
ABSTRACT
We reveal that in a prototypical bcc high-entropy alloy NbMoTaW chemical interactions are long ranged and highly frustrated. We show that this is the reason that bcc solid solutions in NbMoTaW can persist to low temperatures. The ab initio-computed long-ranged interactions strongly impact characteristic thermodynamic properties and ordering temperatures. This highlights the genuine importance of taking long-ranged chemical interactions into account for accurate theoretical predictions of high-entropy alloy properties.
GRAPHICAL ABSTRACT
IMPACT STATEMENT
Long-ranged chemical interactions critically impact the thermodynamics and ordering temperature of NbMoTaW and are responsible that this HEA retains the bcc solid solution up to low temperatures.
High entropy alloys (HEAs) are of great interest due to their excellent mechanical,[Citation1–4] magnetic[Citation5–7] and electronic properties.[Citation8,Citation9] Refractory HEAs, such as bcc NbMoTaW, possess extraordinary mechanical properties, comparable to current state-of-the-art nickel-based superalloys, [Citation4,Citation10–12] making them promising candidates for the next generation of high-temperature applications.
Despite their excellent materials properties, little is known about fundamental physical properties, for example, their ground states or the degree of chemical ordering at elevated temperatures. A fundamental conceptual aspect of HEAs is the presumption of a high degree of configurational disorder. Indeed, for NbMoTaW experimental studies reveal no indication of chemical ordering at room temperature as well as in the annealed state.[Citation11] However, due to slow diffusivity, characteristic for refractory elements, chemical ordering is hardly ever approached under typical experimental conditions but can significantly influence creep properties in practical (long-term) applications. At the same time complementary theoretical studies addressing chemical ordering in these alloys are so far limited due to the challenging nature inherent to the simulation of multi-component alloys.[Citation13–18] In this paper we show that long-ranged chemical interactions, which have so far not been taken into account,[Citation15–17] have an unexpected and dramatic effect on the degree of chemical ordering, causing chemical frustration and implying significant consequences for the ordering temperature as well as thermodynamic properties of HEAs.
NbMoTaW HEA feature very small size mismatch, none of the constituents deviates more than 2.5% in lattice parameter from the average value.[Citation19] Relaxation effects can be presumed to be small, so that treatments taking the disordered bcc solid solution as reference state are a natural choice for studying the tendencies toward chemical order. Such a method is provided by the generalized perturbation method (GPM).[Citation20] Alternately, a cluster expansion (CE) can be attempted. However, as a detailed study of the involved binary alloys indicates, such an expansion is already quite complex when there are just two constituents,[Citation21] let alone when there are four, as is the case here. The efficiency of a GPM-based expansion can be intuited by considering that it pertains to one specific composition while a multi-component CE must describe the whole composition range in a quaternary alloy. Furthermore, a GPM expansion typically features strongly dominant effective pair interactions (EPIs) over effective multi-site interactions. The EPIs can be used to simulate configurational order–disorder processes through real-space Monte Carlo simulations. We will show below that long-ranged EPIs are of crucial importance in multi-component alloys. The long-ranged EPIs cause chemical frustration and affect the characteristics of the order–disorder transition.
To capture the chemical degree of freedom, the configurational ordering energy of the quaternary is mapped onto an Ising Hamiltonian employing EPIs as
(1)
where the second sum runs over all pairs, p, and
are the EPIs between distinct atomic species μ and ν. The first sum runs (in the ‘host’ picture) over the N−1 species μ, ν and
,
denote the concentration variables at lattice sites n and m. The N−1 independent concentration fluctuations
are given as
with concentration
(i.e. in the equi-atomic alloy
≡ 0.25). The
are site-occupation variables which equal 1 (0) if the atom at site i is (is not) occupied by species
. Higher order terms in Equation (1) are found to be small and are neglected.
The EPIs have been computed using a density functional theory method employing the exact muffin-tin orbitals (EMTOs) [Citation22] in the Lyngby version of the code.[Citation23] The Brillouin zone integration has been performed employing a 34 × 34 × 34 k-point mesh according to the Monkhorst-Pack scheme.[Citation24] The lattice constant a0 has been chosen as 3.235 Å being between the reported room temperature experimental and theoretically derived T = 0 K value. Chemical disorder is simulated in the single-site coherent potential approximation.[Citation25–27] Screened Coulomb interactions have been taken into account by means of the screened generalized perturbation method.[Citation28,Citation29] Screening parameters are computed employing a large supercell containing 1024 atoms for the 4-component random alloy (with the first two Warren–Cowley short-range order parameters equal zero and the next 4 being less than 0.007 in absolute value for the 6 unique pairs in the considered alloy) using the locally self-consistent Greens function (LSGF) method [Citation30,Citation31] within the EMTO technique, the EMTO-LSGF (ELSGF).[Citation32] Other technical details are as in [Citation33].
In we show the chemical interactions along three distinct crystallographic directions that is, [001], [011] and [111] for three selected atomic pairs of the HEA. We observe long-ranged and oscillating chemical interactions, particular in [111]-direction.
Figure 1. Pair interactions in the HEA NbMoTaW in [001] (circles), [011] (triangles) and [111] (squares) direction for (a) Mo-Ta, (b) Mo-Nb and (c) Ta-W. Long-ranged chemical interactions in [111] are highlighted in subfigure (a). First nearest-neighbor interactions are not shown to facilitate visualization.
![Figure 1. Pair interactions in the HEA NbMoTaW in [001] (circles), [011] (triangles) and [111] (squares) direction for (a) Mo-Ta, (b) Mo-Nb and (c) Ta-W. Long-ranged chemical interactions in [111] are highlighted in subfigure (a). First nearest-neighbor interactions are not shown to facilitate visualization.](/cms/asset/f136ab86-5e95-4abf-866e-9953ef725c0c/tmrl_a_1198837_f0001_c.jpg)
Long-ranged chemical interactions are, for instance, also observed in fcc CuPd alloys [Citation34] which can be traced back to Fermi surface nesting effects. Although this behavior in the present HEA might also originate from Fermi surface nesting effects, which are known to be present in the pure refractory elements,[Citation35] in a disordered multi-component alloy lifetime effects would weaken such surface nesting characteristics. More intuitive is a tight-binding argument connected to the moments of the Hamiltonian, which favors linear graphs along the strongest hopping integrals [Citation20] in bcc along [111].
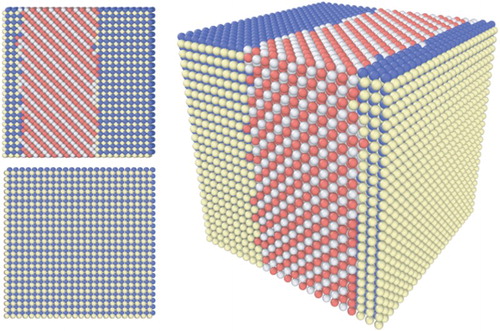
Based on the computed pair interactions, the ground state of the NbMoTaW HEA, being unknown so far, has been predicted. We first employ the MC method to equilibrate the system at a low temperature of 10 K. A snapshot of the equilibrated simulation cell is presented in . We find that the HEA separates at low temperatures into B2(Mo;Ta) and B32(Nb;W) as illustrated in . This is consistent with previous binary considerations.[Citation21] In order to verify our findings obtained by the MC simulations we performed in addition an extensive ground state search over more than a quarter of a million structures by considering all possible ordered structures containing 12 atoms per primitive unit cell and translation vectors
2 a0. No other ordered structure has been found being lower in energy compared to the phase separated B2(Mo;Ta)/B32(Nb;W) alloy.
Figure 2. The ground state of the HEA NbMoTaW consists of B2(Mo;Ta) (blue and yellow) and B32(Nb;W) (red and silver). The simulation box contained 27,648 atoms.
We now turn to the prediction of finite-temperature properties. Our key results are shown in . The MC simulations have been performed for a box containing 5488 atoms with periodic boundary conditions from 2000 K down to 10 K, including 1, 2, 5 and 99 EPIs where the latter can be regarded as the scenario for a well-converged data set with respect to included EPIs. The configurational contribution to the specific heat, evaluated via the energy fluctuations, is presented in the first row in (a)–(d). Two phase transitions are identified for all considered cases. Since B32 ordering requires at least second nearest-neighbor pair interactions, a B2(Mo;Ta) and B2(Nb;W) ground state is observed for the special case of , whereas for
, a B2(Mo;Ta) and B32(Nb;W) separation is found (). Above
K, a B2(Mo,W;Ta,Nb) ordering is observed consistent with previous works.[Citation13–17] Note that the determination of first-order phase transition temperatures could be subject to hysteresis effects within MC simulations, which might thus affect the estimated transition temperature. If only nearest-neighbor interactions are taken into account, the B2 order–disorder transition is found at
K ((a)). The corresponding site occupancies clearly reveal the order–disorder transition as well as the dominant role of Mo-Ta pairs. The computed site occupancies are in excellent agreement with previous calculations by Huhn.[Citation15] The transition temperature changes, however, dramatically if long-ranged interactions () are taken into account. For
(which corresponds to a cutoff radius of 1.73 a0 for the interactions), the critical temperature is already decreased by
to
1050 K. By observing it becomes clear, however, that even five coordination shells cannot provide an adequate representation of the long-ranged nature of chemical interactions in this HEA. Eventually, if the long-ranged tail of interactions is taken into account, the ordering temperature is further reduced down to
K, which is only
of the predicted value based on nearest-neighbor interactions only. We can therefore conclude that the refractory NbMoTaW HEA does not retain a bcc solid solution down to low temperatures because the interactions are so small and short ranged, but because they are long ranged and frustrated. Therefore, their considerable strength does not lead to highly stable ordered states.
Figure 3. Specific heat capacity (a)–(d) and site occupation (e)–(h) for different scenarios of included pair interactions. The B2 ordering temperature is decreased by almost a factor of 2 if long-ranged interactions are taken into account. For nearest-neighbor interactions (pmax = 1), the computed site occupancies (e) are compared with previous results obtained in [Citation15].
![Figure 3. Specific heat capacity (a)–(d) and site occupation (e)–(h) for different scenarios of included pair interactions. The B2 ordering temperature is decreased by almost a factor of 2 if long-ranged interactions are taken into account. For nearest-neighbor interactions (pmax = 1), the computed site occupancies (e) are compared with previous results obtained in [Citation15].](/cms/asset/c89bc935-0f13-4e0a-ad5a-28d20fcba5b1/tmrl_a_1198837_f0003_c.jpg)
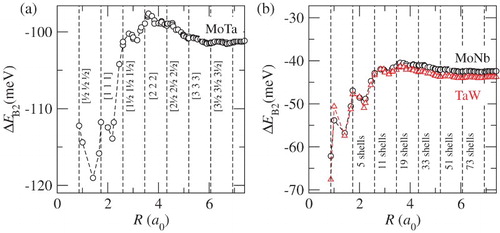
We can also directly relate the observed strong suppression of the B2 order–disorder transition temperature to the dependence of the respective B2 ordering energies, ΔEB2(R), of the involved pairs on the number of included shells. This is highlighted in , where ΔEB2 is shown for the three B2-favoring pairs MoTa (a) as well as MoNb and TaW (b). The suppression of the B2-ordering temperature when increasing the number of included shells goes along with the increase in ΔEB2 for all three individual pairs. In fact, all three ordering energies clearly reveal the fingerprints of the long-ranged nature of the underlying chemical interactions shown in .
Figure 4. B2 ordering energies depending on the range of included interactions for MoTa (a), MoNb and TaW both (b) The suppression of the B2 order–disorder transition temperature is directly related to the increase in the B2 ordering energies of the individual dominant ordering pairs.
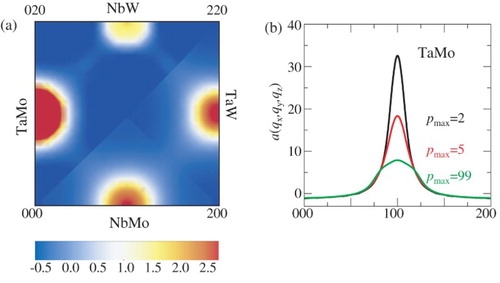
The long-ranged nature of chemical interactions has further consequences for the appearance of short-range order (SRO) at elevated temperatures. We show in (a) the SRO parameters a(qx, qy, qz) at 1300 K computed from the MC simulations including long-ranged interactions (pmax = 99). The four quarters of the SRO still clearly reveal the signature of B2(Mo,W;Ta,Nb) ordering, where Mo-Ta pairs are the most dominant SRO contributors followed by Ta-W, Nb-Mo, and Nb-W. Nb-W pairs also reveal fingerprints of the B32-ordering tendencies causing a second (weaker) maximum at a(½ ½ ½) (not shown). In (b) the SRO parameter in [100] direction is shown for different scenarios of included chemical interactions. The strong impact of the predicted magnitude of SRO on the included number of interactions is consistent with the computed specific heat capacity contribution in (a)–(d) and reveals once more the importance of taking long-ranged interactions into account when simulating HEAs at elevated temperatures.
Figure 5. Pairwise short-range order parameter in the HEA at 1300 K computed from Monte Carlo simulations. (a) SRO reveals signatures of the B2(Mo,W;Ta,Nb) ordering. Ta-Mo pairs show the strongest SRO tendencies. (b) SRO parameters for TaMo for various scenarios of included chemical interactions reveal the necessity of including long-ranged interactions.
We finally note that the employed interactions derived from the homogeneous disordered alloy represent a good choice for simulating the experimentally relevant high-temperature order–disorder transition, where the alloy remains completely homogeneous.
In contrast to common belief that a few chemical pair interactions are sufficient to describe chemical ordering in transition metal alloys,[Citation20] the prototype bcc NbMoTaW HEA reveals long-ranged interactions causing chemical frustration. The ground state of bcc NbMoTaW is predicted to consist of B2(Mo;Ta) and B32(Nb;W) and the alloy reveals a B2(Mo,W;Ta,Nb) ordering at ambient temperatures. At elevated temperatures, chemical interactions cause chemical frustration and suppress the B2 ordering down to low temperatures. We demonstrate that truncated interactions can have serious consequences for the prediction of thermodynamic properties and ordering effects. Surprisingly, the origin of the appearance of an NbMoTaW solid solution at elevated temperatures is not the presence of short-ranged but rather of competing, long-ranged and frustrated interactions. This new and unexpected insight will greatly facilitate theoretical modeling of the new and promising class of HEAs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Gludovatz B, Hohenwarter A, Catoor D, Chang EH, George EP, Ritchie RO. A fracture-resistant high-entropy alloy for cryogenic applications. Science. 2014;345:1153–1158. doi: 10.1126/science.1254581
- Tsai M-H, Yeh J-W. High-entropy alloys: a critical review. Mater Res Lett. 2014;2:107–123. doi: 10.1080/21663831.2014.912690
- Youssef KM, et al. A novel low-density, high-hardness, high-entropy alloy with close-packed single-phase nanocrystalline structures. Mater Res Lett. 2015;3:95–99. doi: 10.1080/21663831.2014.985855
- Zou Y, Ma H, Spolenak R. Ultrastrong ductile and stable high-entropy alloys at small scales. Nat Commun. 2015;6:7748. doi: 10.1038/ncomms8748
- Zhang Y, Zuo T, Cheng Y, Liaw PK. High-entropy alloys with high saturation magnetization, electrical resistivity, and malleability. Sci Rep. 2013;3:1455.
- Lužnik J, Koželj S, Vrtnik A. et al. Complex magnetism of Ho-Dy-Y-Gd-Tb hexagonal high-entropy alloy. Phys Rev B. 2015;92:224201.
- Körmann F, Ma D, Belyea DD, et al. “Treasure maps” for magnetic high-entropy-alloys from theory and experiment. Appl Phys Lett. 2015;107:142404. doi: 10.1063/1.4932571
- Kozelj P, Vrtnik S, Jelen A, et al. Discovery of a superconducting high-entropy alloy. Phys Rev Lett. 2014;113:107001. doi: 10.1103/PhysRevLett.113.107001
- Kozak R, Sologubenko A, Steurer W. Single-phase high-entropy alloys – an overview. Zeitschrift für Kristallographie - Crystalline Materials. 2015;230:55–68.
- Senkov ON, Wilks GB, Scott JM, Miracle DB. Mechanical properties of Nb25Mo25Ta25W25 and V20Nb20Mo20Ta20W20 refractory high entropy alloys. Intermetallics. 2011;19:698–706. doi: 10.1016/j.intermet.2011.01.004
- Senkov ON, Wilks GB, Miracle DB, Chuang CP, Liaw PK. Refractory high-entropy alloys. Intermetallics. 2010;18:1758–1765. doi: 10.1016/j.intermet.2010.05.014
- Zou Y, Maiti S, Steurer W, Spolenak R. Size-dependent plasticity in an Nb25Mo25Ta25W25 refractory high-entropy alloy. Acta Mater. 2014;65:85–97. doi: 10.1016/j.actamat.2013.11.049
- del Grosso MF, Bozzolo G, Mosca HO. Modeling of high entropy alloys of refractory elements. Phys B Condens Matter. 2012;407:3285–3287. doi: 10.1016/j.physb.2011.12.088
- del Grosso MF, Bozzolo G, Mosca HO, Determination of the transition to the high entropy regime for alloys of refractory elements. J Alloy Compd. 2012;534:25–31. doi: 10.1016/j.jallcom.2012.04.053
- Huhn WP. Thermodynamics from first principles: prediction of phase diagrams and materials properties using density functional theory [dissertations]. 2014.
- Widom M, Huhn WP, Maiti S, Steurer W. Hybrid Monte Carlo/molecular dynamics simulation of a refractory metal high entropy alloy. Metall Mater Trans A. 2013;45:196–200. doi: 10.1007/s11661-013-2000-8
- Huhn WP, Widom M. Prediction of A2 to B2 phase transition in the high-entropy alloy Mo-Nb-Ta-W. JOM. 2013;65:1772–1779. doi: 10.1007/s11837-013-0772-3
- Toda-Caraballo I, Wróbel JS, Dudarev SL, Nguyen-Manh D, Rivera-Díaz-del-Castillo PEJ. Interatomic spacing distribution in multicomponent alloys. Acta Mater. 2015;97:156–169. doi: 10.1016/j.actamat.2015.07.010
- Lide DR. Handbook of chemistry and physics 85 Boca Raton (Fla.). London, New York: CRC Press; 2004.
- Ducastelle F. Order and phase stability in alloys. Vol. 3, Cohesion and Structure. Amsterdam: North-Holland/Elsevier Science Publishers; 1991.
- Blum V, Zunger A. Prediction of ordered structures in the bcc binary systems of Mo, Nb, Ta, and W from first-principles search of approximately 3,000,000 possible configurations. Phys Rev B. 2005;72:020104(R). doi: 10.1103/PhysRevB.72.020104
- Vitos L. Computational quantum mechanics for materials engineers: the EMTO method and applications, engineering materials and processes series. London: Springer-Verlag; 2007.
- Ruban AV. Thermal vacancies in random alloys in the single-site mean-field approximation. Phys Rev B. 2016;93:134115. doi: 10.1103/PhysRevB.93.134115
- Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B. 1976;13:5188–5192. doi: 10.1103/PhysRevB.13.5188
- Soven P. Coherent-potential model of substitutional disordered alloys. Phys Rev. 1967;156:809–813. doi: 10.1103/PhysRev.156.809
- Gyorffy BL. Coherent-potential approximation for a nonoverlapping-muffin-tin-potential model of random substitutional alloys. Phys Rev B. 1972;5:2382–2384. doi: 10.1103/PhysRevB.5.2382
- Vitos L, Abrikosov IA, Johansson B. Anisotropic lattice distortions in random alloys from first-principles theory. Phys Rev Lett. 2001;87:156401. doi: 10.1103/PhysRevLett.87.156401
- Ruban AV, Simak SI, Korzhavyi PA, Skriver HL. Screened Coulomb interactions in metallic alloys. II. Screening beyond the single-site and atomic-sphere approximations. Phys Rev B. 2002;66:024202. doi: 10.1103/PhysRevB.66.024202
- Ruban AV, Skriver HL. Screened Coulomb interactions in metallic alloys. I. Universal screening in the atomic-sphere approximation. Phys Rev B. 2002;66:024201. doi: 10.1103/PhysRevB.66.024201
- Abrikosov IA, Simak SI, Johansson B, Ruban AV, Skriver HL. Locally self-consistent Green’s function approach to the electronic structure problem. Phys Rev B. 1997;56:9319–9334. doi: 10.1103/PhysRevB.56.9319
- Abrikosov IA, Niklasson AMN, Simak SI, Johansson B, Ruban AV, Skriver HL. Order-N Green’s function technique for local environment effects in alloys. Phys Rev Lett. 1996;76:4203–4206. doi: 10.1103/PhysRevLett.76.4203
- Peil OE, Ruban AV, Johansson B. Self-consistent supercell approach to alloys with local environment effects. Phys Rev B. 2012;85:165140. doi: 10.1103/PhysRevB.85.165140
- Rahaman M, Johansson B, Ruban AV. First-principles study of atomic ordering in fcc Ni-Cr alloys. Phys Rev B. 2014;89:064103. doi: 10.1103/PhysRevB.89.064103
- Ruban AV, Shallcross S, Simak SI, Skriver HL. Atomic and magnetic configurational energetics by the generalized perturbation method. Phys Rev B. 2004;70:125115. doi: 10.1103/PhysRevB.70.125115
- Nagasako N, Jahnátek M, Asahi R, Hafner J. Anomalies in the response of V, Nb, and Ta to tensile and shear loading: Ab initiodensity functional theory calculations. Phys Rev B. 2010;81:094108. doi: 10.1103/PhysRevB.81.094108