
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The unusual mechanical and radiation properties found in concentrated solid–solution alloys (CSAs) make them promising candidate materials in nuclear applications. In this work, He behavior in a typical CSA, NiCFeCr, is assessed by first-principles calculations. Our results suggest that the energy difference of He in substitutional and interstitial sites is greatly reduced. The energy barriers of He through both interstitial and substitutional diffusion are much higher than those in pure Ni, indicating slower He dynamics in CSAs. The calculated activation energies show that the dissociation mechanism is more favorable in NiCoFeCr, which helps to suppress He growth as observed experimentally.
GRAPHICAL ABSTRACT
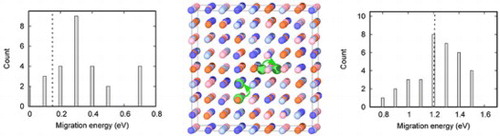
IMPACT STATEMENT
For the first time, ab initio calculations reveal that the ease of He-vacancy cluster dissociation is the main mechanism responsible for the suppression of He bubble growth in concentrated NiCoFeCr alloys.
Concentrated solid–solution alloys (CSAs), which are composed of several different metallic elements in equimolar or near-equimolar fractions, have demonstrated excellent mechanical properties and promising radiation resistance [Citation1–5]. These merits make CSAs promising candidate structural materials applicable in next-generation advanced nuclear systems. In the service environment of nuclear reactors, helium (He) will be inevitably introduced into the matrix by the (n, α) nuclear transmutation reaction. Since He is insoluble in metallic alloys, its diffusion and aggregation usually lead to He precipitation and He bubbles, which ultimately may cause material degradation because of He embrittlement [Citation6]. Therefore, it is of critical significance to understand the behavior of He in CSAs before they can be used in nuclear applications.
Several He implantation experiments have been carried out to explore the He resistance of selective CSAs (predominantly in NiCoFeCr) [Citation7–9]. In general, experiments show that the NiCoFeCr CSA exhibits better resistance to He bubble growth compared to both pure Ni and steels, as evidenced by the observed smaller bubble size and lower bubble density. The small He bubbles in CSAs is reasoned as a result of the low He mobility that influences the nucleation, migration and growth of He bubbles [Citation7,Citation8]. It is argued that similar to the sluggish self-diffusion which is a unique feature of high-entropy alloys [Citation10], diffusion of He will encounter significant local traps and blocks in NiCoFeCr that leads to a high activation energy. However, the diffusion of vacancies, which is believed to have a close relation to the He diffusion, actually possesses low migration barriers in NiCoFeCr [Citation11]. These results highlight the need to look into the detailed diffusion mechanism of He in chemically-disordered CSAs.
In this work, we perform first-principles calculations to study He behavior in a typical CSA of NiCoFeCr. The structure of the CSA is represented by a special quasirandom structure (SQS). The formation and migration energies of He in NiCoFeCr are obtained and compared to those in Ni. Our results suggest that the formation energies of He in substitutional and interstitial sites are similar in NiCoFeCr, thus site preference is greatly reduced due to the disordered states. The migration barriers of He are found much higher than those in pure Ni, indicating a slower He diffusion in CSAs. Based on activation energy analysis, we argue that the dissociation of He-vacancy complex is favorable in NiCoFeCr, making it difficult to nucleate He bubbles compared to that in pure Ni.
The first-principles calculations were performed using the projector augmented wave (PAW) method [Citation12] as implemented in the VASP code [Citation13]. Exchange and correlation were treated by the parametrization proposed by Perdew and Wang [Citation14]. An improved Vosko–Wilk–Nusair scheme [Citation15] was used for spin interpolation of the correlation potential. The energy cutoff of plane waves was 450 eV. Simulations were carried out in a cubic 4 × 4 × 4 supercell containing 256 atoms with a Γ-centered 2 × 2 × 2 k-point mesh. The disordered structure of NiCoFeCr was represented by a SQS that was constructed by optimizing the Warren–Cowley short-range order parameters [Citation16,Citation17] based on a simulated annealing algorithm [Citation18]. The optimal volume of NiCoFeCr was first determined by calculating the energy–volume curve; the volume was then frozen for all the following calculations with additional He impurities. Spin polarizations were considered with the elemental magnetic moments initialized by our previous results [Citation11]. The formation energy of He defects is calculated by:
(1)
(1) where E and E0 are the total energies of defective and perfect supercells, nX is the number of added (−) or removed (+) atoms, and
is the reference energy of atom X. Here, the reference energies of Ni, Co, Fe and Cr were calculated as the energies per atom in their pure bulk metals, i.e. ferromagnetic (FM) face-centered cubic (fcc) Ni, hexagonal close-packed (hcp) FM Co, body-centered cubic (bcc) FM Fe, and antiferromagnetic (AFM) bcc Cr. For He, EHe is the energy of a single He atom, which is calculated to be 0.013 eV/atom by putting a He in a cubic box. Based on the formation energy, the binding energy of a defect complex can be defined as:
(2)
(2) where Ef(A + B), Ef (A) and Ef (B) are the formation energies of defect A + B, A, and B, respectively. To calculate the energy barriers of He diffusion in Ni, a series of nudged elastic band (NEB) [Citation19] calculations were carried out, using a single k-point in the 4 × 4 × 4 supercell. The migration path and saddle structures were analyzed in these NEB calculations, as provided in Supplementary Materials. Once the saddle structures were determined, we calculated the energy barrier by optimizing the corresponding saddle configuration directly, as did in our previous work [Citation11]. We have compared the results obtained through these two different methods for pure Ni and the differences in energy barriers are very small (<0.05 eV). Therefore, we choose to do saddle structure optimization to calculate energy barriers in concentrated NiCoFeCr alloys.
For benchmark purpose, we first study the energetics of He in pure Ni. The lattice constant of Ni is determined to be 3.522 Å. Within this supercell, three possible He positions are considered, namely substitutional (Sub), tetrahedral (Tetra) and octahedral (Octa) positions. The calculated energies are summarized in Table , together with previous results for a comparison.
Table 1. Formation energies of a He defect in pure Ni (in eV/atom). The energy barrier for Tetra–Octa–Tetra transition is given in the last row.
The results in Table show that the substitutional position is the most stable position for a He in pure Ni, with the lowest formation energy of 3.20 eV. It can be understood by the large free volume provided from the missing lattice atom, resulting in smaller relaxation compared to the tetrahedral and octahedral interstitial positions. The tetrahedral position is slightly energy preferable than the octahedral position, with an energy difference of 0.15 eV. Consistent with previous calculations [Citation20], our results suggest that the most stable octahedral interstitial position of He is not symmetric but off-center along the direction in pure Ni. The energy difference between the tetrahedral and octahedral positions (T–O–T) corresponds to the migration energy of an interstitial He in Ni (The migration path is illustrated in Supplementary Materials). Note that the experimental value of interstitial He diffusion in Ni is 0.14 ± 0.03 eV [Citation23]. These results indicate that the migration energy of an interstitial He and a self-interstitial (0.11 eV [Citation24]) in Ni is comparable.
The migration of a substitutional He generally proceeds through dissociative or vacancy mechanisms [Citation20,Citation25,Citation26], depending on vacancy concentrations. For the dissociative mechanism, the substitutional He (can be regarded as a He-Vacancy (HeV) complex) first dissociates, and then the He migrates through interstitial sites until it becomes trapped by another vacancy. This process is unlikely to happen because the energy barrier is high through interstitial sites, as suggested by the large formation energy difference between the substitutional and interstitial sites in Table . The vacancy mechanism is dominant by the migration of the HeV2 complex. We have calculated the energy barrier for this process using both NEB method and single configuration calculations. The saddle point is found in the middle of the migration path of the moving He and lattice atoms. The whole migration path is illustrated in Supplementary materials. The energy barrier of this reaction is determined to be 1.21 eV, in good agreement with previous values [Citation20]. This value is 0.20 eV higher than the migration barrier of a single vacancy [Citation24], which can be interpreted as the additional energy in order to move the He from its central position in the HeV2 complex towards the lattice site during migration.
Because of chemical disorder, He can have different energy states in NiCoFeCr depending on the local environment. Therefore, formation energies of He exhibit distributions, similar to those of vacancies and interstitials in CSAs [Citation24]. To characterize these energies, we have calculated the formation energies of a He in different positions, and the results are shown in Figure . For substitutional positions, the energies obtained by substituting the four different types of lattice atoms are distinguished. Generally, lower formation energies are observed when He occupies Co and Cr vacancy sites, whereas He in Ni and Fe sites exhibits higher energies. Compared to the results in pure Ni (indicated by the dotted line in Figure ), it is found that the formation energies of He in NiCoFeCr at substitutional sites are significantly higher, whereas He at octahedral and tetrahedral sites show slightly lower formation energies. This observation is similar to the formation energies of intrinsic defects, i.e. interstitials show lower whereas vacancies exhibit higher formation energies [Citation24]. Consequently, the site preference of He is not as significant as that in pure Ni. This is a result of the decreased symmetry in the NiCoFeCr CSA due to disorder states. The higher formation energies at substitutional sites shown in Figure (a) is consistent with the higher vacancy formation energies in NiCoFeCr, compared to that in Ni [Citation11].
Figure 1. Formation energies of a single He in NiCoFeCr at different positions: (a) substitutional, (b) octahedral, and (c) tetrahedral positions.
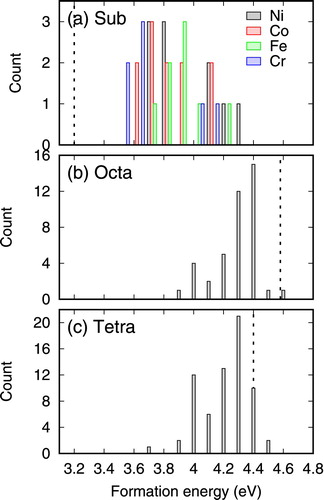
As the substitutional He can be regarded as an interstitial He binds to a vacancy defect, the binding energy can be calculated by Eb(HeV)=Ef(HeI)+Ef(V)−Ef (HeV), where Ef(HeV), Ef(HeI), and Ef(V) are formation energies of a HeV complex, a He interstitial, and a vacancy, respectively. In pure Ni, this formula gives a binding energy of 2.60 eV, indicating strong binding of the HeV defect complex. In NiCoFeCr, the difference between Ef(HeI) and Ef(HeV) decreases significantly, as shown in Figure . Taking into consideration of the higher Ef(V) in NiCoFeCr compared to that in Ni [Citation11], the resultant Eb(HeV) in NiCoFeCr is slightly lower than that in pure Ni, suggesting a weaker binding between an interstitial He and a vacancy in this case. In fact, in NiCoFeCr, substitutional He becomes less favorable with respect to other interstitial sites, as seen by their higher formation energies.
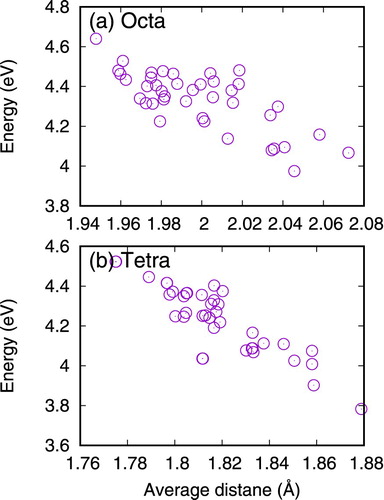
For substitutional sites, the replacement of the original lattice atom by a He only leads to slight relaxation of surrounding neighbor atoms (less than 0.06 Å). On the other hand, He in interstitial sites induces larger relaxations. To study this effect, we have calculated the nearest neighbor distance between the interstitial He and its neighbors. In a perfect fcc lattice structure, the octahedral interstitial site has six nearest neighbors at a distance of 0.5a0 and the tetrahedral interstitial site has four nearest neighbors at 0.433a0, where a0 is the lattice constant. The averaged distance in NiCoFeCr is shown in Figure , together with the obtained formation energies. It can be seen that the formation energies correlate well with the distance, suggesting that larger relaxation around the He interstitial helps to dissipate the energy, and thus lower its formation energy.
Figure 2. Dependence of the formation energy of a He in the interstitial site of NiCoFeCr on the relaxation distance between the He and its nearest neighbors.
Similar to Ni, we consider the migration of He in NiCoFeCr through two different mechanisms, namely interstitially and substitutionally. For diffusion of an interstitial He between tetrahedral sites, the saddle configuration is just the octahedral interstitial site between them. The migration of substitutional He involves a HeV2 complex, and the saddle structure is located at the middle point of the path. We have calculated these barriers through single point energy calculations by sampling migration paths at different local environments. An analysis of the detailed local environments around the migrating He is provided in the Supplementary materials. The obtained migration energies are demonstrated in Figure . It shows that the energy barriers for interstitial diffusion increase significantly in NiCoFeCr compared to that in Ni. Note that there are some barriers with zero energy due to disorder in concentrated NiCoFeCr, as also observed for interstitials [Citation24]. For substitutional diffusion through the HeV2 complex, the energy barriers are slightly higher than 1.21 eV, the value obtained in pure Ni. Therefore, He is more difficult to diffuse both interstitially and substitutionally in concentrated NiCoFeCr alloys compared to pure Ni.
Figure 3. Distribution of the migration energy of a He in NiCoFeCr through different mechanisms: (a) interstitial and (b) substitutional diffusion. The dashed line denotes the results in pure Ni.
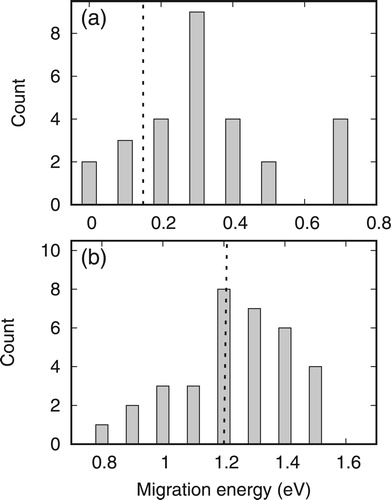
For these diffusion pathways, we have looked into the local environment of the migrating He by analyzing the distribution of the number of different elements surrounding it within a 3 Å cutoff radius. The results are provided in Supplementary materials. Generally, we find that the distribution of different elements is uniform, suggesting that our calculations are not biased and can represent typical behavior of He diffusion through these mechanisms. The relation between the calculated energy barriers and the number changes of different elements in the neighbors of the migrating He indicates that lower migration barriers are often observed when the number of Ni neighbors increase and Fe/Cr neighbors decrease. Therefore, increasing Fe/Cr in the He neighborhood may increase the migration energy barriers of He. In practice, the preferential position and the dominant migration mode of He are related to temperatures and other defects, especially vacancies [Citation27]. When radiation damage is negligible, diffusion of He proceeds interstitially until it is trapped by thermal vacancies. When the vacancy concentration is high (such as high temperature and irradiation conditions), vacancy mechanism in which He diffuses through HeV2 complex start to play important roles.
For substitutional He diffusion, the activation energy of the dissociation and vacancy mechanisms are related to the binding energy of HeV and HeV2 defect complex [Citation20,Citation26]. As discussed above, Eb(HeV) in NiCoFeCr is slightly lower. For Eb(HeV2), it is given by Ef(HeS)+Ef(V)−Ef (HeV2), where Ef(HeS), Ef(V), and Ef(HeV2) are formation energies of a substitutional He, a vacancy, and the HeV2 defect complex, respectively. In a HeV2 defect complex, the He atom is located in the midway between the two vacancies after relaxation. Actually, the He is located exactly at the center between the two vacancies in pure Ni. However, in NiCoFeCr, the He deviates the center position slightly, with a displacement less than 0.3 Å depending on the local environment. For pure Ni, Ef(HeV2) = 4.22 eV and Eb(HeV2) = 0.38 eV. In NiCoFeCr, the formation energies of Ef(HeV2) complex are shown in Figure . The results suggest that Ef(HeV2) in NiCoFeCr is ∼0.6 eV higher than those in pure Ni, almost the same order of the increase of Ef(V). Hence, Eb(HeV2) will be higher in NiCoFeCr due to its higher Ef (He). Knowing these binding energies, we have calculated the activation energies for substitutional He diffusion in both dissociation and vacancy mechanisms at different vacancy concentrations [Citation20,Citation26]. The results in Ni and NiCoFeCr are summarized in Table . It can be seen that the activation energies in NiCoFeCr is widely distributed. Based on the averaged values, it indicates that dissociation mechanism is favorable in NiCoFeCr due to the low Eb(HeV), which may help to suppress the growth of large He bubbles. Since formation energies of single vacancy and HeV2 defect complex are much higher in NiCoFeCr, their concentrations will be lower and contribute less to He diffusion. As a result, substitutional He diffusion through vacancy mechanism will be restrained in NiCoFeCr.
Figure 4. Distribution of formation energies of the HeV2 complex in NiCoFeCr.
Table 2. Activation energy (in eV) for substitutional He diffusion under two different conditions [Citation20,Citation26].
We have shown that He dissociation is the main mechanism for He diffusion in NiCoFeCr CSAs, due to the reduced energy preference of substitutional He sites and the high formation energies of vacancies. The first factor leads to a weak binding inside HeV complex, while the latter causes a low concentration of vacancies. As a result, the nucleation of He bubbles from vacancy sites is greatly suppressed in NiCoFeCr CSAs. This result explains the low number density of He bubble observed experimentally [Citation7,Citation8]. However, with increasing irradiation dose where vacancy supersaturation occurs, the activation energy difference between NiCoFeCr and Ni is very small, an indication of similar bubble size in this case consistent with experimental observations [Citation7]. The results of this work elucidate the correlation between He energetics and He bubble growth in typical CSAs, which provides insight to the irradiation performance of chemically complex alloys, including high-entropy alloys.
To conclude, we have revealed the growth mechanism of He bubbles in a typical CSA of NiCoFeCr through first-principles calculations. Our results indicate that the binding energies of HeV complex decrease significantly in NiCoFeCr, whereas the binding energies of HeV2 are higher. Accordingly, the dissociation mechanism is dominant in NiCoFeCr at low vacancy concentrations, leading to low He bubble densities. When vacancy concentration supersaturates, He bubble grows through the vacancy mechanism. These results indicate that CSAs are effective in resisting He bubble growth, especially at the early stage.
Supplemental Material
Download MS Word (889.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Shijun Zhao http://orcid.org/0000-0003-0870-8153
Additional information
Funding
Related Research Data
References
- Yeh J-W, Chen S-K, Lin S-J, et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv Eng Mater. 2004;6:299–303. doi: 10.1002/adem.200300567
- Cantor B. Multicomponent and high entropy alloys. Entropy. 2014;16:4749–4768. doi: 10.3390/e16094749
- Zhang Y, Zhao S, Weber WJ, et al. Atomic-level heterogeneity and defect dynamics in concentrated solid-solution alloys. Curr Opin Solid State Mater Sci. 2017;21:221–237. doi: 10.1016/j.cossms.2017.02.002
- Zhang Y, Jin K, Xue H, et al. Influence of chemical disorder on energy dissipation and defect evolution in advanced alloys. J Mater Res. 2016;31:2363–2375. doi: 10.1557/jmr.2016.269
- Gludovatz B, Hohenwarter A, Catoor D, et al. A fracture-resistant high-entropy alloy for cryogenic applications. Science. 2014;345:1153–1158. doi: 10.1126/science.1254581
- Zinkle SJ, Was GS. Materials challenges in nuclear energy. Acta Mater. 2013;61:735–758. doi: 10.1016/j.actamat.2012.11.004
- Yan Z, Liu S, Xia S, et al. He behavior in Ni and Ni-based equiatomic solid solution alloy. J Nucl Mater. 2018;505:200–206. doi: 10.1016/j.jnucmat.2018.04.009
- Chen D, Tong Y, Li H, et al. Helium accumulation and bubble formation in FeCoNiCr alloy under high fluence He+ implantation. J Nucl Mater. 2018;501:208–216. doi: 10.1016/j.jnucmat.2018.01.041
- Chen D, Tong Y, Wang J, et al. Microstructural response of He+ irradiated FeCoNiCrTi0.2 high-entropy alloy. J Nucl Mater. 2018;510:187–192. doi: 10.1016/j.jnucmat.2018.08.006
- Tsai K-Y, Tsai M-H, Yeh J-W. Sluggish diffusion in Co–Cr–Fe–Mn–Ni high-entropy alloys. Acta Mater. 2013;61:4887–4897. doi: 10.1016/j.actamat.2013.04.058
- Zhao S, Egami T, Stocks GM, et al. Effect of d electrons on defect properties in equiatomic NiCoCr and NiCoFeCr concentrated solid solution alloys. Phys Rev Mater. 2018;2:013602. doi: 10.1103/PhysRevMaterials.2.013602
- Blöchl PE. Projector augmented-wave method. Phys Rev B. 1994;50:17953. doi: 10.1103/PhysRevB.50.17953
- Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci. 1996;6:15–50. doi: 10.1016/0927-0256(96)00008-0
- Perdew JP, Chevary JA, Vosko SH, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys Rev B. 1992;46:6671–6687. doi: 10.1103/PhysRevB.46.6671
- Vosko SH, Wilk L, Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can J Phys. 1980;58:1200–1211. doi: 10.1139/p80-159
- Cowley JM. Short-range order and long-range order parameters. Phys Rev. 1965;138:A1384. doi: 10.1103/PhysRev.138.A1384
- Cowley JM. An approximate theory of order in alloys. Phys Rev. 1950;77:669. doi: 10.1103/PhysRev.77.669
- Zhao S, Stocks GM, Zhang Y. Stacking fault energies of face-centered cubic concentrated solid solution alloys. Acta Mater. 2017;134:334–345. doi: 10.1016/j.actamat.2017.05.001
- Henkelman G, Uberuaga BP, Jónsson H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J Chem Phys. 2000;113:9901–9904. doi: 10.1063/1.1329672
- Hepburn DJ, Ferguson D, Gardner S, et al. First-principles study of helium, carbon, and nitrogen in austenite, dilute austenitic iron alloys, and nickel. Phys Rev B. 2013;88:024115. doi: 10.1103/PhysRevB.88.024115
- Zu XT, Yang L, Gao F, et al. Properties of helium defects in bcc and fcc metals investigated with density functional theory. Phys Rev B. 2009;80:054104. doi: 10.1103/PhysRevB.80.054104
- Connétable D, Andrieu É, Monceau D. First-principles nickel database: energetics of impurities and defects. Comput Mater Sci. 2015;101:77–87. doi: 10.1016/j.commatsci.2015.01.017
- Philipps V, Sonnenberg K. Interstitial diffusion of He in nickel. J Nucl Mater. 1983;114:95–97. doi: 10.1016/0022-3115(83)90076-4
- Zhao S, Stocks GM, Zhang Y. Defect energetics of concentrated solid-solution alloys from ab initio calculations: Ni0.5Co0.5, Ni0.5Fe0.5, Ni0.5Fe0.5 and Ni0.5Cr0.5. Phys Chem Chem Phys. 2016;18:24043–24056. doi: 10.1039/C6CP05161H
- Mansur LK, Lee EH, Maziasz PJ, et al. Control of helium effects in irradiated materials based on theory and experiment. J Nucl Mater. 1986;141–143:633–646. doi: 10.1016/0022-3115(86)90066-8
- Fu C-C, Willaime F. Ab initio study of helium in α − Fe : Dissolution, migration, and clustering with vacancies. Phys Rev B. 2005;72:064117. doi: 10.1103/PhysRevB.72.064117
- Trinkaus H, Singh B. Helium accumulation in metals during irradiation—where do we stand? J Nucl Mater. 2003;323:229–242. doi: 10.1016/j.jnucmat.2003.09.001