
ABSTRACT
This letter is motivated by an apparent paradox, in that some quaternary systems (particularly in the Ti–Zr–Al–C system) have been shown to exhibit M-site out-of-plane ordering, while prior work and calculations by the present authors suggest endothermic interactions between Zr and Ti. In this letter we provide a resolution to this issue and provide a more extended analysis on the out-of-plane and in-plane ordering in the M sites of quaternary MAX alloys. The results provide further insights to develop criteria to predict potential out-of-plane ordering tendencies in other MAX systems.fx1
GRAPHICAL ABSTRACT
IMPACT STATEMENT In this work, we resolve a recently evident contradiction between theoretical predictions for phase separation in the Ti–Zr–Al–C 312 MAX system and experimental observations that indicate out-of-plane ordering.
1. Introduction
MAX
phases have intermediate properties to those of ceramics and metals due to their unique crystal chemistries and layered atomic structures [Citation1–6]. While the number of pure ternary MAX phases synthesized and characterized thus far is relatively small—i.e. around 70 [Citation7]—the MAX design space becomes much larger if one considers possible different stacking sequences and deviations from stoichiometries in the M, A and X sites [Citation8]. Moreover, when one considers quaternary and higher-order systems, the number of possible stable configurations grows factorially. Until recently, only a very small fraction of these stable quaternary compounds has been observed [Citation9] but research by the present authors [Citation10,Citation11] and others [Citation12] suggest the existence of a vast, yet to be explored, materials space.
Quaternary (and higher order) additions to one (or more) sublattices in the MAX crystal system have important consequences from the perspective or phase stability. Exothermic interactions can stabilize ordered phases [Citation13–15] and in some cases these energetically (enthalpically)-stabilized compounds can have very different (and potentially useful) functionalities compared to the end-members (i.e. ternary) compounds [Citation16]. When the interactions are only weakly exothermic, entropic contributions to the free energies of the MAX alloys may be sufficient to stabilize solid solutions. The ability to stabilize these quaternary compounds or alloys is important from a technological perspective as the further degree of compositional control provides the means to further tune and improve the properties of MAX systems [Citation17–20]. Moreover, moving into quaternary directions within the MAX design space provides the means to stabilize MAX phases that otherwise would not be thermodynamically stable, as exemplified by the work by Horlait et al., who synthesized quaternary Zr(Al,Bi)C MAX phases even through the end members Zr
AlC and Bi
AlC are not (apparently) thermodynamically stable [Citation21].
Within the body of work dedicated to investigating the synthesis and properties of quaternary MAX systems, so-called out-of-plane ordered compounds has received significant attention recently. Liu et al. where among the first ones to report ordered MAX
structures in which a Ti layer was sandwiched between outer carbide layers, as in the Cr
TiAlC
compound [Citation22]. Anasori et al. [Citation23] later discovered the ordered Mo
TiAlC quaternary compound. Moreover, Anasori et al. [Citation24] showed that the unique Mo–Ti–Mo 2D carbide configuration (i.e. MXene) obtained by selective etching of A element from Mo
TiAlC results in electrochemical properties that are significantly different from those achieved from conventional MXene compounds (e.g. TiC
). Meshkian et al. [Citation25] very recently provided yet one more example of an out-of-plane MAX compound, Mo
ScAlC
, which is thermodynamically stable, as opposed to the ternary end-members Mo
AlC
and Sc
AlC
. This work is significant as they demonstrated the synthesis of the 2D variant through selective etching of the Al layer, thus expanding the MXene repertoire.
2. Motivation and description of this work
Notably, all the recent experimental investigations on out-of-plane ordering in quaternary MAX systems have gone hand-in-hand with DFT-based computations that have verified the thermodynamic stability of these ordered compounds against decomposition into combinations of lower order compounds from the constituent ternary, binary and unary systems. This agreement between experiments and calculations was initially reassuring as it suggests that high-throughput approaches pioneered by the present authors [Citation10,Citation11] can be used to screen in an efficient manner vast regions of the MAX materials space. This positive outlook, however, was partially put into question by the recent work of Tunca et al. [Citation26], who observed strong evidence for out-of-plane ordering in samples with TiZrAlC
stoichiometry, while Dahlqvist and Rosen [Citation27] predicted that such an ordering is metastable relative to the end members Ti
AlC
and Zr
AlC
. The primary motivation of this work is to resolve this controversy.
While the discrepancy between the experiments by Tunca et al. and the predictions by Dahlqvist et al. may be dismissed, the apparent inability of theoretical approaches to predict ordering tendencies in the Ti–Zr–Al–C system would imply that recent computational approaches are not as effective at making the search for new quaternary solid solutions and/or ordered compounds more efficient and robust. The primary objective of this work is the systematic investigation of the alloying behavior of a system in which both theory and experiments suggest strong ordering trends—Mo-Ti-Al-C [Citation23]—in comparison with a system that is predicted to phase separate [Citation27] but in which solid solutions—(Zr,Ti)AlC—and ordered compounds—TiZrAlC
—have been observed [Citation26]. We extend our analysis to the (Ti,Hf)
AlC
and (Ti,Nb)
AlC
in order to attempt to de-convolute the different factors (atomic radii, charge transfer, etc.) responsible for the stability of different configurations in these systems.
Contrary to prior approaches in which an arbitrary set of crystal structures are used to estimate the nature of mixing in specific sublattices of an MAX compound, in this work we employed a more systematic approach first introduced by the present authors [Citation10,Citation11] in which alloy theoretic approaches [Citation28], in combination with a high-throughput DFT framework are used to investigate alloying trends within the M sublattice in 312 systems. In this work, we do not explicitly investigate the stability of the systems studied against decomposition into lower order phases as our objective is merely to compare observed trends with theoretical predictions.
3. Results and discussion
To carry out the present alloy theoretic investigation, we followed an approach already employed by the present authors [Citation10,Citation11], whereby the energetics of the configuration space in the systems under study was investigated through the Cluster Expansion (CE) formalism [Citation28,Citation29], as described in more detail in the Supplementary Material.
3.1. Alloying trends in (Ti,Mo) AlC MAX alloys
AlC MAX alloys
Figure shows the calculated energies (per M-site) for different ordered structures derived from different ordering sequences within the M sublattice in the system, as determined by the exhaustive search using the MAPS code in the ATAT package. A systematic and exhaustive search over the configuration space yielded a set of configurations that were most stable at a given composition and that, together, form the so-called convex hull, along the Ti-M composition range. This of course, ignores competing phases coming regions in the composition space outside the
pseudo-binary region.
Figure 1. (color online) Energetics of Mo-Ti mixing in (Ti,Mo)AlC
MAX alloys predicted through the combination of cluster expansion lattice searches with DFT calculations—for a definition of the energy units (energy per M-site) refer to Supplementary Material. Calculated ground states are indicated by the blue markers and convex hull is indicated by solid blue line. Non-ground state structures are depicted by red markers with random energy—defined in Eq. S2 of Supplementary Material—shown as green dashed line. Structure marked with magenta circle corresponds to Mo
TiAlC
synthesized by Anasori et al. [Citation24]. (insets) Model crystal structure and high-resolution TEM—reproduced (adapted) with permission from [Citation23].
![Figure 1. (color online) Energetics of Mo-Ti mixing in (Ti,Mo)AlC MAX alloys predicted through the combination of cluster expansion lattice searches with DFT calculations—for a definition of the energy units (energy per M-site) refer to Supplementary Material. Calculated ground states are indicated by the blue markers and convex hull is indicated by solid blue line. Non-ground state structures are depicted by red markers with random energy—defined in Eq. S2 of Supplementary Material—shown as green dashed line. Structure marked with magenta circle corresponds to MoTiAlC synthesized by Anasori et al. [Citation24]. (insets) Model crystal structure and high-resolution TEM—reproduced (adapted) with permission from [Citation23].](/cms/asset/9e4a9af0-8cae-4064-b743-45aae0a0da38/tmrl_a_1380723_f0001_c.jpg)
The figure also includes insets corresponding to the structural model and corresponding high-resolution TEM image of the MoTiAlC
compound as synthesized by Anasori et al. [Citation24]. According to the calculations, the ground state of this 312 system consists of a series of ordered structures at the fraction of M site occupied by Mo (x
) equal to 1/3, 1/2, 7/12, 2/3 and 10/12. We further compared the calculated energy-per-M site of the Mo
TiAlC
compound calculated by Anasori et al. [Citation30] from their reported energies for the end-members and their most stable configuration at the same stoichiometry and found excellent agreement—within 7 meV/M-site—with our calculations.
As shown in Figure , the convex hull is clearly non-symmetrical, exhibiting a deep minimum at =2/3, which corresponds to the layered, out-of-plane ordered Mo
TiAlC
compound synthesized by Anasori et al. [Citation23]. In this compound, Ti sits on the Wyckoff 2a-sites (
sublattice), while Mo sits on the Wyckoff 4f-sites (
sublattice) of the MAX lattice (see Figure S1 in the Supplementary Material). In this structure Ti is surrounded by C in a face-centered configuration that is highly favorable relative to other possible stacking sequences [Citation27]. In fact, the calculations show that, at this composition, there is no degeneracy in the energetics of different stacking sequences, with the second most stable stacking sequence lying almost 150 meV/site above the most stable configuration. Incidentally, this latter structure consists of Mo and Ti alternatively occupying different 2a-sites along the c-axis of the structure.
The stabilization of this configuration has been previously investigated by Dahlqvist and Rosen [Citation27] and it has been determined that the local configuration consisting of Mo atoms surrounded by C atoms in an fcc arrangement (NaCl-prototype) is energetically unfavorable as the binary carbide MoC favors instead arrangements analogous to the WC prototype. Moreover, the large electronegativity difference between Mo and Al makes Al atoms neighboring Mo (in the 4f-sites/ sublattice) more positively charged, reducing the number of electrons populating Al–Al anti-bonding states, the net result being that the ordered structure with Ti in 2a- and Mo in 4f-sites is considerably stabilized relative to not only the end members Ti
AlC
and Mo
AlC
but also relative to competing configurations with mixed occupancy of Ti and Mo in the 2a- and 4f-sites.
The situation is very different for the configuration. In this case, the most stable structure consists of a combination of in-plane and out-of-plane ordering (see Figure S2 in the Supplementary Material). Degeneracy, however, is significant as there is a large number of ordered states relatively close in energy. In fact, our calculations suggest that for a maximum size of two unit cells (24 atoms), there are a dozen or so configurations within 40 meV of the ground state. Such degeneracy suggests that at temperatures within the 600–1000 K range, configurational entropy would be sufficient to stabilize random solid solutions with Mo and Ti mixing in the M sublattice, provided no competition with other phases would render this structure metastable.
Results from Dahlqvist and Rosen [Citation27] suggest that the MoTiAlC
composition lies above the convex hull of this system, competing with TiC, Mo
Al and Mo
Al
. The distance to the convex hull, however, is only 9 meV/atom or 18 meV/M-site. This metastability is rather small, and it may be possible that random solutions in the (Ti,Mo)
AlC
become stabilized at elevated temperatures. We should note, however, that even though Mo
Al and Mo
Al
are line compounds [Citation31] without much configurational entropy, TiC exhibits a large degree of non-stoichiometry [Citation32] and is likely to be further stabilized by entropy at elevated temperatures. Further analysis of the high-temperature phase stability of this system incorporating combined DFT, CALPHAD and experimental data [Citation33,Citation34] may be necessary to determine whether it is likely that (Ti,Mo)
AlC
solid solutions are indeed stabilized at high temperatures.
3.2. Alloying Trends in (Ti,Zr)AlC MAX alloys
Recently, Tunca et al. [Citation26] considered additions of Ti—over the entire composition range—into the Zr–Al–C system with the focus on synthesizing (ZrTi
)
AlC
and (Zr
Ti
)
AlC MAX phase solid solutions. Precursor powders (ZrH
, TiH
, Al and C) were used to synthesize the target MAX alloys through both hot-pressing and pressureless sintering methods. XRD analysis of the hot-pressed sintered samples as a function of Ti content and hot-pressing temperature (ranging from 1300
C to 1700
C) indicate that in the entire composition-temperature synthesis space there was no condition that resulted in a single phase.
In samples with Ti content close to 50% the dominant phase was (ZrTi
)
AlC, with a significant amount of secondary phases as well as (Zr
Ti
)
AlC
when hot pressing at intermediate temperatures. At elevated temperatures and at Ti amounts close to 0% or to 100%, (Zr
Ti
)
AlC
was the dominant MAX solid solution, although the samples characterized contained most of the times significant amounts of non-MAX secondary phases [Citation26]. Analysis by Dahlqvist et al. [Citation27] indicates that (Zr
Ti
)
AlC competes with binary subsystems (TiC, Zr
Al
,Zr
Al
), while (Zr
Ti
)
AlC
is definitely metastable with regards to the end-members Zr
AlC
and Ti
AlC
.
The considerable amounts of non-MAX secondary phases during hot-pressing synthesis were attributed to liquid losses arising from invariant reactions involving the liquid phase and intermetallics in the Zr–Al system [Citation26]. Presureless sintering, however, did not yield more phase-pure samples, potentially due to the extreme sensitivity of phase stability of MAX phases to even the smallest deviations in stoichiometry and the disrupted diffusion in highly porous samples since during pressureless reaction sintering the samples are highly porous so thus it is hard to achieve chemical homogeneity. Presureless sintered samples with overall 50% Ti, however, offered a significant yield of (ZrTi
)
AlC
. Rietveld refinement suggests that samples with x
exhibited significant deviations from Vegard's Law, which was interpreted as possible ordering, with Zr and Ti occupying (preferentially) the 2a- and 4f- Wyckoff sites, respectively. HAADF STEM analysis provided strong confirmation of the previously hypothesized ordered configuration.
Figure shows the energetics of mixing in the M sublattice in the (ZrTi
)
AlC
system, which exhibits rather strong endothermic interactions between Zr and Ti, in perfect agreement with Dahlqvist et al. [Citation27] but, unfortunately, in perfect disagreement with the experimental results of Tunca et al. [Citation26]. Here, we would like to clarify that while Tunca provides evidence for out-of-plane ordering, partitioning among the 2a and 4f-sites may not be perfect and in fact Tunca reports this ordering as a tendency for preferential occupancy in one or the other sublattice. From the theoretical perspective, such partial partitioning could be accounted for through Monte Carlo simulations [Citation35] but this is outside the scope of the present work.
Figure 2. (color online) Energetics of Zr–Ti mixing in (Ti,Zr)AlC
MAX alloys predicted through the combination of cluster expansion lattice searches with DFT calculations—for a definition of the energy units (energy per M-site) refer to Supplementary Material. Calculated ground states are indicated by the blue markers and convex hull is indicated by solid blue line. Non-ground state structures are depicted by red markers with random energy—defined in Eq. S2 of Supplementary Material—shown as green dashed line. Structure marked with magenta circle corresponds to the Ti
ZrAlC
observed by Tunca et al. [Citation26]. (insets) Model crystal structure and the HAADF STEM intensity profile and HRTEM micrographs, with atomic layers enclosed by green rectangle corresponding to 413 stacking faults—reproduced (adapted) with permission from Tunca et al. [Citation26], copyright (2015) American Chemical Society.
![Figure 2. (color online) Energetics of Zr–Ti mixing in (Ti,Zr)AlC MAX alloys predicted through the combination of cluster expansion lattice searches with DFT calculations—for a definition of the energy units (energy per M-site) refer to Supplementary Material. Calculated ground states are indicated by the blue markers and convex hull is indicated by solid blue line. Non-ground state structures are depicted by red markers with random energy—defined in Eq. S2 of Supplementary Material—shown as green dashed line. Structure marked with magenta circle corresponds to the TiZrAlC observed by Tunca et al. [Citation26]. (insets) Model crystal structure and the HAADF STEM intensity profile and HRTEM micrographs, with atomic layers enclosed by green rectangle corresponding to 413 stacking faults—reproduced (adapted) with permission from Tunca et al. [Citation26], copyright (2015) American Chemical Society.](/cms/asset/2aaaced8-dbf8-45cf-a698-8386c67b3dbf/tmrl_a_1380723_f0002_c.jpg)
To gain a better understanding of the predicted positive mixing enthalpies in this system, one can consider the following: Ti and Zr are isoelectronic, with minimal chemical interaction between them. On the other hand, the (metallic) atomic radii of Zr (1.60Å [Citation36]) is about 9% larger than that of Ti (1.47Å [Citation36]). This difference falls within the Hume-Rothery limit for solid solutions (15%) and when including comparisons of electronegativity (1.54 (Zr) vs 1.33 (Ti) [Citation36]) and valency, a cursory analysis would suggest that this system would tend to form random mixtures in the M sublattice. Yet, it seems that a 9% difference in atomic radii is sufficient to result in positive/endothermic strain energy contributions when Zr and Ti randomly mix in the and
sublattices.
Ignoring for a moment chemical/electronic effects—Zr and Ti are isoelectronic and there should not be strong tendency towards charge exchange between them [Citation27]—the mixing energy in the M sublattices, per M site, of (ZrTi
)
AlC
has a significant contribution from elastic interactions and, ignoring crystal anisotropy, can be expressed as—see [Citation37,Citation38]:
(1) where
is the (average) atomic volume of (Zr
Ti
)
AlC
,
is (average) Young's modulus and ν is Poisson's ratio. Using Vegard's Law, we can calculate the variation in the mismatch ϵ with Zr content
:
(2) where
corresponds to the lattice parameter of the solution assuming Vegard's Law and
is the atomic volume of Zr
AlC
. Equation (Equation1
(1) ) shows that if one equates the mixing energy to the strain energy resulting from lattice mismatch, the former is expected to be symmetrical around
, which is consistent with Figure .
Yet, while one can use these arguments to rationalize our and others'[Citation27] calculations, the fact remains that these results are at odds with the observations by Tunca et al. [Citation26], who have convincingly showed out of plane ordering in this system, at the TiZrAlC
stoichiometry. Whenever encountering such discrepancies between theory/calculations and experiments, one is compelled to either resolve the controversy or to establish limits to the predictive abilities of the theories used.
3.3. Order vs. phase separation energetics
As indicated by our calculations, in the (Ti,Zr)AlC
system, the predicted equilibrium state (phase-separating mixture of Ti
AlC
and Zr
AlC
domains) does not correspond to the observed state (out-of-plane ordered compound). While in some instances it is safe to invoke kinetic considerations as the reason for this discrepancy, a better approach is to consider possible changes to the thermodynamic boundary conditions prevalent during the formation of the system under study.
To be more specific about this, our alloy theoretic calculations allowed for each configuration to relax (see Supplementary Material) to the zero pressure volume (at 0 K). On the other hand, the synthesis by Tunca et al. [Citation26] may have resulted in conditions that differed from these isobaric/isothermal bulk conditions. In fact, there is considerable literature on the influence of non-isothermal/isobaric thermodynamic constraints on phase stability [Citation39–43]. These constraints can arise from strain, size, interfacial effects, etc. and they can result in dramatic changes in the phase stability of a system. Just recently, for example, one of the present authors and collaborators showed how epitaxial constraints in multi-layered Mg–Nb films were sufficient to stabilize Mg in its bcc form, while under bulk conditions pressures above 20 GPa are necessary to do the same [Citation42].
In order to make sense of these findings, a formal analysis of how changes in thermodynamic boundary conditions modify a system's phase stability is necessary. A decade ago, Liu and Zunger [Citation44] developed a theory (see Figure ) to rationalize the influence of epitaxial conditions on the phase stability of AB
C semiconductor alloys grown into ultra-thin films. This analysis was applied to the cubic (Ga,In)N phase-separating system, which exhibits strong tendencies towards the formation of ordered superlattices when epitaxial constraints are imposed. In fact, Liu and Zunger [Citation44] predicted that, under such constraints, (Ga,In)N would form a series of ordered structures instead of undergoing phase separation (at low temperatures).
Figure 3. (color online) Schematic plot for alloy energetics in bulk-incoherent and bulk-coherent alloys. Due to the large difference in atomic radii between Ti and Zr, an out-of-plane ordered structure with TiZrAlC
stoichiometry would have a higher energy than the incoherent phase-separated state, Ti
AlC
+Zr
AlC
, i.e.
. On the other hand, when coherent constraints are imposed in the bulk, the strain energy cost that must be incurred in order to maintain lattice coherence (
) makes the ordered state favorable, relative to the coherent, phase-separated state,
. Note that a
corresponds to the lattice parameter of the coherent configurations.
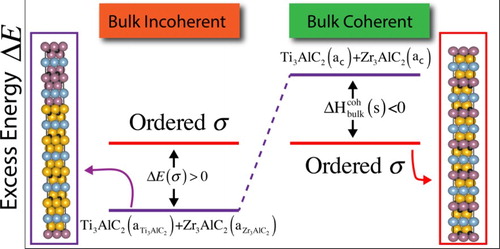
Following the analysis by Liu and Zunger [Citation44], we start by defining two characteristic energies: the excess energy of the unconstrained system, , and that of the system under constraints,
.
is the total energy difference between the configuration σ—in this case the out-of-plane ordered Ti
ZrAlC
compound—at its equilibrium lattice parameters, aσ, and the pure end members, Ti
AlC
and Zr
AlC
, at their own, unconstrained lattice parameters:
(3) where
for this very specific case. The excess energy,
, is the formation energy (positive in this case) of configuration σ relative to the end members and in this very specific case has a value of approximately 60 meV/M-site—this state corresponds to the circled marker in Figure . At this level of energy, one would need to have approximately a thermal energy of about 800
C to stabilize a disordered state. One can think of this
as a measure of the cohesive tendency for the configuration σ. In this case, this is positive because of the positive energy contributions arising from the large mismatch in atomic radii between Ti and Zr.
on the other hand, corresponds to the relative energy difference between the compound/configuration σ under constraints, relative to the corresponding end-members under the same constraining conditions:
(4)
As in Liu and Zunger [Citation44], we use this to compare the thermodynamics of two possible states, described graphically in Figure :
Bulk-incoherent states: when there are no constraints whatsoever in the growth of theTi
Bulk-coherent states: When, for whatever reason, a 3D bulk alloy system maintains its coherence or is forced to grow under constraints, the constituents of the pseudo-binary, Ti
To test whether the latter scenario could be used to explain the ordering behavior in the (Ti,Zr
)
AlC
system observed by Tunca et al. [Citation26], we proceeded to carry out the following analysis: assuming that the system was constrained to have the lattice parameters as prescribed by Vegard's Law—applied to the lattice parameters of the end members as calculated in this work—we calculated the total energies of two configurations. One configuration corresponded to two unit cells of Ti
AlC
stacked atop a unit cell of Zr
AlC
, while the second configuration corresponded to three unit cells of an ordered structure,Ti
ZrAlC
, in which Zr occupied the 2a-sites (
sublattice) while Ti occupied the 4f-sites (
sublattice), as reported by Tunca et al. [Citation26]. These two configurations are shown in Figure .
Table shows the results of the calculations. In this case, we assume that the system was constrained to have the lattice parameters as prescribed by Vegard's Law with the correct 2:1 stoichiometry between Ti and Zr. As indicated in the table, our calculations suggest that the difference in energy between a constrained ordered configuration and a constrained configuration in which the two compositions are phase separated is negative, with a value of −27 meV/M-site, or –162 meV per formula unit. This difference in energy is quite significant, and represents a shift in energy of approximately 90 meV/M-site, relative to the formation energy for the ordered TiZrAlC
configuration considering incoherent end-members as shown in Figure . While there may be questions with regard to the assumed constraints (Tunca et al. [Citation26] report deviations from Vegard's Law at x
), the calculations provide at least a plausible explanation for the observed out-of-plane ordering in the (Ti
,Zr
)
AlC
system may be induced by coherency constraints.
Table 1. DFT-derived energetics as well as M′-X and M′′-X layer separation in phase-separated (2 Ti3AlC2 + 1 Zr3AlC2) and ordered (3 Ti2AlC2) constrained configurations, assuming lattice parameters derived from Vegard's Law, a/c = 2/3 a/cTi3AlC2 + 1/3 a/cZr3AlC2
To gain a better understanding of the structural/atomic relaxation basis for the energy difference between the constrained ordered and phase-separated configurations, we have analyzed the interlayer separation between the (2a-sites) and
(4f-sites) sublattices and the X layer in both configurations. Table shows that, for the case of the ordered configuration, the 2a-X and 4f-X interlayer separation, corresponding to Zr–X and Ti–X layer pairs, is 1.475 and 1.033 Å, respectively. Since Ti and Zr are isoelectronic, and theoretical analysis by Dahlqvist et al. [Citation27] indicates that the electron charge per atom is almost identical in the case of TiZr
AlC
and Ti
Zr
AlC
compounds, regardless of the local environment (i.e. Wyckoff site), the difference in interlayer separation can be attributed mostly to differences in atomic radii. Under the constrained conditions, these interlayer distances are to be considered the most stable separation necessary to maintain the c-lattice parameter at the value corresponding to Vegard's Law.
In the case of the phase-separating configuration, on the other hand, we observe two sets of 2a-X and 4f-X interlayer separation. When the 2a site is occupied by Ti, the distance is 1.292 Å, which is considerably larger than the 1.033 Å distance separating Ti and C when Ti occupies the 4f-sites in the ordered compound. In the case of Zr occupancy of the 2a-site, the distance is instead 1.441 Å, which is only slightly shorter than the distance of 1.475 Å f or the Zr-X layer pair in the ordered compound. The 4f-X interlayer separation distances in the phase-separating configuration are 1.033 and 1.278 Å when Ti and Zr occupy the 4f-sites, respectively. In this case, the Zr-X distance is shorter than the Zr-X distance when Zr occupies the 2a-site in both the phase-separating and ordering configuration. We note, however, that the average M-X interlayer distance is identical in both configurations, when considering -X and
-X layer pairs in an equal footing.
From this analysis it seems that local deviations from the stable 2a-X and 4f-X interlayer separation observed in the ordered compound are responsible for the destabilization of the phase-separated state relative to the ordered configuration under constrained conditions. These results, while not conclusive, seem to suggest that the observed ordered TiZrAlC
configuration is the result of constrained conditions during the synthesis of these materials.
3.4. On the possible origin of coherency constraints
While our analysis presented above provides convincing evidence that a constrained ordered state at the TiZrAlC
composition is more stable than a phase-separating state under the same constraints, we have not discussed where these constraints may come from.
From the synthesis conditions reported by Tunca et al. [Citation26], one could assume that there was a possibility for the reacted powders to form a random solid solution, provided the disordering temperature is below the 1300–1700C range used in the sintering process.
The disordering temperature can be calculated as follows:
(5) where
is the mixing energy (or enthalpy at 0K) at the
composition, while
is given by
(6) with
and
given per M-site.
From the calculated values for the (random) energy of mixing at (
meV/M-site), one can estimate a disordering temperature of about 1350
C, which lies at the lower bound of the sintering temperatures [Citation26].
After reacting to form a quaternary (Ti,Zr)AlC
random solid solution, there would be a driving force for phase separation as the samples are cooled to room temperature. The average lattice parameter, though, would correspond to the one given by Vegard's Law, which is the constraint assumed in our calculations. Under this constraint, phase separation would be energetically unfavorable and instead out-of-plane ordering would be the preferred (metastable) configuration. An implication of this (admittedly ad hoc) model is that over longer times, long-range diffusion would relax the coherency constraint and the system would begin to phase separate.
Before moving on, we would like to point out that one further prediction that arises from the strain-ordering theory is the necessary presence of planar defects that may accommodate some of the significant strains associated with coherency between 2a-Zr and 4f-Ti layers. Tunca et al. [Citation26] in fact observed such defects in the so-called 312 grains composed of ordered TiZrAlC
configurations. Figure shows one observed 413 stacking fault in a 312 grain, likely originating from the need to relax some of the strain associated with coherency constraints.
3.5. Further investigation of ordering trends
In order to arrive at a clearer picture of the ordering trends in systems with possible out-of-plane ordering between the and
sublattices in (
,
)
AlC
systems, we proceeded to investigate the lattice system involving mixing between Ti and Nb, Hf, respectively.
Figure shows the alloy energetics for the (Ti,Hf)AlC
MAX alloy system. The system, as in the case of mixing between Ti and Zr, exhibits endothermic interactions, with the mixtures of the end members Ti
AlC
and Hf
AlC
being the most stable configurations for all compositions. These results are to be expected given the fact that Hf is isoelectronic to Ti and Zr. What is unexpected, however, is the fact that even though the
ratio is similar to
(see Table ), the energy of the Ti
HfAlC
ordered configuration is actually much closer to the convex hull formed by Ti
AlC
-Hf
AlC members, indicating a much lower tendency for phase separation, corresponding to about 182 K. Table shows that the difference in (Pauling) electronegativity between Ti and Zr and Ti and Hf is identical.
Figure 4. (color online) Energetics of Hf-Ti mixing in (Ti,Hf)AlC
MAX alloys predicted through the combination of cluster expansion lattice searches with DFT calculations—for a definition of the energy units (energy per M-site) refer to Supplementary Material. Calculated ground states are indicated by the blue markers and convex hull is indicated by solid blue line. Green dashed line corresponds to the random mixing energy derived from the cluster energies. Bottom-left inset shows the pair cluster energies as a function of radius, while bottom-right inset provides an indication of the goodness of fit.
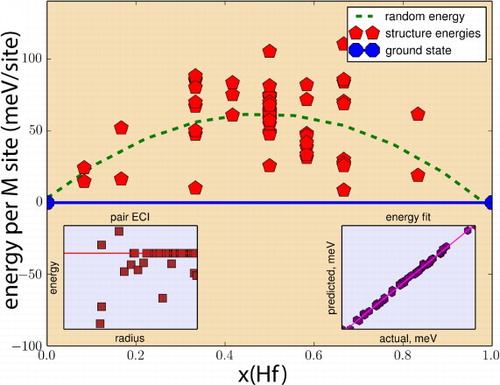
Table 2. Atomic properties of Ti, Zr, Hf [Citation36].
Tunca et al. [Citation26] put forward two possible reasons for the preference for out-of-plane ordered configurations in TiMAlC
MAX quaternary compounds: (a) differences in electronegativity and/or (b) differences in atomic radii, the latter possibility termed as the steric-based hypothesis. In the case of the Zr–Ti, the differences are somewhat significant (although the difference in atomic radii is well within the Hume–Rothery limit). Their analysis of ordered compounds reported to date suggests that M elements with the smallest(largest) atomic radii (
) and highest(lowest) electronegativity (
) would tend to partition preferentially to the 4f-sites(2a-sites). While these results seem to hold for the observed compounds, the comparison with the Hf–Ti system is confounding because even though differences in atomic radii and electronegativities are very similar, the (Ti,Hf)
AlC
exhibits a much weaker tendency to phase separate than (Ti,Zr)
AlC
.
Table 3. Energetics for different stacking configurations in the Ti--Zr, Ti--Hf, Ti--Nb and Ti--Mopseudo-binary systems for Ti2MAlC2 and TiM2AlC2 stoichiometries. The table only contains stacking sequence involving 2a- and 4f-M sites, with the 2a sites enclosed by parentheses. Energy/M-site is calculated as per definition in Supplementary Material.
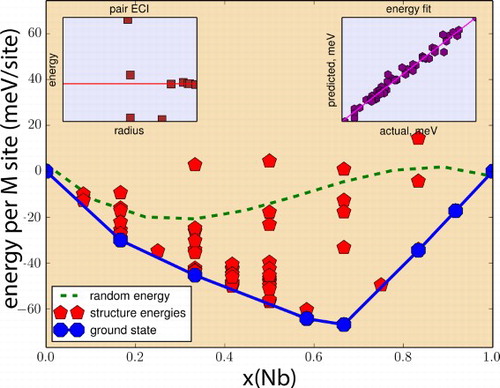
Things are more complicated, however, when considering the (Ti,Zr), (Ti,Nb) and (Ti,Mo) sequence. In this case, the three systems have almost identical atomic radii ratios (see Table ) and yet, when comparing Figures , and , it is clear that for similar atomic radii ratios, the alloying trends go from phase-separation to strong ordering as one moves up in periodic groups. Examining the differences in electronegativity (Ti–1.54, Zr–1.33, Nb–1.6, Mo–2.16) also confound the results since the difference in electronegativity between Ti and Zr is higher than the difference in electronegativity between Ti and Nb. Yet, as Figure shows, the ordering trend in the Ti–Nb system is intermediate to those of the Ti–Zr and Ti–Mo system.
Figure 5. (color online) Energetics of Nb–Ti mixing in (Ti,Nb)AlC
MAX alloys predicted through the combination of cluster expansion lattice searches with DFT calculations—for a definition of the energy units (energy per M-site) refer to Supplementary Material. Calculated ground states are indicated by the blue markers and convex hull is indicated by solid blue line. Green dashed line corresponds to the random mixing energy derived from the cluster energies. Top-left inset shows the pair cluster energies as a function of radius, while top-right inset provides an indication of the goodness of fit.
The analysis by Dahlqvist et al. [Citation27] provides an explanation for these trends. As shown in Figure S2 (Supplementary Material), the analysis of the site-projected electronic charges shows that for TiMAlC
configurations, the difference in atomic charges between 2a-sites (occupied by Ti) and 4f-sites (occupied by Zr, Nb and Mo) increases as one moves along the Zr → Nb → Mo direction. The (electro-positive) charge per atom for Ti occupying the 2a-site remains the same and the loss of charge in the 4f-sites according to the Zr → Nb → Mo sequence results in more electro-positive Al, which in turn reduces the number of electrons available for populating the anti-bonding Al–Al states, increasing the stability of the system. Comparison of the atomic charges in the TiZr
AlC
and TiHf
AlC
and the alloy energetics as shown in Figures and shows, however, that this bonding argument is not the entire picture.
To close the present discussion, Table lists the energetics for different M-stacking configurations for stoichiometries TiMAlC
and TiM
AlC
in the Ti–Zr, Ti–Hf, Ti–Nb and Ti–Mo system. The results indicate that the most stable configurations for the Ti
MAlC
and TiM
AlC
stoichiometries in the Ti–Zr and the Ti–Hf system have M in the 2a-sublattice as the most stable configuration. The energy landscape, however, is very different when comparing the configuration degeneracy in the TiM
AlC
Ti–Zr/Hf systems to the rather sharp energy separation between the most stable configuration and those following in the case of the Ti
MAlC
system. This makes sense as excess of Zr/Hf relative to Ti in the Ti
MAlC
systems has to be accommodated, by force, by placing M atoms in 4f-sites as opposed to exclusively 2a-sites. Any energetic advantage derived from exclusive occupancy of 4f-sites by Ti is lost and therefore several configurations have essentially the same energy.
The behavior is qualitatively different when examining the Ti–Mo and Ti–Nb systems. In this case, the most stable configuration with the TiMAlC
stoichiometry is much more stable (twice in absolute temperature) than the next following configuration. In this case, there is a very strong preference for partitioning of Ti into 2a-sites, with Nb or Mo occupying exclusively the 4f-sites. This ordering sequence is more complicated in the case of the Ti
MAlC
stoichiometry, when it is evident that the energy separation between the most stable state and the next following tends to be much closer. More importantly, in both cases, the most stable configuration for the Ti
MoAlC
and Ti
NbAlC
stoichiometries correspond to compounds with both in-plane and out-of-plane ordering.
Finally, we return to the discussion of the effect of constraints on the stability of phase-separated states relative to out-of-plane ordered configurations but extend the analysis to the Ti–Hf, Ti–Mo and Ti–Nb systems. Table compares the energetics of phase-separated and out-of-plane ordered states under Vegard's Law constraints for all the TiMAlC
systems under study in this work. The table indicates the following: in the case of Ti
ZrAlC
and Ti
HfAlC
, under constraints, the ordered states are more stable than the phase-separated states. The Ti
NbAlC
and Ti
MoAlC
systems are also shown to be more stable than the phase-separated states, although in this case these systems are less stable than when comparing the fully relaxed ordered structures relative to the fully relaxed end-members. This is more noticeable for Ti
MoAlC
, in which, under constraints, the ordered configuration is more stable than the phase-separated one by approximately 100 meV/M-site, while in the unrelaxed states, the stability of this compound relative to its end members is more than 350 meV/M-site. In the case of the fully relaxed states, as shown in Table S1 of the Supplementary Material, the Ti
MoAlC
undergoes a considerable volume reduction upon ordering and the Vegard's Law constraints subject the configuration to considerable tensile strains.
Table 4. Comparison between phase-separated and layered (out-of-plane ordered) states in Ti2MAlC2 systems under Vegard's Law constraints.
Table 5. Comparison between phase-separated and layered (out-of-plane ordered) states in TiM2AlC2 systems under Vegard's Law constraints
This behavior is not seen in the case of the TiMAlC
systems as shown in Table 5. In this case, the ordered TiZr
AlC
and TiHf
AlC
states are less stable than the phase-separated states, implying that the constraints as considered in this work do not always result in stabilization of ordered states relative to phase separation. In the case of the Ti
HfAlC
system, this result is surprising as in the relaxed state, the ordered configuration lies only about 10 meV/M-site above the Convex Hull. Table 5 shows that the TiMo
AlC
and TiNb
AlC
are, again, more stable than the corresponding phase-separated states. In this case, the stability is very similar to the formation energies of the relaxed ordered states with respect to the relaxed end-members.
These results point to the need to further investigate, in a more systematic manner, the effects of constraints on the relative stability of phase-separated and ordered states in MAX quaternary and higher order compounds. Further analysis, however, is beyond the scope of this work.
4. Conclusions
In this paper, we have used a DFT-based alloy theoretic approach to investigate the out-of-plane ordering phenomenon already observed in several quaternary MAX alloy systems. The following can be concluded from the results presented above:
The cluster expansion-based alloy theoretic analysis confirmed the strong tendency for TiMo
By invoking a thermodynamic analysis based on the comparison of constrained vs. non-constrained systems, we have explained the apparent discrepancy between the predicted (by us and others) tendency towards phase separation in the (Ti,Zr)
Different theories for the physical underpinning for the observed ordering tendencies were examined in light of the results not only for the Ti–Zr and Ti–Mo systems, but also by including Ti–Hf and Ti–Nb. In the case of the isoelectronic systems Ti–Zr and Ti–Hf, differences in atomic radii were sufficient to favor phase separation over ordering, although the results are inconclusive when considering the much weaker tendency towards phase separation of Ti–Hf as compared to Ti–Zr.
The results are important as they show that it is not possible to rely only on incoherent alloy theoretic approaches when attempting to confirm or rule out the possibility for out-of-plane ordering in MAX phases.
Supplementary_Material
Download PDF (422.5 KB)Acknowledgments
First-principles calculations by were carried out at the Texas A& M Supercomputing Facility at Texas A&M University as well as the Texas Advanced Computing Center (TACC) at the University of Texas at Austin. The Texas A&M Materials Modeling Automation Library (TAMMAL) developed by R.A., A.T. and collaborators (soon to be released to the general scientific community) was used to carry out the HT cluster expansion calculations. The ATAT package [Citation28] was used to carry out the cluster expansions.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
R. Arróyave http://orcid.org/0000-0001-7548-8686
A. Talapatra http://orcid.org/0000-0002-6446-2437
T. Duong http://orcid.org/0000-0001-8694-814X
W. Son http://orcid.org/0000-0002-4158-7181
M. Radovic http://orcid.org/0000-0003-4571-2848
Additional information
Funding
Related Research Data
References
- Barsoum MW. MAX phases: properties of machinable ternary carbides and nitrides. Weinhem, Germany: Wiley-VCH Verlag GmbH; 2013.
- Radovic M, Barsoum MW. MAX phases: bridging the gap between metals and ceramics. Am Ceram Soc Bull. 2013;92(3):20–27.
- Barsoum MW. The MAX phases: A new class of solids: thermodynamically stable nanolaminates. Prog Solid State Chem. 2000;28(1):201–281. doi: 10.1016/S0079-6786(00)00006-6
- Barsoum MW, Radovic M. Mechanical properties of the MAX phases. In: Buschow KHJ, Cahn R, Flemings M, Ilschner B, Kramer E, Mahajan S, Veyssiere P, editors. Encyclopedia of Materials: Science and Technology. Amsterdam: Elsevier; 2004. p. 1–16.
- Barsoum MW, Radovic M. Elastic and mechanical properties of the MAX phases. Ann Rev Mater Res. 2011;41:195–227. doi: 10.1146/annurev-matsci-062910-100448
- Sun Z. Progress in research and development on MAX phases: a family of layered ternary compounds. Int Mater Rev. 2011;56(3):143–166. doi: 10.1179/1743280410Y.0000000001
- Barsoum M. The MAX Phases and Their Properties. In: Ceramics Science and Technology Volume 2: Materials and Properties. Wiley Online Library; 2010. p. 299–347.
- Aryal S, Sakidja R, Barsoum MW, et al. A genomic approach to the stability, elastic, and electronic properties of the MAX phases. Phys Status Solidi (B). 2014;251(8):1480–1497. doi: 10.1002/pssb.201451226
- Naguib M, Bentzel G, Shah J, et al. New solid solution MAX phases:(Ti,V)AlC, (Nb,V)AlC, (Nb, V)AlC and (Nb,Zr)AlC. Mater Res Lett. 2014;2(4):233–240. doi: 10.1080/21663831.2014.932858
- Talapatra A, Duong T, Son W, et al. High-throughput combinatorial study of the effect of m site alloying on the solid solution behavior of MAlC MAX phases. Phys Rev B. 2016;94(10):104106. doi: 10.1103/PhysRevB.94.104106
- Arróyave R, Talapatra A, Duong T, et al. Does aluminum play well with others? intrinsic al-a alloying behavior in 211/312 MAX phases. Mater Res Lett. 2017;5(3):170–178.
- Ashton M, Hennig RG, Broderick SR, et al. Computational Discovery of Stable MAX phases. Phys Rev B. 2016;94(5):054116. doi: 10.1103/PhysRevB.94.054116
- Burr PA, Horlait D, Lee WE. Experimental and DFT investigation of (Cr, Ti)AlC MAX phases stability. arXiv preprint arXiv:160706162. 2016.
- Li Y, Ding Y, Xiao B, et al. Anisotropic electrical and lattice transport properties of ordered quaternary phases CrTiAlC and MoTiAlC. A first principles study. Phys Lett A. 2016;380(44):3748–3755. doi: 10.1016/j.physleta.2016.09.015
- Qing-He G, Zhi-Jun X, Ling T, et al. Evidence of the stability of MoTiAlC from first principles calculations and its thermodynamical and optical properties. Comput Mater Sci. 2016;118:77–86. doi: 10.1016/j.commatsci.2016.03.010
- Mockute A, Dahlqvist M, Emmerlich J, et al. Synthesis and ab initio calculations of nanolaminated (Cr, Mn)AlC compounds. Phys Rev B. 2013;87(9):094113. doi: 10.1103/PhysRevB.87.094113
- Cabioc'h T, Eklund P, Mauchamp V, et al. Tailoring of the thermal expansion of Cr 2 (Al,Ge)C phases. J Eur Ceram Soc. 2013;33(4):897–904. doi: 10.1016/j.jeurceramsoc.2012.10.008
- Bei G, Pedimonte BJ, Fey T, et al. Oxidation behavior of MAX phase TiAlSnC solid solution. J Am Ceram Soc. 2013;96(5):1359–1362. doi: 10.1111/jace.12358
- Yu W, Li S, Sloof WG. Microstructure and mechanical properties of a CrAl(Si) C solid solution. Mater Sci Eng: A. 2010;527(21):5997–6001. doi: 10.1016/j.msea.2010.05.093
- Gao H, Benitez R, Son W, et al. Structural, physical and mechanical properties of Ti(AlSi)C solid solution with x=0–1. Mater Sci Eng: A. 2016;676:197–208. doi: 10.1016/j.msea.2016.08.098
- Horlait D, Middleburgh SC, Chroneos A, et al. Synthesis and DFT investigation of new bismuth-containing MAX phases. Sci Rep. 2016;6(1):195. doi: 10.1038/srep18829
- Liu Z, Wu E, Wang J, et al. Crystal structure and formation mechanism of (CrTi)AlC MAX phase. Acta Mater. 2014;73:186–193. doi: 10.1016/j.actamat.2014.04.006
- Anasori B, Halim J, Lu J, et al. MoTiAlC: a new ordered layered ternary carbide. Scripta Mater. 2015;101:5–7. doi: 10.1016/j.scriptamat.2014.12.024
- Anasori B, Xie Y, Beidaghi M, et al. Two-dimensional, ordered, double transition metals carbides (MXenes). ACS Nano. 2015;9(10):9507–9516. doi: 10.1021/acsnano.5b03591
- Meshkian R, Tao Q, Dahlqvist M, et al. Theoretical stability and materials synthesis of a chemically ordered MAX phase, MoScAlC, and its two-dimensional derivate MoScC MXene. Acta Mater. 2017;125:476–480. doi: 10.1016/j.actamat.2016.12.008
- Tunca B, Lapauw T, Karakulina OM, et al. Synthesis of MAX phases in the Zr-Ti-Al-C system. Inorg Chem. 2017;56(6):3489–3498. doi: 10.1021/acs.inorgchem.6b03057
- Dahlqvist M, Rosen J. Order and disorder in quaternary atomic laminates from first-principles calculations. Phys Chem Chem Phys. 2015;17(47):31810–31821. doi: 10.1039/C5CP06021D
- Van de Walle A, Asta M, Ceder G. The alloy theoretic automated toolkit: a user guide. Calphad. 2002;26(4):539–553. doi: 10.1016/S0364-5916(02)80006-2
- Connolly J, Williams A. Density-functional theory applied to phase transformations in transition-metal alloys. Phys Rev B. 1983;27(8):5169–5172. doi: 10.1103/PhysRevB.27.5169
- Anasori B, Dahlqvist M, Halim J, et al. Experimental and theoretical characterization of ordered MAX phases MoTiAlC and MoTiAlC. J Appl Phys. 2015;118(9):094304. doi: 10.1063/1.4929640
- Saunders N. The Al-Mo system (Aluminum–Molybdenum). J Phase Equilib. 1997;18(4):370–378. doi: 10.1007/s11669-997-0063-1
- Cadoff I, Nielsen JP. Titanium–Carbon phase diagram. J Met. 1953;5:1564.
- Duong TC, Talapatra A, Son W, et al. On the stochastic phase stability of TiAlC–CrAlC. Sci Rep. 2017;7:5138. doi: 10.1038/s41598-017-05463-1
- Duong TC, Talapatra A, Son W, et al. On the stochastic phase stability of TiAlC-CrAlC. arXiv preprint arXiv:170d402500, 2017.
- Arróyave R, Radovic M. Ab initio investigation of TiAl(C,N) solid solutions. Phys Rev B. 2011;84(13):134112. doi: 10.1103/PhysRevB.84.134112
- Winter M. Webelements, 1993. Available from: http://www.webelements.com/.
- Barabash S, Ozolins V, Wolverton C. First-principles theory of the coherency strain, defect energetics, and solvus boundaries in the PbTe–AgSbTe system. Phys Rev B. 2008;78(21):214109. doi: 10.1103/PhysRevB.78.214109
- Balluffi RW, Allen S, Carter WC. Kinetics of materials. Hoboken, NJ: Wiley; 2005.
- Van der Ven A, Garikipati K, Kim S, et al. The role of coherency strains on phase stability in Li FePO. Needle crystallites minimize coherency strain and overpotential. J Electrochem Sco. 2009;156(11):A949–A957. doi: 10.1149/1.3222746
- Wagemaker M, Mulder FM, Van der Ven A. The role of surface and interface energy on phase stability of nanosized insertion compounds. Adv Mater. 2009;21(2526):2703–2709. doi: 10.1002/adma.200803038
- Ham B, Junkaew A, Arroyave R, et al. Hydrogen sorption in orthorhombic Mg hydride at ultra-low temperature. Int J Hydrogen Ener. 2013;38(20):8328–8341. doi: 10.1016/j.ijhydene.2013.04.098
- Junkaew A, Ham B, Zhang X, et al. Stabilization of bcc mg in thin films at ambient pressure: experimental evidence and ab initio calculations. Mater Res Lett. 2013;1(3):161–167. doi: 10.1080/21663831.2013.804218
- Mayrhofer PH, Hörling A, Karlsson L, et al. Self-organized nanostructures in the ti–al–n system. Appl Phys Lett. 2003;83(10):2049–2051. doi: 10.1063/1.1608464
- Liu JZ, Zunger A. Thermodynamic states and phase diagrams for bulk-incoherent, bulk-coherent, and epitaxially-coherent semiconductor alloys: Application to cubic (Ga, In) N. Phys Rev B. 2008;77(20):205201.