
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
To investigate the effects of silencing neuropilin-2(NRP-2) on the proliferation, migration, and invasion of colorectal cancer(CRC) HT-29. Lipofectamine 2000 was used to transfect specific siRNA for NRP-2 and nonspecific control siRNA into human colorectal cancer HT-29 as the transfection group and meaningless sequence group. HT-29 cultured in a medium was used as the blank control group. The expression levels of NRP-2 mRNA in the cells were detected by real-time fluorescence quantitative PCR. The expressions of proliferation-associated protein Ki-67 in the cells were detected by immunochemical staining. Migration ability was assessed by a monolayer cell scratch wound damage and repair experiment. The Transwell chamber invasion experiment was adopted to determine invasive ability by measuring the number of tumor cells crossing the chamber membrane. Compared with the meaningless sequence group and blank control group, real-time fluorescence quantitative PCR showed that the relative expression level of NRP-2 mRNA in the transfection group was significantly decreased(P < 0.05). Results of immunochemical staining revealed that the expression of Ki-67 protein in the transfected cells was significantly reduced, and the proliferation ability was decreased(P < 0.05). The results further showed that the scratch healing rate of the transfected cells decreased after 24 h of healing(P < 0.05). Results of Transwell invasion assay showed that the number of cells passing through the stromal membrane of the upper chamber to the back of the chamber was significantly reduced in the transfection group(p < 0.05). Silencing NRP-2 could inhibit the proliferation, migration, and invasion of colorectal cancer HT-29.
Graphical abstract
Highlights
Silencing neuropilin-2 can inhibit the proliferation, migration and invasion ofHT-29 cells.
NRP-2 plays an important role in the biological behavior of colon cancer, and can
provide a new breakthrough in the molecular targeted therapy of colon cancer in the future.
This study may provide a theoretical basis for NRP-2 to become a new research direction of targeted therapy for colon cancer.
1. Introduction
Colorectal cancer (CRC) is the third most common malignant tumor in the world, ranking third and second in the incidence of malignant tumors in males and females in China, respectively [Citation1]. In 2015, the number of new-onset malignant tumors in China was 4.292 million, and the number of deaths caused by malignant tumors was 2.814 million [Citation2]. In recent years, there exists an incremental increase in the number of deaths caused by CRC [Citation3]. With the rapid development of tumor molecular biology and further research on gene expression, a better understanding of the occurrence and development of CRC has been achieved. It has been established that the occurrence of CRC is not caused by a single gene change, but by a process involving multiple events. Through multiple gene changes, the normal colonic mucosa gradually develops into atypical hyperplasia, adenoma, carcinoma in situ, and advanced cancer [Citation4].
NRP-2 belongs to one subtype of the Neuropilin (NRP) family, and the other subtype is Neuropilin-1 (NRP-1). NRP-2 has the same domain as NRP-1 and approximately 44% of homologous amino acids overlapping with NRP-1 [Citation5]. NRP-2 may be expressed in lower amounts in normal human cells. A number of studies have proven that NRP-2 exists and is highly expressed in a variety of solid cell tumors such as breast cancer [Citation6], thyroid cancer [Citation7], oral squamous cell carcinoma [Citation8], and gastric cancer [Citation9]. In our previous studies, it was found that NRP-1 had a role in promoting malignant biological behaviors in humans [Citation10]. However, there are few studies on NRP-2 in CRC. In this study, NRP-2 was used in the cellular study of CRC cells to investigate the changes in the proliferation, migration, and invasion of CRC HT-29 cells.
In this study, we compared the expression of NRP-2 in HT-29 silenced by RNAi with normal HT-29 at the level of colon cancer cells in order to observe the biological behavior changes such as apoptosis, proliferation, migration and invasion of cells after silencing of messenger RNA (mRNA) of NRP-2 in HT-29 cells. Real-time polymerase chain reaction was used to detect the expression level of NRP-2 mRNA in cells of different groups to verify the expression of NRP-2 in cells; immunocytochemical staining was used to detect the expression of Ki-67 protein in cells of different groups; monolayer scratch injury repair assay was used to detect the scratch healing area of cells of different groups to detect the migration ability of cells; transwell chamber invasion assay was used to determine the invasion ability of cells by detecting the number of tumor cells crossing the chamber membrane. By these results, we aim to provide some theoretical basis for NRP-2 that may become a new research direction for the treatment of targeted therapy for colon cancer and its future combination with NRP-2 small interfering RNA (siRNA) on the basis of chemotherapy alone, which may improve the prognosis of patients with NRP-2 overexpression.
2. Materials and methods
2.1 General materials and main reagents
Human CRC cell line HT-29 was purchased from Shanghai Cell Bank, Chinese Academy of Sciences. The siRNA sequence of NRP-2 was synthesized by Guangzhou Ruibo Biotechnology Co., Ltd. The forward sequence was 5'-CAGAGAUCGCGAGCGAGGCA dTdT-3' and the backward sequence was 3'-dTdT GUCUCUAGCGCUCGCUCCGU- 5'. Lipofectamine 2000 was purchased from Invitrogen Co. NRP-2 PCR primers and internal reference β-actin were synthesized by Invitrogen Co. The forward sequence of NRP-2 primers was 5'- CGGCTTTTGCAGTGGACATC −3', and the backward sequence was 5'-TTTCTTTGTCGGTCGAGGGG −3'. The forward sequence of β-actin primers was 5'-GTCACCTTCACCGTTCCAGTTTT-3', and the backward sequence was 5'- CTTAGTTGCGTTACACCCTTTCTT-3'. Kits for PCR reverse transcription and amplification were purchased from Invitrogen Co. McCoy’s 5A medium and fetal bovine serum (FBS) were purchased from BI Co. Mouse anti-Human Ki-67 monoclonal antibody was purchased from Abnova Co. Immunochemical staining kit was purchased from Zhongshan Jinqiao Co. Matrigel glue and Transwell chamber were purchased from Corning Co.
2.2 Methods
2.2.1 Cell culture
HT-29 cells were cultured and subcultured according to the norms: Complete McCoy’s 5A medium with a mixture of penicillomycin and 10% FBS was adopted for cell culture in a standard incubator for routine cell passage procedures.
2.2.2 Fluorescent siRNA transfection
Fluorescent siRNA was transfected into the HT-29 cells: The cells were digested and planted in 6-well plates for cell culture. After the cells in each plate grew to an appropriate density, the medium in the cell plate was discarded before transfection and 1 ml of McCoy’s 5A blank medium was added to starve the cells. Lipofectamine 2000 and fluorescent siRNA, at 5 ul each, were mixed with 500 ul of McCoy’s 5A blank medium, respectively, and placed in a 1 ml EP tube to stand still for 5 min at room temperature. After mixing the above two mediums and standing at room temperature for 20 min, the mixture was added to the cells starved for 1 h. After incubation at 37 and 5% CO2 for 6 h, the liquid in the plate was discarded and replaced with fresh complete medium. Fluorescence microscopy was used to observe the fluorescence transfection rate of HT-29 cells after 48 h of standard culture. The condition for successful transfection was set at a transfection rate of above 60%.
2.2.3 Cell grouping
Transfection of siRNA into HT-29 cell lines and cell grouping: As mentioned above, the cells were digested according to the routine steps of cell digestion. The cells were planted in 6-well plates and transfected at the logarithmic growth stage. The medium in the cell plate was discarded, and 1 ml of McCoy’s 5A blank medium was added and the cells were starved for 1 h in the cell incubator. Lipofectamine 2000 and NRP-2-siRNA, at 5 ul each, were added into 500 ul of McCoy’s 5A blank medium, respectively, and incubated for 5 min. The two mediums were then mixed and incubated for 20 min. The mixture was added to previously starved cells, blended evenly, and labeled as the transfection group. Based on the above procedures, the control siRNA was transfected into cells to set a negative control group. The cells cultured in medium only were labeled as the blank control group.
2.2.4 Real-time PCR
Determination of the NRP-2 mRNA expression in HT-29 cells by real-time PCR: HT-29 cells were cultured to an appropriate planting density and then grouped. The Trizol method was used to extract the total RNA of the cells 48 h after grouping. The concentration and purity of total mRNA were measured using Quawell 5000 trace ultraviolet-visible spectrophotometer. According to the instructions in the M-MLV reverse transcriptase kit, the volume of 1 μg mRNA was obtained, the reagents were successively added into the enzyme-free trace EP tube, and reverse transcription of the extracted mRNA was conducted. The cDNA (2 ul) obtained by reverse transcription was mixed with the forward and backward primers of NRP-2 (each 1 ul), the SYBR Green (10 ul) and DEPC water (6 ul) in eight parallel tubes and then placed on the Rotor-Gene 3000 fluorescence quantitative PCR for amplification. The 2 – ΔΔCt method was used to measure the NRP-2 mRNA levels in cells.
2.2.5 Immunochemical staining
Detection of the Ki-67 protein expression in HT-29 cells by immunochemical staining: The cells were cultured to a suitable density and grouped. The cells were planted in a 24-well cell culture plate with a density of 1 × 105 cells/ml. When the cells were firmly adhered to the wall and grew to an appropriate density, the cells were fixed with 4% formaldehyde at room temperature. The cells were incubated with 0.25% TritonX-100 at room temperature, washed with phosphate buffered saline (PBS), and then incubated with endogenous peroxidase blockers. Goat serum working solution was used for sealing, and an appropriate dose of the primary antibody was added. The mixture was left to stand still overnight at 4°C. According to the instructions, an appropriate amount of biotin-labeled goat anti-mouse IgG polymer and horseradish enzyme-labeled streptavidin working solution were successively added, followed by DAB staining after washing with PBS. Hematoxylin staining was conducted together with differentiation by hydrochloric acid ethanol. Cell sections were prepared and observed under a microscope.
2.2.6 Monolayer cell scratch wound damage and repair experiment
Detection of cell migration ability by monolayer cell scratch wound damage and repair experiment: A single cell suspension was prepared based on the groups in the experiment. According to the groups, 1 × 105 cells were inoculated in each well on the 6-well plates and cultured for 12 h to form monolayer cells. A horizontal trace was made on the monolayer cells with the tip of a 200 µl pipette and washed with PBS three times. McCoy’s 5A medium containing 10% fetal bovine serum (FBS) was added and cultured at 37°C with 5% CO2 for 24 h. The culture medium was discarded by aspiration and washed with PBS three times. The cells were observed under an inverted microscope and photographed.
2.2.7 Transwell
Detection of the invasive ability of tumor cells by Transwell assay: Before the experiment, 20 ul of Matrigel glue was prepared and spread on the upper layer of the upper chamber, which was placed in the cell incubator overnight for solidification of the glue. The cells were digested, transfected, and grouped. Then, 48 h after transfection, the cells were inoculated into the upper chamber with a density of 1 × 105 cells/ml and cultured with the McCoy’s 5A blank medium. A volume of 600 ul of 10% FBS complete medium was added to the lower chamber. After culture for 18 h in the incubator, the chamber was taken out and gently washed with PBS 2–3 times and fixed with 4% formaldehyde for at least 30 min. After drying the chamber, crystal violet was added to the well plate for staining for 15–20 min, and the cells in the upper chamber were gently wiped with cotton swabs. Cell sections were prepared and observed under a microscope.
2.3 Statistical analysis
All data were processed by SPSS 20.0 software. The results were expressed as means ± standard deviations ( ±s). One-way analysis of variance (ANOVA) was adopted to analyze the results of scratch wound and cell invasion experiments. The results of MTT were analyzed by repeated measurement data ANOVA. α = 0.05 was set as the significant test level.
3 Results
Our results showed that the relative expression level of NRP-2 mRNA in the transfection group was significantly decreased (P < 0.05). Results of immunochemical staining revealed that the expression of Ki-67 protein in the transfected cells was significantly reduced, and the proliferation ability was decreased (P < 0.05). The results further showed that the scratch healing rate of the transfected cells decreased after 24 h of healing (P < 0.05). Results of Transwell invasion assay showed that the number of cells passing through the stromal membrane of the upper chamber to the back of the chamber was significantly reduced in the transfection group (p < 0.05). These results provided theoretical basis for NRP-2 that may become a new research direction for the treatment of targeted therapy for colon cancer and its future combination with NRP-2 small interfering RNA (siRNA) on the basis of chemotherapy alone.
3.1 Detection of the transfection efficiency
Lipofectamine 2000 was used as a carrier to transfer the NRP-2 fluorescent siRNA into the CRC HT-29 cell line. Fluorescent siRNA was red when excited by a green light (A). The percentage of red cells in total cell number (B) under transmission microscope was the transfection efficiency of cells. The transfection results were observed by fluorescence microscope 48 h after transfection. The transfection efficiency was more than 80% in the present study (). It was suggested that Lipofectamine 2000 successfully transfected the NRP-2-siRNA into the CRC cell line, which proved the feasibility of the following experiments.
Figure 1. Fluorescent siRNA under the fluorescence microscope after 48 h transfection (×100). a: Fluorescence detection; b: Transmittance tests; c: Composite image.

3.2 Determination of the NRP-2 mRNA content in HT-29 by real-time quantitative PCR
The relative mRNA expression of β-actin was set to 1. The mRNA content of NRP-2 in the blank control group was 1.00 ± 0.00. The NRP-2 mRNA content was 1.01 ± 0.02 in the meaningless sequence group and 0.36 ± 0.02 in the transfection group. The results were statistically significant when compared among the three groups (F = 2999.66, P < 0.001). The results showed that the differences in the mRNA content of NRP-2 were statistically significant between the transfection group and the blank control group and between the transfection group and the meaningless sequence group (P < 0.001) ().
Table 1. The relative mRNA expressions in each group by qPCR (n = 9, )
Figure 2. The expression of Ki-67 protein in HT-29 cells by immunochemical staining (×100). a: The blank control group; b: The meaningless sequence group; c: The transfection group.
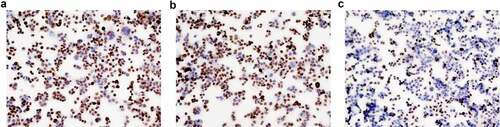
3.3 The detection of Ki-67 expression in cells by immunochemical staining ()
The staining index score in the blank control group was 8.60 ± 0.55. The staining index score in the meaningless sequence group was 8.40 ± 0.55. The staining index score in the transfection group was 3.40 ± 0.89. The differences in the staining index score were statistically significant among the three groups (F = 93.00, P < 0.001) ().
Table 2. The staining index score of Ki-67 protein in each group (n = 10, )
3.4 The detection of cell migration ability by monolayer cell scratch wound damage and repair experiment
After 24 hours of scratch repair, the scratch healing rate was 0.17 ± 0.02, 0.18 ± 0.02, and 0.11 ± 0.01 in the blank control group, the meaningless sequence group, and the transfection group, respectively. The differences in the scratch healing rate were statistically significant among the groups (F = 33.34, P < 0.001) (, ). Thus, it could be suggested that the scratch repair speed in the transfection group was significantly slower than that in the blank control group and the meaningless sequence group (P < 0.001).
Figure 3. Scratch area in each group at 0 h and 24 h (×40). a: The blank control group at 0 h; b: The meaningless sequence group at 0 h; c: The transfection group at 0 h; d: The blank control group at 24 h; e: The meaningless sequence group at 24 h; f: The transfection group at 24 h.

Table 3. Scratch healing rate in each group (n = 5, )
3.5 Detection of invasiveness of tumor cells in vitro
Chamber invasion experiments showed that the mean number of cells from the upper compartment crossing the stromal membrane to the back of the upper compartment were 48.20 ± 1.92, 45.80 ± 3.56, and 21.20 ± 4.15 cells/field of vision in the blank control group, meaningless sequence group, and transfection group, respectively (). Comparison between the blank control group and meaningless sequence group showed the difference was not statistically significant (P = 0.28), while the differences were statistically significant when compared between the blank control group and the transfection group, as well as when compared between the meaningless sequence group and transfection group (P < 0.001) ().
Figure 4. The number of cells penetrating into the back of the upper chamber in each group (×100). A: The blank control group; B: The meaningless sequence group; C: The transfection group.
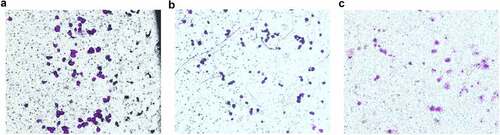
Table 4. Ber of cells penetrating into the back of the upper chamber in each group (n = 30)
4. Discussion
A recent study indicates that by 2030, the number of new cases of CRC in the world will be approximately 2.2 million and the number of deaths will be approximately 1.1 million, with an increase of 60% from the current level. The study also points out that the pattern and trend of the morbidity and mortality in CRC are correlated with the current level of human development, and the increase is closely correlated with an increasingly westernized lifestyle [Citation11]. The conventional treatments for CRC include surgery, chemotherapy, and radiotherapy. Molecular targeted therapy has become another therapeutic method, after conventional therapy.
One of the main causes of death in patients with cancer is tumor metastasis [Citation12]. Tumor cells break through the vascular walls or the walls of lymphatic vessels from the primary site into the systemic circulation, and then migrate to other organs for continued growth. The main reason for failure of tumor treatment is closely correlated with tumor metastasis. Recent studies have suggested that metastasis of tumor cells may be correlated with NRPs. NRPs are now recognized as important intermediaries for other physiological and pathophysiological processes. After binding with the NRP-1 or NRP-2 receptors on endothelial cells, the vascular endothelial growth factor (VEGF) family is involved in the formation of new vessels in both malignant and nonmalignant conditions. NRPs play a key role in the growth of vascular endothelial cells. NRP-1 not only acts as a receptor for the VEGF-A subtype in vascular endothelial cells, but also acts as a signal receptor in lymphatic endothelial cells. NRP-2 can promote lymphatic vessel growth under the induction of VEGF-C [Citation13]. Relevant studies have shown that NRP-2 is highly expressed in solid tumor tissues and plays a role in tumor progression and metastasis [Citation14,Citation15].
Based on previous national and international research results, it was hypothesized that the expression of silenced NRP-2 could affect biological behavior changes such as proliferation, migration, and invasion of tumor cells. In the present study, NRP- 2- siRNA was transfected into the human CRC HT-29 cell line by Lipofectamine 2000, and a series of experiments on tumor cell proliferation, migration, and invasion were conducted. In the present study, the NRP-2 mRNA expressions in the CRC cells in the three groups were detected by real-time quantitative PCR. The results showed that the expression of NRP-2 in the transfection group was significantly suppressed after the silencing of NRP-2, which also indicated that NRP-2 was highly expressed in CRC cells. The expression of Ki-67 protein in the transfection group was significantly reduced compared with that in the meaningless sequence group and the control group, which proved that silencing NRP-2 could inhibit the proliferation of CRC cells. It was found through a monolayer cell scratch wound damage and repair experiment that 24 h after scratch formation, the area of cell repair in the transfection group was much smaller than that in the meaningless sequence group and the blank control group. These results indicated that the transfection of silenced NRP-2 into the CRC cells could significantly reduce the migration ability of cancer cells. The results of Transwell chamber invasion experiments showed that the number of CRC cells passing through the stromal membrane to the back of the chamber after transfection of NRP-2-siRNA was less than that in the other two groups. These results indicated that NRP-2-siRNA could significantly reduce the invasion ability of the transfected CRC cells.
5. Conclusion
In summary, silencing NRP-2 could inhibit the proliferation of CRC HT-29 and reduce the invasion ability and migration ability of tumor cells. The above experimental results indicated that NRP-2 might play a key role in the biological behavior of CRC. It might provide a new breakthrough point for molecular targeted therapy for CRC in the future. It also provided a theoretical basis for the application of NRP-2-siRNA-related drugs combined with systemic therapy in clinical practice for CRC with a high expression of NRP-2.
Acknowledgements
We are particularly grateful to all the people who have given us help on our article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Torre LA, Siegel RL, Ward EM, et al. Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol Biomarkers Prev. 2016;25(1):16–27.
- Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132.
- Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):177–193.
- Audet-Walsh É, Giguére V. The multiple universes of estrogen-related receptor α and γ in metabolic control and related diseases. Acta Pharmacol Sin. 2015;36(1):51–61.
- Fujisawa H. Discovery of semaphorin receptors, neuropilin and plexin, and their functions in neural development. J Neurobiol. 2004;59(1):24–33.
- Yasuoka H, Kodama R, Tsujimoto M, et al. Neuropilin-2 expression in breast cancer: correlation with lymph node metastasis, poor prognosis, and regulation of CXCR4 expression. BMC Cancer. 2009;9(1):220.
- Tu DG, Chang WW, Jan MS, et al. Promotion of metastasis of thyroid cancer cells via NRP-2-mediated induction. Oncol Lett. 2016;12(5):4224–4230.
- Zhang B, Gao Z, Sun M, et al. Prognostic significance of VEGF-C, semaphorin 3F, and neuropilin-2 expression in oral squamous cell carcinomas and their relationship with lymphangiogenesis. J Surg Oncol. 2015;111(4):382–388.
- Nie N, Wu XL, Yu HH, et al. Expressions and clinical significance of Neuropilin-1 and Neuropilin-2 in human gastric cancer. Chin J Gastroenterol Hepatol. 2017;26(12):1340–1344. Articlein Chinese. https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFDLAST2018&filename=WCBX201712006&uniplatform=NZKPT&v=Yd7E9B7EzfbTNVYIC9tujWzrsmEjvucUEFVdAuk-qZ0LhfTTs-cEb5nNR0DBnzdg
- Xu WG, Yang XQ, He SZ, et al. The expression of neuropilin-1 and its correlation with angiogenesis in colorectal cancer. Chin Clin Oncol. 2009;14(1):29–33. [Articlein Chinese]. https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFD2009&filename=LCZL200901008&uniplatform=NZKPT&v=iSYu6zbM4u2TLyhnua45BJcH_UHPU1AiVaHItzmXibVivnyvI3uknv71luyBtvb3
- Arnold M, Sierra MS, Laversanne M, et al. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683–691.
- Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–458.
- West DC, Rees CG, Duchesne L, et al. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J Biol Chem. 2005;280(14):13457–13464.
- Yasuoka H, Kodama R, Hirokawa M, et al. Neuropilin-2 expression in papillary thyroid carcinoma: correlation with VEGF-D expression, lymph node metastasis, and VEGF-D-induced aggressive cancer cell phenotype. J Clin Endocrinol Metab. 2011;96(11):E1857–E1861.
- Rushing EC, Stine MJ, Hahn SJ, et al. Neuropilin-2: a novel biomarker for malignant melanoma? Hum Pathol. 2012;43(3):381–389.