
ABSTRACT
The search for further control over the properties of MAX phases as well as the promise of discovering compounds with new functionalities has resulted in an increased interest in MAX solid solutions resulting from mixing in either the M, A, or X sublattices. The possibility of alloying MAX compounds not only enables finer tuning of their properties but can also be used to stabilize compounds that may otherwise be metastable in their pure state. In this letter, we present an ab initio-based investigation of the intrinsic alloying behavior in the A sublattice of Ti(Al,A)C, Zr
(Al,A)C and Ti
(Al,A)C
MAX compounds.
GRAPHICAL ABSTRACT

IMPACT STATEMENT
In this work, we present for the first time a comprehensive study of the intrinsic alloying tendencies in MAX solid solutions with (Al,A) mixing in the A sublattice.
1. Introduction and motivation
MAX
phases are layered ternary carbides and nitrides that, due to their unique crystal chemistry, share properties that are—in general—somewhat between those of ceramics and metals [Citation1–6]. To date, less than a hundred pure MAX phases have been observed experimentally [Citation7]. However, the potential MAX phase space is considerably larger if one considers the different possible layering sequences (211, 312, 413, etc.) as well distinct occupancies in the M, A and X sites [Citation8]. If we further consider the possibility of mixing of more than one unique species in the different lattices, the MAX phase space expands astronomically to potentially billions of possibilities. Depending on the intrinsic mixing energies (and temperatures) within each sublattice, potential MAX alloys could form as true solid solutions or ordered compounds [Citation9]. To date, however, only an infinitesimal fraction of this enormous space has been explored, with at most a few dozen solid solutions [Citation10] having been synthesized and characterized.
Mixing of more than one species in one (or more) sublattices in the MAX phase crystal structure potentially has important consequences from the point of view of phase stability. Sufficiently exothermic interactions may lead to the stabilization of ordered phases, as exemplified by the (CrTi
)
AlC
system [Citation11–13]. These compounds in some cases have functionalities that are dramatically different from the parent compounds [Citation14]. When the chemical interactions within a single sublattice are less exothermic, solid solutions can be stabilized through contributions from the resulting configurational (i.e. mixing) entropy [Citation9]. Entropic stabilization has been explored as a strategy to synthesize specific MAX phases that are metastable in their pure state, as the work by Horlait et al. [Citation15] on the attempted stabilization of (Zr, M)
AlC and Zr
(Al, A)C illustrates.
The possibility of using sublattice mixing to stabilize desirable (but thermodynamically unattainable) phases or extending the functional performance envelope of MAX systems provides ample motivation to further investigate this expanded MAX phase design space. In prior work [Citation9] the present authors investigated the phase stability (at 0 K) of a large number of pseudo-binary systems belonging to the MAlC class, in which two species at a time were considered to mix in the M sublattice. In this work, we focus instead on mixing in the A sublattice.
Mixing in the A sublattice has been recently investigated experimentally and computationally [Citation16]. Bei et al. [Citation17], for example, investigated TiAl
Sn
C solid solutions and concluded that the addition of low melting elements in the A sublattice of aluminum-containing MAX phases could lead to reduction of oxidation-induced crack healing temperatures. Cabioch et al. [Citation18] proposed the partial substitution of Al for Ge in the Cr
Al
Ge
C
system as a strategy to tune the thermal expansion coefficient in this system. Yu et al. [Citation19] investigated Cr
(Al,Si)C solid solutions and concluded that partial substitution of Al for Si had positive effects on the strength of the material. Gao et al. [Citation20] recently investigated solid solutions in the Ti
(Al
Si
)C
system and found not only stable solid solutions through the entire composition range but also significant solid-solution hardening without compromising oxidation resistance. Finally, Horlait et al. [Citation16] provided the first evidence for Bi-containing MAX phases (Zr
Al
Bi
C with
), while Lapauw recently reported the synthesis of Zr
AlC, albeit in a two-phase microstructure with ZrC being the secondary phase [Citation21].
2. Description of this work
While the examples provided above do not constitute an exhaustive review of the many recent investigations of sublattice mixing in MAX phases, the fact is that only a few dozen or so studies on these systems have been carried out. The considerable resources and ingenuity that must be devoted to the synthesis of quaternary compositions [Citation15] makes a purely experimental plan of attack prohibitive. While one may be tempted to then resort to computational approaches—such as those illustrated by Aryal et al. [Citation8]—the combinatorial explosion arising from all the possible permutations brought about by multiple occupancy in MAX phase lattice sites, as well as the need to take into account not only the ground state phase stability—involving all possible competing phases in the ternary, binary and unary subsystems—but also its extension to finite temperatures make computational approaches almost equally intractable.
In this work, we pursue a more modest plan of attack and focus instead on the intrinsic alloying tendencies in the A sublattice in select MAX Phase systems. Specifically, we study the Ti(Al,A)C, Zr
(Al,A) as well as the Ti
(Al,A)C
systems, in which the A species was taken from Groups 12, 13, 14, 15 and 16 (in modern notation), or IIB, IIIA, IVA, VA, VIA groups (in traditional notation)—in the remainder we will be using modern notation. The selection of the groups was based on experimental findings pointing at the synthesis of a MAX phase with an A constituent belonging to this group. The total number of pseudo-binary systems studied was 51.
Through the use of alloy theoretic approaches [Citation22], in combination with a high-throughput DFT framework developed by the present authors, we investigate alloying trends between Al and A species in the A sublattice of select 211 and 312 systems. The rationale for selecting Al as the common A species is the fact that many Al-containing MAX phases have very good oxidation resistance [Citation23–25] as well as oxidation-induced crack healing properties [Citation26–28]. The Ti(Al,A)C
was selected due to its overall technological importance. Studying the effect of A mixing in Ti
(Al, A)C enables the comparison of alloying trends in light of different atomic environments (due to different layering in 211 and 312 variants), while the study of Zr
(Al,A) enables the comparison between MAX compounds with mixing in the A sublattice and different elements in the M site. The selection of Zr
(Al,A) is also motivated by the potential use of Zr-based MAX phases as novel cladding materials in nuclear fuel technologies [Citation15,
Citation16].
The present work is limited to examining the intrinsic alloy energetics involving Al-A interactions within the A sublattice. As such, the present work does not explicitly address issues related to the stability of quaternary solid solutions or ordered MAX phases relative to competing ternary, binary and unary phases in each of the systems in question. The value of this work rests on the fact that it provides valuable information on the systems that are more likely to become stabilized with respect to competing phases, in comparison with other systems.
3. Methods
3.1. Alloy theoretic approaches to mixing energies
One approach to examine alloying trends in specific sublattices of a crystal structure is through so-called special quasi-random structures (SQS) [Citation29]. SQS work because they mimic (with a relatively small supercell) random configurations at least within a limited range around any given site. This approach has been previously used to look into mixing in the M, A and X sublattices of MAX phases [Citation30–36]. SQS, however, are limited in that they assume a random(-like) state, precluding access to possibly ordered states.
In this work we instead use cluster expansions (CEs) [Citation22,Citation37] to parameterize the energetics of MAX phases with mixing in the A sublattice as a function of arbitrary configurational states. Formally, a CE is defined by assigning occupation variables, to each site i of a lattice that has configurational degrees of freedom. The occupation variables are assigned specific values—
in a (pseudo) binary system—depending on the identity of the atom occupying the site. A particular arrangement of these ‘spins’ corresponds to a configuration which is then represented as a vector
of spins
. The CE then parametrizes the energy (or any other scalar property, such as bulk, shear modulus, etc.) of the alloy in terms of the so-called correlation functions of the occupation variables:
(1) where α is a cluster (a collection of sites i). The sum is taken over all clusters α which are not equivalent by a symmetry operation of the space group of the lattice and the average is taken over all
symmetry-equivalent clusters. The coefficients
are called the effective cluster interactions (ECIs) and relate a given configurational state to a particular energy. The multiplicities
correspond to the number of equivalent clusters (by symmetry) in a given configuration. The ECIs are obtained through the solution of an overdetermined algebraic problem where the (known, calculated) energy of a set of compounds is expressed as functions of the (known) correlation functions and the (unknown) ECIs for every symmetry-unique cluster considered [Citation22,Citation37]. Once the cluster expansion is found—the convergence of the expansion is highly system-dependent—the energy of any configuration can be calculated by simply applying Equation (Equation1
(1) ).
Once the ECIs have been determined, the energy of an equivalent random configuration can be obtained from [Citation38]:
(2) where the sum is over all the possible clusters α,
is the multiplicity of the cluster,
is the number of sites in the cluster and
correspond to the ECIs.
is the average spin concentration, related to the alloy concentration through
.
In this work, we use the MIT Ab-initio Phase Stability (MAPS) [Citation39] code as implemented in the Alloy Theoretic Automated Toolkit (ATAT) [Citation22] package to construct the CEs of the pseudo-binary systems, with the energies of ordered configurations used to obtain the ECIs obtained from DFT methods. In all cases, the CEs were calculated until a cross-validation (CV) score of less than 1–10 meV was obtained. Specific stopping criteria varied depending on the energy scale of the calculated mixing energies. Total energies of 2,356 crystal structures across the 51 pseudo-binaries were necessary in order to achieve this level of overall convergence.
3.2. DFT calculation parameters
The total energy calculations were performed within the density functional theory (DFT) [Citation40] framework, as implemented in the Vienna ab initio simulation package (VASP) [Citation41,Citation42]. The generalized gradient approximation (GGA) [Citation43] is used in the form of the parameterization proposed by Perdew, Burke, and Ernzerhof (PBE) [Citation44,Citation45]. The electronic configurations of the appropriate elements were realized using the projector augmented-wave (PAW) [Citation46] pseudo-potentials. Brillouin zone integrations were performed using a Monkhorst-Pack mesh [Citation47] with at least 5000 k points per reciprocal atom. Full relaxations were realized by using the Methfessel-Paxton smearing method [Citation48] of order one and a final self-consistent static calculation with the tetrahedron smearing method with Blöchl corrections [Citation49]. A cutoff energy of 533 eV was set for all of the calculations and the spin polarizations were taken into account. The relaxations were carried out in three stages: first stage by allowing change in shape and volume—corresponding to the tag—, second stage by additionally allowing the relaxation of ions —corresponding to the
tag— and a final self- consistent static calculation run. We would like to note that spin–orbit coupling (SOC) was neglected. Since none of the systems studied exhibited magnetism, neglect of SOC is justified. Moreover, even in moderately magnetic MAX phases, such as (Mn
Cr
)GaC SOC has not been found to be significant [Citation50].
4. Results
4.1. An example of a CE calculation: the Zr2(Al,Sn)C system
As mentioned above, in a forthcoming study we will be presenting a more detailed analysis of the phase stability of the 51 pseudo-binary studies presented in this work. For the sake of completeness, in Figure we present a representative calculation, corresponding to the CE analysis of the Zr(Al,Sn)C pseudo-binary system. This system is relevant to recent attempts by Horlait et al. [Citation15] to synthesize Zr
AlC-based MAX phases via doping of the A sublattice. Figure shows the energies (per spin or per A-atom) of structures, indicates the ground state and also shows the random mixing energy resulting from application of Equation (Equation2
(2) ).
Figure 1. Representative CE ground state exploration of mixing in the A sublattice of a 211 MAX Phase: Zr(Al,Sn)C. Inset at bottom left corresponds to the energy (multiplied by the cluster multiplicity) of the pair ECIs resulting from the CE fit. Inset at bottom right represents the ‘goodness of fit’, comparing the calculated energies with the predicted energies from the CE.
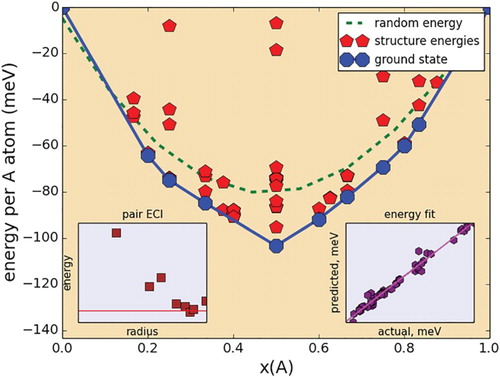
The figure suggests that this system has a tendency toward ordering, with an ordering energy roughly equivalent to the thermal energy of room temperature when considering a per-atom basis (energy per A-atom, divided by four). While not significant, this exothermic mixing energy, combined with the configurational mixing entropy suggests stabilization of Zr(Al,Sn)C solid solutions. When considering that the Zr-Sn-C region of the Zr-Al-Sn-C quaternary is likely to be populated by less stable binary phases (Sn has a low melting point and carbides in the Zr-Sn-C system are likely to be less stable than their counterparts in the Zr-Al-C system), the synthesis of Zr2(Al
Sn
)C by Horlait et al. [Citation15] can be rationalized. A more in-depth study of the competition of the entire solid-solution range against all stable unary, binary
and ternary phases, at finite temperatures in this quaternary system, however, would be necessary to fully compare with experimentally determined phase stabilities.
4.2. Random mixing energies in Ti (Al,A)C, Zr(Al,A)C and Ti(Al,A)C pseudo-binaries
(Al,A)C, Zr(Al,A)C and Ti(Al,A)C pseudo-binaries
Figures show the calculated random mixing energies for the Ti(Al,A)C, Zr
(Al,A)C and Ti
(Al,A)C
pseudo-binary sets, respectively. We would like to note that the fact that in some cases the predicted mixing energies of the end members are not zero and reflect the model discrepancy of the CEs. This error, however, is less significant than the trends observed. Examining the three figures, several observations can be made:
Figure 2. Random mixing energies (per number of spins/A sites) in Ti2(Al,A)C pseudo-binary systems. The inset corresponds to a zoom in of the plots within the −100,100 meV/spin range. Left: 100% Al. Right: 100% A Dopant.
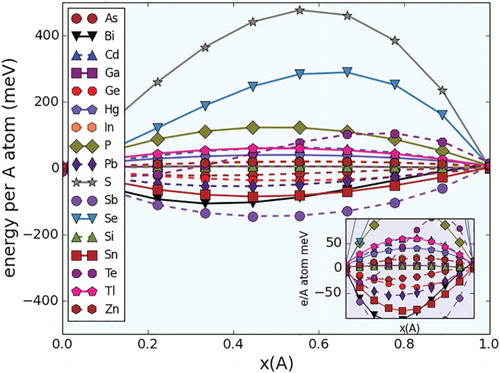
Figure 3. Random mixing energies (per number of spins/A sites) in Zr2(Al,A)C pseudo-binary systems. The inset corresponds to a zoom in of the plots within the −100,100 meV/spin range. Left: 100% Al. Right: 100% A Dopant.
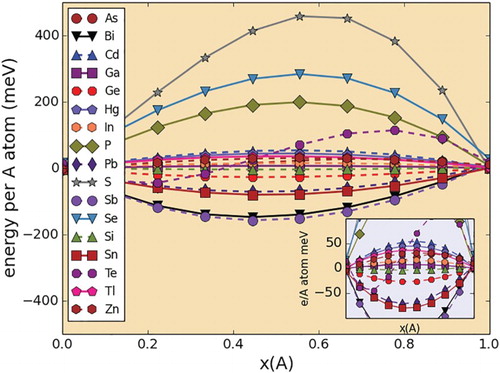
Figure 4. Random mixing energies (per number of spins/A sites) in Ti(Al,A)C
pseudo-binary systems. The inset corresponds to a zoom in of the plots within the −100,100 meV/spin range. Left: 100% Al. Right: 100% A Dopant.
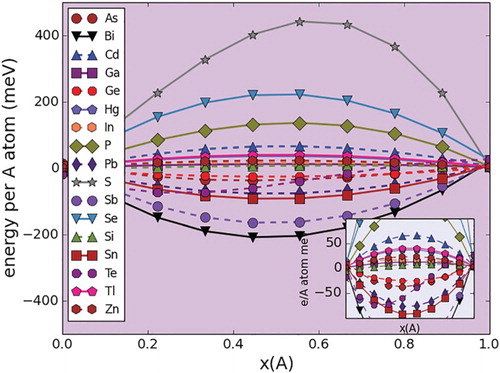
In all three pseudo-binary sets the mixing energies are within the [−150, 450 meV/A-atom] range. This range of energies is significant as this shows that the systems studied span the entire alloying tendency range, from ordering to phase separation, passing through a region where there is a clear tendency to form almost ideal solutions.
In all the three cases, it is evident that approximately 60% of the systems exhibited mixing energies within the [−50,50 meV/spin] range. Notably, in all three crystal systems studied, the A dopants that tend to have almost negligible chemical interactions with Al are Si, Ga and to a lesser extent In. A priori, Ga and In are likely to have almost no chemical interaction with Al in a MAX chemical environment due to the fact that they are isoelectronic, and that the A layer is loosely bound to the rest of the structure, minimizing the effect of differences in ionic radii on the energetics of mixing in the A sublattice.
The case of Al-Si, however, is not as straightforward. The Al-Si binary phase diagram [Citation51] shows a system in which the solid phases are mutually insoluble but in which there is a relatively deep eutectic close to the Al side. While mutual insolubility would suggest a tendency for phase separation (endothermic Al-Si chemical interactions), the calculations for the Ti2(Al,Si)C, Ti(Al,Si)C
and Zr
(Al,Si)C systems suggest instead almost no interaction between Al and Si in the MAX chemical environment. This is a significant result that indicates that one cannot glean trends for possible doping strategies by simply looking into the types of interactions that occur between A-type species in bulk binary crystal systems. The almost ideality of Al–Si interactions is supported by the recent synthesis of Ti2(Al,Si)C over the entire composition range [Citation20]. A more detailed analysis of the electronic structure of this system close to the Fermi level would be necessary to elucidate the nature of the (lack of) chemical interactions within the A layer in this system, but this is beyond the scope of this work.
The rest of the A dopants (Zn, Hg, Tl, As) that show moderate endothermic and exothermic chemical interactions with Al in all the three crystal systems are good candidates for synthesis of close-to-ideal (Al,A) solid solutions. While it may be questionable the inclusion of Zn and Hg in this list, the fact that TiCdC has reportedly been synthesized [Citation52] provides justification for proposing the possible existence of MAX solid solutions involving Al and Zn,Hg, provided the competition with neighboring phases in their respective quaternary systems is favorable. With regards to Al-As, while the As-bearing end members are yet to be synthesized, the study suggests an easy incorporation of As into the A sublattice, provided thermodynamics (and kinetics) allows this to happen.
Only a very small fraction of the systems exhibit clear tendencies for phase separation. According to the calculations for all the three pseudo-binary sets, Se, P and S, in that order, have the most endothermic interactions with Al within the A layer. The tendency for phase separation is not trivial, with a mixing energy of close to 0.5 eV/A-atom in the case of the M
(Al,S)C
systems. Ti
SC has been synthesized already [Citation53] and the calculations suggest that attempts to co-synthesize Ti
AlC and Ti
SC are likely to yield a microstructure consisting of two distinct iso-structural phases. In the same crystal system, the fact that Ti
PC seems to be at best metastable relative to lower order competing phases in its ternary [Citation54], coupled to the clear tendency for phase separation with Al indicates that P (as well as Se) constitute poor choices for solid-solution stabilizing dopants in Al-bearing MAX systems.
A similarly small fraction of the systems () exhibit clear exothermic tendencies. While the fraction of these systems is small, they are rather interesting as some of the same dopants have already been proven to stabilize solid solutions, at least in the Zr
(Al,A)C system [Citation15,Citation16]. The calculated mixing energy for the Zr
(Al,Bi)C system is in remarkable agreement with the recent calculations by Horlait et al. [Citation16], although the previous calculations were based on a limited set of relatively small supercell calculations with random (Al,Si) configurations. The Zr
(Al,Bi)C system is notable due to the fact that while the end members are at best metastable with competing phases, the exothermic nature of the Al–Bi chemical bonds within the A sublattice is sufficient to stabilize intermediate composition ranges. Based on our calculations, similar results are to be expected from attempts to dope Zr
AlC with Sn, Pb and Sb, as was recently demonstrated by Horlait et al. [Citation15]. Further validation of the predictions of this work is the fact that Sn has been shown to stabilize solid solutions in the Ti
(Al,Sn)C
system [Citation55].
Although this letter does not concern itself with the phase stability of the systems studied, an analysis of the stability of the ordered compounds in the Zr(Al,A)C systems with A=Sn, Sb, Pb, Bi against all competing phases available in the Materials Project dataset [Citation56] suggests that all the (quaternary) ordered compounds in these systems in fact belong to the convex hull, i.e. they are thermodynamically stable at 0 K. This suggests that exothermic energies above 50 meV/A-atom are sufficient to stabilize quaternary compounds in the Zr
(Al,A)C system. A more in-depth analysis of phase competition in this and the other systems studied will be forthcoming in future work by the present authors. Most importantly, all pseudo-binary sets behave in a remarkably similar manner. This is perhaps the most unexpected and surprising result from this work. As can be seen by comparing Figures –, the values for the random mixing energies (on a per A-site basis) as well as the rank order of the different Al-A chemical subsystems is almost identical—the background color of the figures was deliberately changed to reinforce this fact. As is already known by the community, the remarkable properties of MAX phases lie on the fact that the A layer is relatively chemically isolated from the rest of the lattice, due to the fact that most of the bonding occurs within the MX layers. What is notable from the present results is the fact that this chemical isolation is such that the alloying trends are
virtually independent of the chemical/structural environment surrounding the A layer.
The practical consequence of this observation is that for other M(A1,A2)
systems, chemical trends observed for example in 211 subsystems can be plausibly expected to be similar in 312 and 413 stackings, as long as the identity of the M element remains the same. While our results show identical trends for Ti
(Al,A)C and Zr
(Al,C), Ti and Zr are isoelectronic and further studies comparing alloying trends in M
(A1,A2)
varying the M element along the transition metal columns need to be carried out.
4.3. Global mixing trends
Having established the almost identical nature of the alloying tendencies in all the pseudo-binary sets considered, we can use a more in-depth analysis of one of them to highlight some general trends and elucidate some of the underlying reasons for the observations. Here we would like to note that, as supplementary material, we are providing the calculated mixing energies for all the 51 pseudo-binary systems.
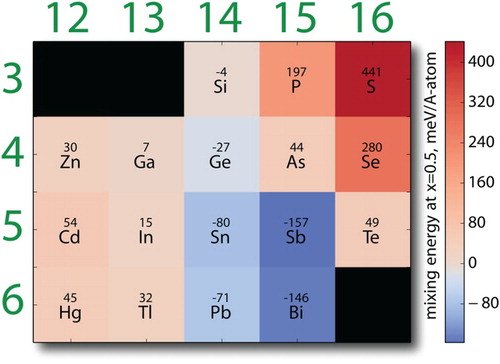
Figure shows the calculated mixing energy at x=0.5 for all the 17 pseudo-binaries in the Zr(Al,A)C crystal system. The figure shows the following overall trends:
In the case of dopants belonging to Groups 14 (Si, Ge, Sn, Pb) and 15 (P, As, Sb, Bi), there is a tendency toward more exothermic interactions as one moves down the rows of the periodic table. An illustrative case is the Group 14, where Si shows an almost ideal interaction with Al, while this interaction becomes more exothermic with Ge and remarkably so for Sn and Pb. At first glance, this result is somewhat counterintuitive as the difference in ionic radii between Al and (Si,Ge,Sn,Pb) increases as the molecular weight of the element increases. However, the A layer is loosely bound to the crystal structure and it is not likely that size effects play an important role. The trend, however, correlates somewhat with the fact that valence electrons are increasingly screened from the core as one moves down the periodic table, and are thus more available to form chemical bonds with the 3p valence electrons of Al.
In the case of Group 13 elements (Ga, In, Tl), and contrary to the case of Groups 14 and 15 dopants, the mixing energies tend to be more exothermic as one moves down the column. The change in mixing energy varies (exactly) linearly with the relative difference in atomic radii (
Closer examination of Group 15 is remarkable as this system shows the entire range of alloying behavior, ranging from highly endothermic to highly exothermic, suggesting that in this case, both size and chemical effects due to screening of the core play a very important role in determining the nature of the alloying interactions.
While Group 12 (Zn, Cd, Hg) belongs to the transition metal family and thus by definition should occupy the M site of MAX phases, if at all, there is at least one example of a synthesized MAX phase with the A site occupied by an element belonging to this group (i.e. Cd). In this case, the trends are somewhat similar to those observed in Group 13, although the change in mixing energy is not monotonic and not as pronounced. Mixing energies, while endothermic, are comparable to the energy of a thermal bath between 300 and 600 K and it may be thermodynamically likely to observe solid solutions involving these elements in the A sublattice, particularly at high temperatures.
Finally, looking into the trends in Group 16 (S, Se, Te) one concludes that the initial very endothermic interaction with Al tends to become remarkably less so as one moves down the period. While at least one MAX phase involving elements from this group have been synthesized before (Ti
Figure 5. Calculated random mixing energies for Zr(Al
,A
)C systems at x=0.5.
5. Conclusions
The alloying trends in the Ti
Results for some of the pseudo-binaries are in agreement with available (but scarce) experimental (and computational) prior work, providing some indication of the validity of the predictions.
The calculations suggest that the chemical and/or structural local environment surrounding the A layer plays a rather minor role—provided the M element is Ti, Zr—in determining the alloying trends as the three systems studied are virtually identical in the ranges and rank ordering of the predicted mixing energies.
Some of the trends observed can be elucidated using simple size and bonding arguments, although a more in-depth analysis of chemical bonding is necessary to generalize the findings of this work to other systems.
The results in this work can potentially provide some guidelines for the synthesis of novel (Al,A) solid solutions that can be used to stabilize previously unattainable MAX forms or to tune the properties of already synthesized systems.
As to the question posed in the title of the present work, the answer is: ‘it depends…’
Supplementary_material
Download MS Excel (73 KB)Acknowledgments
First-principles calculations by R.A., T.D, W. S. and A.T. were carried out at the Texas A& M Supercomputing Facility at Texas A&M University as well as the Texas Advanced Computing Center (TACC) at the University of Texas at Austin. Preparation of the input files and analysis of the data have been performed using AFLOW [Citation57] and OQMD [Citation58]. The Texas A&M Materials Modeling Automation Library (tammal) developed by R.A., A.T. and collaborators (soon to be released to the general scientific community) was used to carry out the HT CE calculations. The ATAT package [Citation22] was used to carry out the CEs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Barsoum MW. MAX phases: properties of machinable ternary carbides and nitrides. Weinheim, Germany: Wiley-VCH; 2013.
- Radovic M, Barsoum MW. Max phases: bridging the gap between metals and ceramics. Amer Ceram Soc Bull. 2013;92(3):20–27.
- Barsoum MW. The MAX phases: a new class of solids: thermodynamically stable nanolaminates. Progress Solid State Chem. 2000;28(1):201–281. doi: 10.1016/S0079-6786(00)00006-6
- Barsoum MW, Radovic M. Mechanical properties of the MAX phases. In: Buschow KHJ, Cahn R, Flemings M, Ilschner B, Kramer E, Mahajan S, Veyssiere P, editors. Encyclopedia of materials: science and technology. Amsterdam: Elsevier; 2004. p. 1–16.
- Barsoum MW, Radovic M. Elastic and mechanical properties of the MAX phases. Annu Rev Mater Res. 2011;41:195–227. doi: 10.1146/annurev-matsci-062910-100448
- Sun ZM. Progress in research and development on MAX phases: a family of layered ternary compounds. Int Mater Rev. 2011;56(3):143–166. doi: 10.1179/1743280410Y.0000000001
- Barsoum MW. The M(n+1)AX(n) phases and their properties. In: Riedel R, Chen IW, editors. Ceramics Science and Technology Volume 2: Properties. Weinheim, Germany: Wiley-VCH; 2010. doi:10.1002/9783527631735.ch7
- Aryal S, Sakidja R, Barsoum MW, et al. A genomic approach to the stability, elastic, and electronic properties of the MAX phases. Phys Status Solidi (B). 2014;251(8):1480–1497. doi: 10.1002/pssb.201451226
- Talapatra A, Duong T, Son W, et al. High-throughput combinatorial study of the effect of M site alloying on the solid solution behavior of MAlC max phases. Phys Rev B. 2016;94(10): 104106. doi: 10.1103/PhysRevB.94.104106
- Naguib M, Bentzel GW, Shah J, et al. New solid solution MAX phases:(Ti,V)AlC, (Nb,V)AlC, (Nb, V)AlC and (Nb,Zr)AlC. Mater Res Lett. 2014;2(4):233–240. doi: 10.1080/21663831.2014.932858
- Burr PA, Horlait D, Lee WE. Experimental and DFT investigation of (Cr, Ti)AlC max phases stability. arXiv preprint arXiv:160706162. 2016.
- Liu Z, Wu E, Wang J, et al. Crystal structure and formation mechanism of (CrTi)AlC max phase. Acta Mater. 2014;73:186–193. doi: 10.1016/j.actamat.2014.04.006
- Horlait D, Grasso S, Al Nasiri N, et al. Synthesis and oxidation testing of max phase composites in the Cr–Ti–Al–C quaternary system. J Amer Ceram Soc. 2016;99:682–690. doi: 10.1111/jace.13962
- Mockute A, Dahlqvist M, Emmerlich J, et al. Synthesis and Ab initio calculations of nanolaminated (Cr, Mn)AlC compounds. Phys Rev B. 2013;87(9):094113. doi: 10.1103/PhysRevB.87.094113
- Horlait D, Grasso S, Chroneos A, et al. Attempts to synthesise quaternary max phases (Zr,M)AlC and Zr(Al,A)C as a way to approach ZrAlC. Mater Res Lett. 2016;1–8.
- Horlait D, Middleburgh SC, Chroneos A, et al. Synthesis and DFT investigation of new bismuth-containing max phases. Scient Rep. 2016;6.
- Bei G, Pedimonte B-J, Fey T, et al. Oxidation behavior of max phase TiAlSnC solid solution. J Amer Ceram Soc. 2013;96(5):1359–1362. doi: 10.1111/jace.12358
- Cabioch T, Eklund P, Mauchamp V, et al. Tailoring of the thermal expansion of Cr(Al,Ge)C phases. J Eur Ceram Soc. 2013;33(4):897–904. doi: 10.1016/j.jeurceramsoc.2012.10.008
- Yu W, Li S, Sloof WG. Microstructure and mechanical properties of a Cr(Al,S)C solid solution. Mater Sci Eng: A. 2010;527(21):5997–6001. doi: 10.1016/j.msea.2010.05.093
- Gao H, Benitez R, Son W, et al. Structural, physical and mechanical properties of Ti(AlSi)C solid solution with . Mater Sci Eng: A. 2016;676:197–208. doi: 10.1016/j.msea.2016.08.098
- Lapauw T, Lambrinou K, Cabioc'h T, et al. Synthesis of the new max phase ZrAlC. J Eur Ceram Soc. 2016;36(8):1847–1853. doi: 10.1016/j.jeurceramsoc.2016.02.044
- Van de Walle A, Asta M, Ceder G. The alloy theoretic automated toolkit: a user guide. Calphad. 2002;26(4):539–553. doi: 10.1016/S0364-5916(02)80006-2
- Sundberg M, Malmqvist G, Magnusson A, et al. Alumina forming high temperature silicides and carbides. Ceram Int. 2004;30(7):1899–1904. doi: 10.1016/j.ceramint.2003.12.046
- Tallman DJ, Anasori B, Barsoum MW. A critical review of the oxidation of TiAlC, TiAlC and CrAlC in air. Mater Res Lett. 2013;1(3):115–125. doi: 10.1080/21663831.2013.806364
- Basu S, Obando N, Gowdy A, et al. Long-term oxidation of TiAlC in air and water vapor at 1000–1300C temperature range. J Electrochem Soc. 2011;159(2):C90–C96. doi: 10.1149/2.052202jes
- Song GM, Pei YT, Sloof WG, et al. Oxidation-induced crack healing in TiAlC ceramics. Scr Mater. 2008;58(1):13–16. doi: 10.1016/j.scriptamat.2007.09.006
- Li S, Song G, Kwakernaak K, et al. Multiple crack healing of a TiAlC ceramic. J Eur Ceram Soc. 2012;32(8):1813–1820. doi: 10.1016/j.jeurceramsoc.2012.01.017
- Sloof WG, Pei R, McDonald SA, et al. Repeated crack healing in max-phase ceramics revealed by 4D in situ synchrotron X-ray tomographic microscopy. Scient Rep. 2016;6:
- Zunger A, Wei S-H, Ferreira LG, et al. Special quasirandom structures. Phys Rev Lett. 1990;65(3):353–356. doi: 10.1103/PhysRevLett.65.353
- Mockute A, Lu J, Moon EJ, et al. Solid solubility and magnetism upon Mn incorporation in the bulk ternary carbides CrAlC and CrGaC. Mater Res Lett. 2015;3(1):16–22. doi: 10.1080/21663831.2014.944676
- Anasori B, Dahlqvist M, Halim J, et al. Experimental and theoretical characterization of ordered max phases MoTiAlC and MoTiAlC. J Appl Phys. 2015;118(9):094304. doi: 10.1063/1.4929640
- Thore A, Dahlqvist M, Alling B, et al. Temperature dependent phase stability of nanolaminated ternaries from first-principles calculations. Comput Mater Sci. 2014;91:251–257. doi: 10.1016/j.commatsci.2014.04.055
- Thore A, Dahlqvist M, Alling B, et al. Phase stability of the nanolaminates VGaC and (MoV)GaC from first-principles calculations. Phys Chem Chem Phys. 2016;18(18):12682–12688. doi: 10.1039/C6CP00802J
- Dahlqvist M, Rosen J. Order and disorder in quaternary atomic laminates from first-principles calculations. Phys Chem Chem Phys. 2015;17(47):31810–31821. doi: 10.1039/C5CP06021D
- Arróyave R, Radovic M. Ab initio investigation of TiAl(C,N) solid solutions. Phys Rev B. 2011;84(13):134112. doi: 10.1103/PhysRevB.84.134112
- Aryal S, Sakidja R, Ouyang L, et al. Elastic and electronic properties of TiAl(CN) solid solutions. J Eur Ceram Soc. 2015;35(12):3219–3227. doi: 10.1016/j.jeurceramsoc.2015.03.023
- Connolly JWD, Williams AR. Density-functional theory applied to phase transformations in transition-metal alloys. Phys Rev B. 1983;27(8):5169–5172. doi: 10.1103/PhysRevB.27.5169
- Liu JZ, Van De Walle A, Ghosh G, et al. Structure, energetics, and mechanical stability of Fe-Cu bcc alloys from first-principles calculations. Phys Rev B. 2005;72(14):144109.
- van de Walle A, Ceder G. Automating first-principles phase diagram calculations. J Phase Equilib. 2002;23(4):348–359. doi: 10.1361/105497102770331596
- Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140(4A):A1133–A1138. doi: 10.1103/PhysRev.140.A1133
- Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54(16):11169–11186. doi: 10.1103/PhysRevB.54.11169
- Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci. 1996;6(1):15–50. doi: 10.1016/0927-0256(96)00008-0
- Perdew JP, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B. 1992;45(23):13244–13249. doi: 10.1103/PhysRevB.45.13244
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–3868. doi: 10.1103/PhysRevLett.77.3865
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys Rev Lett. 1997;78:1396–1396. doi: 10.1103/PhysRevLett.78.1396
- Blöchl PE. Projector augmented-wave method. Phys Rev B. 1994;50(24):17953–17979. doi: 10.1103/PhysRevB.50.17953
- Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B. 1976;13(12):5188–5192. doi: 10.1103/PhysRevB.13.5188
- Methfessel M, Paxton AT. High-precision sampling for Brillouin-zone integration in metals. Phys Rev B. 1989;40(6):3616–3621. doi: 10.1103/PhysRevB.40.3616
- Blöchl PE, Jepsen O, Andersen OK. Improved tetrahedron method for Brillouin-zone integrations. Phys Rev B. 1994;49(23):16223–16233. doi: 10.1103/PhysRevB.49.16223
- Salikhov R, Semisalova AS, Petruhins A, et al. Magnetic anisotropy in the (CrMn)GaC max phase. Mater Res Lett. 2015;3(3):156–160. doi: 10.1080/21663831.2015.1036324
- Murray JL, McAlister AJ. The Al-Si (Aluminum-Silicon) system. Bull Alloy Phase Diagrams. 1984;5(1):74–84. doi: 10.1007/BF02868729
- Jeitschko W, Nowotny H, Benesovsky F. Die h-phasen: TiCdC, TiGaC, TiGaN, TiInN, ZrInN und NbGaC. Monatshefte für Chemie und verwandte Teile anderer Wissenschaften. 1964;95(1):178–179. doi: 10.1007/BF00909264
- Amini S, Barsoum MW, El-Raghy T. Synthesis and mechanical properties of fully dense TiSc. J Amer Ceram Soc. 2007;90(12):3953–3958.
- Hug G. Electronic structures of and composition gaps among the ternary carbides TiMC. Phys Rev B. 2006;74(18):184113. doi: 10.1103/PhysRevB.74.184113
- Dubois S, Bei GP, Tromas C, et al. Synthesis, microstructure, and mechanical properties of TiSnAlC max phase solid solutions. Int J Appl Ceram Technol. 2010;7(6):719–729. doi: 10.1111/j.1744-7402.2010.02554.x
- Jain A, Ong SP, Hautier G, et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 2013;1(1):011002. doi: 10.1063/1.4812323
- Curtarolo S, Setyawan W, Wang S, et al. AFLOWLIB.ORG: a distributed materials properties repository from high-throughput Ab initio calculations. Comput Mater Sci. 2012;58:227–235. doi: 10.1016/j.commatsci.2012.02.002
- Saal JE, Kirklin S, Aykol M, et al. Materials design and discovery with high-throughput density functional theory: the open quantum materials database (OQMD). JOM. 2013;65(11):1501–1509. doi: 10.1007/s11837-013-0755-4