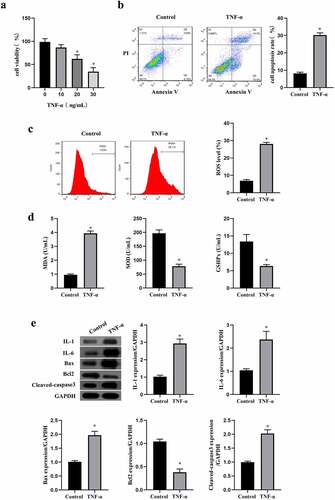
MEG3 silence reduced oxidative stress and apoptosis induced by TNF-α in ICC
MEG3 has been identified as a tumor inhibitor and can also promote inflammation, oxidative stress, and cell apoptosis of non-tumor cells in previous studies [Citation12,Citation16,Citation17]. As shown in , the MEG3 expression was significantly elevated by TNF-α treatment in ICC (p < 0.05), which suggested that MEG3 might be involved in the stress process of ICC with TNF-α stimulation. Therefore, we knockdown the expression of MEG3 by transfecting siRNA-MEG3 to ICC, and assessed oxidative stress, inflammation, and apoptosis under the intervention of TNF-α. The data in showed that silencing MEG3 rescued cell viability and inhibited cell apoptosis induced by TNF-α in ICC (p < 0.05). Furthermore, we found that MEG3 silence also reduced the oxidative stress level and enhanced the antioxygenic ability in TNF-α-treated ICC (p < 0.05, ). These data were confirmed by the results of western blot, and knockdown of MEG3 reduced the expression of inflammatory factor and pro-apoptotic factor in ICC (p < 0.05, ). These findings suggested that MEG3 silence reduced oxidative stress and apoptosis induced by TNF-α in ICC.
Figure 2. MEG3 silence reduced inflammation and apoptosis induced by TNF-α in ICC. a: MEG3 expression was detected by qRT-PCR in ICC treated with or without 20 ng/mL of TNF-α. ICC was transfected with siRNA-MEG3 and treated with 20 ng/mL of TNF-α. b: Cell viability was detected by MTT method, c-d: Flow cytometry was performed to detect cell apoptosis rate (c) and ROS level (d). e: Oxidative stress reaction markers were detected by ELISA method with a microplate reader. f:Western blot was used to determine the expression of IL-1, IL-6, Bax, Bcl2 and cleaved caspase3.‘*’ means P < 0.05 compared with control group. ‘#’ means P < 0.05 compared with TNF-α group.
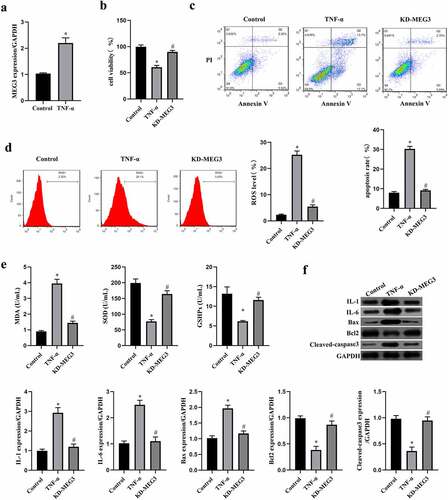
MiR-21 was a target of MEG3 and IKKB was directly targeted by miR-21
Previous evidences have demonstrated that miR-21 is a negative regulator of cell apoptosis and is a target of MEG3 [Citation13,Citation37]. To investigate the downstream regulatory mechanism of MEG3 in ICC, we examine the relationship between miR-21 and MEG3 through luciferase reporter gene assay first (). MiR-21 mimic decreased the luciferase activity in cells transfected with wild type of MEG3 sequence (P < 0.05), but not in cells transfected with mutant of MEG3 sequence (P > 0.05). Then, transfection with siRNA-MEG3 increased the expression of miR-21 in ICC, while pcDNA-MEG3 decreased miR-21 expression (P < 0.05, ). In ICC treated with TNF-α, we observed miR-21 expression was decreased compared with cells without TNF-α treatment, and MEG3 silence reversed the expression of miR-21 in TNF-α-treated ICC (P < 0.05, ). The data verified that miR-21 was a target of MEG3 in ICC.
Figure 3. MiR-21 was a target of MEG3 and IKKB was directly targeted by miR-21. a: Luciferase reporter gene assay was conducted to verify the targeting relationship of miR-21 and MEG3. ‘*’ means P < 0.05 compared with vector group. b: The regulatory effect of MEG3 on the expression of miR-21 was measured using western blot. ‘*’ means P < 0.05 compared with siRNA-NC group. ‘#’ means P < 0.05 compared with pcDNA-NC group. c: The expression of miR-21 in ICC transfected with siRNA-MEG3 and treated with TNF-α. ‘*’ means P < 0.05 compared with control group. ‘#’ means P < 0.05 compared with TNF-α group. d: The binding site between miR-21 and IKKB was predicted on the miRwalk website, and then luciferase reporter gene assay was conducted to verify the targeting relationship. ‘*’ means P < 0.05 compared with vector group. e: The regulatory effect of miR-21 on the expression of IKKB was measured using qRT-PCR and western blot. ‘*’ means P < 0.05 compared with NC group. f: The expression of IKKB mRNA and protein in ICC transfected with siRNA-MEG3 and treated with TNF-α. ‘*’ means P < 0.05 compared with control group. ‘#’ means P < 0.05 compared with TNF-α group.
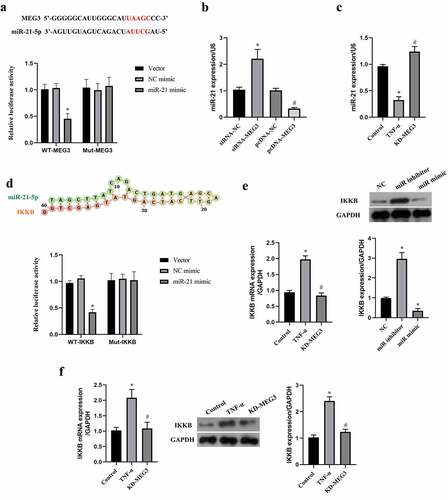
Subsequently, the downstream gene of miR-21 was predicted by miRwalk website, and data suggested that miR-21 binds to the 5’-UTR of IKKB (). Similarly, the targeting relationship between miR-21 and IKKB was confirmed by the luciferase reporter assay (). MiR-21 mimic decreased the expression of IKKB, while knockdown of miR-21 by miR-21 inhibitor elevated IKKB expression in ICC (P < 0.05, ). In addition, we observed that TNF-α increased IKKB expression, and which could be reversed by MEG3 silence (P < 0.05, ). The data suggested that IKKB was a target of miR-21 in ICC.
MEG3 activated NF-κB pathway by miR-21 in ICC
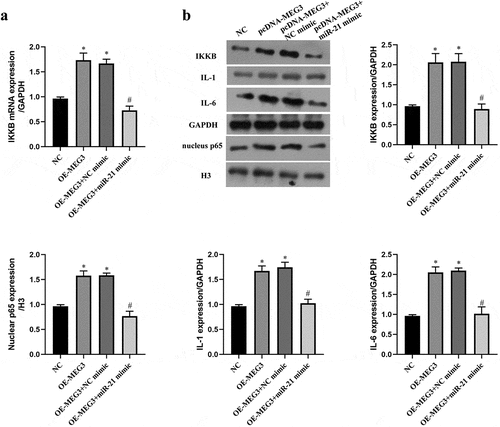
IKKB is a key upstream kinase of the NF-κB pathway, and the NF-κB pathway plays a critical role in oxidative stress and apoptosis [Citation29,Citation38]. To verify whether MEG3 regulated the NF-κB pathway by miR-21/IKKB axis, we upregulated the expression of MEG3 and miR-21 at the same time. The results indicated that upregulation of MEG3 elevated the expression of IKKB mRNA and protein, which was reversed by upregulation of miR-21 (P < 0.05, ). It reflected that MEG3 upregulated the expression of IKKB via sponging miR-21. With the raise of IKKB level by pcDNA-MEG3, the expression of nuclear NF-κB p65 and main effector of the NF-κB pathway were increased in ICC, which was also counteracted by miR-21 mimic (P < 0.05, ). These data evidenced that MEG3 activated NF-κB pathway by miR-21 in ICC.
Figure 4. MEG3 activated NF-κB pathway by miR-21 in ICC. ICC was transfected with pcDNA-MEG3 and miR-21 mimic. a: IKKB mRNA was detected by qRT-PCR. b:The expression of IKKB and NF-kB pathway related protein was detected by western blot. ‘*’ means P < 0.05 compared with NC group. ‘#’ means P < 0.05 compared with OE-MEG3 group.
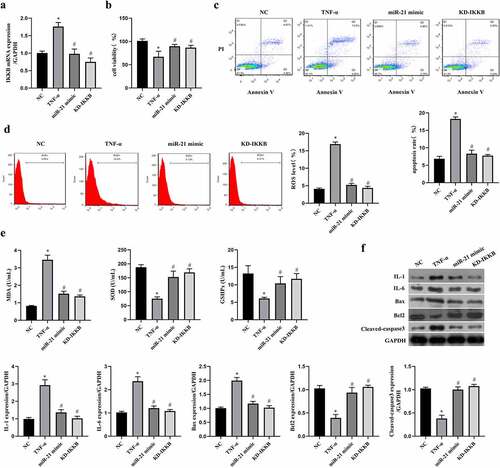
Regulation of the miR-21/IKKB axis reversed TNF-α-induced oxidative stress, inflammation and apoptosis in ICC
In order to ensure the role of the miR-21/IKKB axis in TNF-α-stimulated ICC, the function verification experiment was conducted by transfection with miR-21 mimic or siRNA-IKKB. By performing qRT-PCR to verify the transfection efficiency, we observed both miR-21 mimic and siRNA-IKKB decreased the expression of IKKB mRNA in TNF-α-treated ICC (P < 0.05, ). Both miR-21 mimic and siRNA-IKKB protected cell viability in TCC (P < 0.05, ), reduced cell apoptosis (P < 0.05, ), oxidative stress (P < 0.05, ) and inflammation (P < 0.05, ) induced by TNF-α. Collecting all data together, we can conclude that MEG3 mediated TNF-α-induced oxidative stress, inflammation, and apoptosis in ICC by the miR-21/IKKB axis.
Figure 5. MiR-21/IKKB axis reversed TNF-α-induced oxidative stress, inflammation and apoptosis in ICC. ICC was transfected with miR-21 mimic and siRNA-IKKB, and then treated with TNF-α. a: MEG3 expression was detected by qRT-PCR in ICC treated with or without 20 ng/mL of TNF-α. ICC was transfected with siRNA-MEG3 and treated with 20 ng/mL of TNF-α. b: Cell viability was detected by MTT method, c-d: Flow cytometry was performed to detect cell apoptosis rate (c) and ROS level (d). e: Oxidative stress reaction markers were detected by ELISA method with a microplate reader. f: Western blot was used to determine the expression of IL-1, IL-6, Bax, Bcl2 and cleaved caspase3. ‘*’ means P < 0.05 compared with control group. ‘#’ means P < 0.05 compared with TNF-α group.
Discussion
ICC is proved to regulate ureteropelvic peristalsis by maintaining pulse propagation [Citation2,Citation4,Citation7,Citation39]. The loss of ICC closely relates to the hypodynamic lesions (such as UPJO), hence drawing a lot of attention [Citation40–42]. It is of great significance for the management of related diseases to investigate the mechanism of ICC loss. Recently, MEG3 is reported to promote inflammation, apoptosis, and oxidative stress in various cells, while its known target miR-21 plays the opposite role [Citation22,Citation43–45]. However, their functions in ICC have not been revealed yet. Thus, we hypothesized MEG3/miR-21 axis involved in the apoptosis of ICC. Accordingly, our study used TNF-α stimulation to simulate the pressure environment of ICC, and a set of function verification experiments around the MEG3/miR-21 axis were carried out, which aimed to provide a potential target for the treatment of ICC dysfunction and loss-related diseases.
In the present study, we extracted ICCs from small intestine of mouse, as its abundance of ICC. Then, we used TNF-α to stimulate ICC to produce stress, and we found that TNF-α-induced apoptosis in ICC. Meanwhile, we also observed that ROS and MDA level were elevated, while SOD and GSHPx level were reduced by TNF-α. ROS and MDA directly reflect the level of cellular oxidative stress, but SOD and GSHPx content represent the cellular antioxidant capacity [Citation46]. More than that, we found the (IL-1 and IL-6) were upregulated by TNF-α in ICC [Citation47]. In most tissues, cells are vulnerable to oxidative stress and inflammation, and these two kinds of reaction mutually influence and mutually stimulation [Citation48]. Normally, the body can achieve anti-inflammatory and antioxidant effects through self-regulation, relieve moderate pressure, while excessive stress will cause apoptosis in cells.
MEG3 plays a tumor-suppressive role in many types of cancer, and in these cancer cells, overexpression of MEG3 can inhibit cell proliferation and induce apoptosis [Citation10,Citation11,Citation49]. Many previous studies have demonstrated that MEG3 participates in the inflammation progression and apoptosis in different types of non-cancer cell [Citation15,Citation50,Citation51]. A study by Xie B et al. suggested that MEG3 downregulation suppresses the inflammation cytokines and oxidative stress level [Citation17]. In agreement with these reporters, our study verified MEG3 was upregulated by TNF-α in ICC, and knockdown of MEG3 reduced inflammation, oxidative stress, and apoptosis in TNF-α-treated ICC. MEG3 has been reported to sponge multiple miRNAs to play its role in post-transcriptional regulation of some effector genes [Citation15,Citation50,Citation51]. Our study found that miR-21 could target of MEG3 in ICC. A study by Zhou W et al. proved that miR-21 could inhibit periodontitis by downregulating inflammation [Citation52]. A study by Qi H et al. reported miR-21 protects cardiomyocytes from ROS oxidative stress [Citation53]. Another study by Li H et al. indicated that the regulation of miR-21-5p suppresses hypoxia/reoxygenation injury-induced apoptosis of type II alveolar epithelial cells [Citation54]. All these evidences support the possibility of miR-21 becoming a downstream target of MEG3 in our study.
Subsequently, we demonstrated that miR-21 binds to the 5’-UTR of IKKB, and TNF-α elevated IKKB expression and the nuclear NF-κB p65 by the interaction between MEG3 and miR-21. IKKB is the upstream kinase of the NF-κB pathway [Citation29]. IKKB phosphorylates NF-κB inhibitor (IκBα) and promotes its degradation, thereby activating NF- κB p65 and promoting its translocation into the nucleus [Citation55]. The nuclear p65 acts as a transcription factor to activate transcription of related inflammatory genes, such as IL-1 and IL-6 [Citation56]. TNF-α is well known as one of the effective activators of NF-κB. The NF-κB pathway is usually identified as a major contributor of inflammation [Citation57]. Actually, in response to acute inflammation effectively, NF-κB also promotes mitochondrial activity and NADPH oxidase expression, which are the main source of endogenous ROS [Citation58,Citation59]. The inflammation induced by NF-κB activation advances oxidative stress in cells [Citation26,Citation28]. Furthermore, the final function verification experiments suggested the miR-21/IKKB axis was also involved in the regulation of cell apoptosis of ICC treated by TNF-α.
Besides the findings mentioned above, there are some limitations in our study. First, we only conducted the in vitro experiment of ICC, and whether the molecular regulatory mechanism is correct in vivo remains to be determined. Moreover, our present study focused on the viability of ICC, but the decrease in the number of ICC is only one of the hypomotility inducements. As ICC plays an important role in peristaltic and contractile function, its mechanism in related diseases is worthy of further exploration.
Conclusion
In summary, lncRNA MEG3 mediates TNF-α-induced inflammation, oxidative stress, and apoptosis in ICC via sponging miR-21 to activate the IKKB-NF-κB axis. This molecular signaling cascade might be a potential target for the intervention of ICC dysfunction and loss-related diseases.
Highlights
• LncRNA MEG3 is upregulated in ICC by TNF-α.
• MEG3 mediates inflammation, oxidative stress, and apoptosis in TNF-α-treated ICC.
• MEG3 elevates IKKB expression to activate NF-κB pathway via sponging miR-21 in ICC.
• Activation of NF-κB pathway leads to inflammation, oxidative stress, and apoptosis in ICC.
Data and materials availability
The data were all included in this paper. The original data are available from the corresponding author upon reasonable request.
Authors’ contributions
All authors contributed equally to the work from research design, experiment, data curation, paper writing to submission of papers.
Ethics approval and consent to participate
The experimental protocol was approved by the animal experiment ethics committee of The First Affiliated Hospital of Xinjiang Medical University, and all the operations are in compliance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and ARRIVE Guidelines.
Supplemental Material
Download Zip (84 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Huizinga JD, Hussain A, Chen JH. Interstitial cells of Cajal and human colon motility in health and disease[J]. Am J Physiol Gastrointest Liver Physiol. 2021;321(5):G552–g575.
- Drumm BT, Koh SD, Andersson K-E, et al. Calcium signalling in Cajal-like interstitial cells of the lower urinary tract[J]. Nat Rev Urol. 2014;11(10):555–564.
- Wishahi M, Mehena AA, Elganzoury H, et al. Telocyte and Cajal cell distribution in renal pelvis, ureteropelvic junction (UPJ), and proximal ureter in normal upper urinary tract and UPJ obstruction: reappraisal of the aetiology of UPJ obstruction[J]. Folia Morphol (Warsz). 2021;80(4):850–856.
- Ma Y, Chen Y, Zheng Y, et al. SCF/c-kit signaling pathway participates in ICC damage in neurogenic bladder[J]. Cell Cycle. 2020;19(16):2074–2080.
- Foong D, Zhou J, Zarrouk A, et al. Understanding the biology of human interstitial cells of cajal in gastrointestinal Motility[J]. Int J Mol Sci. 2020;21(12):4540.
- Huang ZP, Qiu H, Yu BP. Distribution changes of interstitial cells of Cajal during cholesterol gallstone formation in Guinea pigs fed a high cholesterol diet[J]. Int J Clin Exp Pathol. 2018;11(3):1653–1659.
- Bayrak O, Basmaci I, Bozdag Z, et al. Immunohistochemical evaluation of Cajal-like cells and Caveolin-1 levels at ureteropelvic junction obstruction[J]. Arch Esp Urol. 2019;72(7):690–696.
- Kaji N, Horiguchi K, Iino S, et al. Nitric oxide-induced oxidative stress impairs pacemaker function of murine interstitial cells of Cajal during inflammation[J]. Pharmacol Res. 2016;111:838–848.
- Gu L, Zhang J, Shi M, et al. lncRNA MEG3 had anti-cancer effects to suppress pancreatic cancer activity[J]. Biomed Pharmacother. 2017;89:1269–1276.
- Wei GH, Wang X. lncRNA MEG3 inhibit proliferation and metastasis of gastric cancer via p53 signaling pathway[J]. Eur Rev Med Pharmacol Sci. 2017;21(17):3850–3856.
- Wu M, Huang Y, Chen T, et al. LncRNA MEG3 inhibits the progression of prostate cancer by modulating miR-9-5p/QKI-5 axis[J]. J Cell Mol Med. 2019;23(1):29–38.
- Sun Y, Cao FL, Qu LL, et al. MEG3 promotes liver cancer by activating PI3K/AKT pathway through regulating AP1G1[J]. Eur Rev Med Pharmacol Sci. 2019;23(4):1459–1467.
- Zhu M, Wang X, Gu Y, et al. MEG3 overexpression inhibits the tumorigenesis of breast cancer by downregulating miR-21 through the PI3K/Akt pathway[J]. Arch Biochem Biophys. 2019;661:22–30.
- Meng J, Ding T, Chen Y, et al. LncRNA-Meg3 promotes Nlrp3-mediated microglial inflammation by targeting miR-7a-5p[J]. Int Immunopharmacol. 2021;90:107141.
- Song B, Ye L, Wu S, et al. Long non-coding RNA MEG3 regulates CSE-induced apoptosis and inflammation via regulating miR-218 in 16HBE cells[J]. Biochem Biophys Res Commun. 2020;521(2):368–374.
- Tong P, Peng QH, Gu LM, et al. LncRNA-MEG3 alleviates high glucose induced inflammation and apoptosis of retina epithelial cells via regulating miR-34a/SIRT1 axis[J]. Exp Mol Pathol. 2019;107:102–109.
- Xie B, Qiao M, Xuan J. lncRNA MEG3 Downregulation Relieves Intracerebral Hemorrhage by Inhibiting Oxidative Stress and Inflammation in an miR-181b-Dependent Manner[J]. Med Sci Monit. 2021;27:e929435.
- Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease[J]. RNA Biol. 2011;8(5):706–713.
- Nasci VL, Chuppa S, Griswold L, et al. miR-21-5p regulates mitochondrial respiration and lipid content in H9C2 cells[J]. Am J Physiol Heart Circ Physiol. 2019;316(3):H710–h721.
- Wei C, Li L, Kim IK, et al. NF-κB mediated miR-21 regulation in cardiomyocytes apoptosis under oxidative stress[J]. Free Radic Res. 2014;48(3):282–291.
- Yuan M, Yang X, Duscher D, et al. Overexpression of microRNA-21-5p prevents the oxidative stress-induced apoptosis of RSC96 cells by suppressing autophagy[J]. Life Sci. 2020;256:118022.
- Deng D, Liang H. Silencing MEG3 protects PC12 cells from hypoxic injury by targeting miR-21[J]. Artif Cells Nanomed Biotechnol. 2020;48(1):610–619.
- Wu L, Zhu L, Li Y, et al. LncRNA MEG3 promotes melanoma growth, metastasis and formation through modulating miR-21/E-cadherin axis[J]. Cancer Cell Int. 2020;20(1):12.
- Xu G, Meng L, Yuan D, et al. MEG3/miR‑21 axis affects cell mobility by suppressing epithelial‑mesenchymal transition in gastric cancer[J]. Oncol Rep. 2018;40(1):39–48.
- Dang X, He B, Ning Q, et al. Alantolactone suppresses inflammation, apoptosis and oxidative stress in cigarette smoke-induced human bronchial epithelial cells through activation of Nrf2/HO-1 and inhibition of the NF-κB pathways[J]. Respir Res. 2020;21(1):95.
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling[J]. Cell Res. 2011;21(1):103–115.
- Wardyn JD, Ponsford AH, Sanderson CM. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways[J]. Biochem Soc Trans. 2015;43(4):621–626.
- Zhu MM, Wang L, Yang D, et al. Wedelolactone alleviates doxorubicin-induced inflammation and oxidative stress damage of podocytes by IκK/IκB/NF-κB pathway[J]. Biomed Pharmacother. 2019;117:109088.
- Nathan S, Ma Y, Tomita YA, et al. BRCA1-mimetic compound NSC35446.HCl inhibits IKKB expression by reducing estrogen receptor-α occupancy in the IKKB promoter and inhibits NF-κB activity in antiestrogen-resistant human breast cancer cells[J]. Breast Cancer Res Treat. 2017;166(3):681–693.
- Ren K, Yong C, Yuan H, et al. TNF-α inhibits SCF, ghrelin, and substance P expressions through the NF-κB pathway activation in interstitial cells of Cajal[J]. Braz J Med Biol Res. 2018;51(6):e7065.
- Zhao K, Tan JY, Mao QD, et al. Overexpression of long non-coding RNA TUG1 alleviates TNF-α-induced inflammatory injury in interstitial cells of Cajal[J]. Eur Rev Med Pharmacol Sci. 2019;23(1):312–320.
- Jia HY, Zhang K, Lu WJ, et al. LncRNA MEG3 influences the proliferation and apoptosis of psoriasis epidermal cells by targeting miR-21/caspase-8[J]. BMC Mol Cell Biol. 2019;20(1):46.
- Guo J, Zhang N, Liu G, et al. Upregulated expression of long non-coding RNA MEG3 serves as a prognostic biomarker in severe pneumonia children and its regulatory mechanism[J]. Bioengineered. 2021;12(1):7120–7131.
- Wang J, Wu M. The up-regulation of miR-21 by gastrodin to promote the angiogenesis ability of human umbilical vein endothelial cells by activating the signaling pathway of PI3K/Akt[J]. Bioengineered. 2021;12(1):5402–5410.
- Kim T, Wahyudi LD, Gonzalez FJ, et al. Nuclear Receptor PPARα Agonist Wy-14,643 Ameliorates Hepatic Cell Death in Hepatic IKKβ-Deficient Mice[J]. Biomol Ther (Seoul). 2017;25(5):504–510.
- Li C, Zhao B, Lin C, et al. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-κB signaling[J]. Cell Biol Int. 2019;43(4):360–372.
- Zhang J, Yao T, Wang Y, et al. Long noncoding RNA MEG3 is downregulated in cervical cancer and affects cell proliferation and apoptosis by regulating miR-21[J]. Cancer Biol Ther. 2016;17(1):104–113.
- Tanaka H, Fujita N, Tsuruo T. 3-Phosphoinositide-dependent protein kinase-1-mediated IkappaB kinase beta (IkkB) phosphorylation activates NF-kappaB signaling[J]. J Biol Chem. 2005;280(49):40965–40973.
- Wolnicki M, Aleksandrovych V, Gil K. Interstitial cells of Cajal and telocytes in the urinary system: facts and distribution[J]. Folia Med Cracov. 2016;56(4):81–89.
- Koleda P, Apoznanski W, Wozniak Z, et al. Changes in interstitial cell of Cajal-like cells density in congenital ureteropelvic junction obstruction[J]. Int Urol Nephrol. 2012;44(1):7–12.
- Pande T, K DS, Chand K, et al. Influence of Interstitial Cells of Cajal in Congenital Ureteropelvic Junction Obstruction[J]. J Indian Assoc Pediatr Surg. 2020;25(4):231–235.
- Tokat E, Gurocak S, Akdemir O, et al. Relation between Cajal Cell Density and Radiological and Scintigraphic Outcomes in Patients with Ureteropelvic Junction Obstruction[J]. Urol Int. 2021;105(11–12):1046–1051.
- Pan X, Cao YM, Liu JH, et al. MEG3 Induces Cervical Carcinoma Cells’ Apoptosis Through Endoplasmic Reticulum Stress by miR-7-5p/STC1 Axis[J]. Cancer Biother Radiopharm. 2021;36(6):501–510.
- Shi Y. MEG3 regulates apoptosis of adipose‑derived stem cells[J]. Mol Med Rep. 2020;21(6):2435–2442.
- Xiang Y, Zhang Y, Xia Y, et al. LncRNA MEG3 targeting miR-424-5p via MAPK signaling pathway mediates neuronal apoptosis in ischemic stroke[J]. Aging (Albany NY). 2020;12(4):3156–3174.
- Chen Y, Zhao C, Liu X, et al. Plumbagin ameliorates liver fibrosis via a ROS-mediated NF-кB signaling pathway in vitro and in vivo[J]. Biomed Pharmacother. 2019;116:108923.
- Chen T, Zhang X, Zhu G, et al. Quercetin inhibits TNF-α induced HUVECs apoptosis and inflammation via downregulating NF-kB and AP-1 signaling pathway in vitro[J]. Medicine (Baltimore). 2020;99(38):e22241.
- Reuter S, Gupta SC, Chaturvedi MM, et al. Oxidative stress, inflammation, and cancer: how are they linked?[J]. Free Radic Biol Med. 2010;49(11):1603–1616.
- Chen L, Wang J, W LJ, et al. LncRNA MEG3 inhibits proliferation and promotes apoptosis of osteosarcoma cells through regulating Notch signaling pathway[J]. Eur Rev Med Pharmacol Sci. 2020;24(2):581–590.
- Liang J, Wang Q, Q LJ, et al. Long non-coding RNA MEG3 promotes cerebral ischemia-reperfusion injury through increasing pyroptosis by targeting miR-485/AIM2 axis[J]. Exp Neurol. 2020;325:113139.
- Yan L, Liu Z, Yin H, et al. Silencing of MEG3 inhibited ox-LDL-induced inflammation and apoptosis in macrophages via modulation of the MEG3/miR-204/CDKN2A regulatory axis[J]. Cell Biol Int. 2019;43(4):409–420.
- Zhou W, Su L, Duan X, et al. MicroRNA-21 down-regulates inflammation and inhibits periodontitis[J]. Mol Immunol. 2018;101:608–614.
- Qi H, Zhang J, Shang Y, et al. Argon inhibits reactive oxygen species oxidative stress via the miR-21-mediated PDCD4/PTEN pathway to prevent myocardial ischemia/reperfusion injury[J]. Bioengineered. 2021;12(1):5529–5539.
- Li H, Yao C, Shi K, et al. Astragaloside IV attenuates hypoxia/reoxygenation injury-induced apoptosis of type II alveolar epithelial cells through miR-21-5p[J]. Bioengineered. 2021;12(1):7747–7754.
- McNamara AJ, Danthi P. Loss of IKK Subunits Limits NF-κB Signaling in Reovirus-Infected Cells[J]. J Virol. 2020;94(10). DOI:10.1128/JVI.00382-20
- Min Y, J KM, Lee S, et al. Inhibition of TRAF6 ubiquitin-ligase activity by PRDX1 leads to inhibition of NFKB activation and autophagy activation[J]. Autophagy. 2018;14(8):1347–1358.
- Chen M, Chen Z, Huang D, et al. Myricetin inhibits TNF-α-induced inflammation in A549 cells via the SIRT1/NF-κB pathway[J]. Pulm Pharmacol Ther. 2020;65:102000.
- Park J, S MJ, Kim B, et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways[J]. Neurosci Lett. 2015;584:191–196.
- Xiao L, Zhong M, Huang Y, et al. Puerarin alleviates osteoporosis in the ovariectomy-induced mice by suppressing osteoclastogenesis via inhibition of TRAF6/ROS-dependent MAPK/NF-κB signaling pathways[J]. Aging (Albany NY). 2020;12(21):21706–21729.