
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In this work, a degenerate diffusion state is identified for common L12 intermetallics in well-defined five-frequency model (FFM) and anti-site bridge model (ASB). The relative energy barrier (
in FFM and
in ASB) of the degenerate state serves as a decomposer which not only bifurcates the degenerate pathways, but also determines the most probable diffusion model. The diffusion coefficients obtained from the most probable diffusion model coincides the experimental value. We find the proposed decomposer complies with the Onsager phenomenological theory and atomistic correlation understanding, and is also supported by the relative work
done by electronic wind force.
GRAPHICAL ABSTRACT
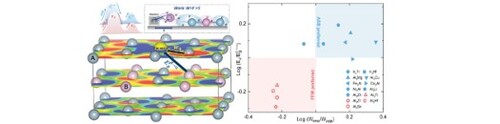
1. Introduction
The diffusion of atoms in gases or liquids is generally Brownian, as proposed by Einstein, which is determined by the root-mean-square of total displacements [Citation1]. The diffusion pathways in solids, on the other hand, are considered finite and restricted by the crystallography, thermodynamics, kinetics, and statistics. The pursuit of possible pathways in solid-state diffusion usually takes years or decades [Citation2–4]. Lacking in situ monitoring and a clear physical picture of the atomic movement in solids makes those proposed pathways ambiguous from one to another [Citation5–7].
In past decades, the atomic behaviors (jump frequency, transition states, thermodynamic factors, etc.) associated with each pathway can be accurately determined by density function theory [Citation8–10]. Many stochastic methods, such as kinetic Monte Carlo calculations [Citation11–13], path probability methods [Citation14,Citation15], mean-field methods [Citation16–18], Ritz variational methods [Citation19–21], and Green function methods [Citation22–24] have been developed to compute the diffusion coefficient from calculated atomic mechanisms, and used to validate the proposed diffusion pathways together with experimental data [Citation25]. Analytically, correlation, drag ratio, escape frequency, etc. have been proposed to connect the possible pathways in different diffusion models. In this endeavor, the important role of some particular pathways in the resulting diffusion coefficient has been emphasized. In some cases, bifurcations caused by the same pathway in different diffusion models may result in distinct outcomes. For example, Trinkle et al. found the asymmetrical solute-vacancy diffusion in the first-nearest neighbor leading to quite different drag ratio in hexagonal close packed (HCP) Mg through Green function methods [Citation23]. Most stochastic methods work well for solute diffusion in dilute solid solution phase as its movement is short ranged [Citation23,Citation26]. However, they strongly depended on the input transition state energies, and often be trapped to the pathway with the lowest energy barrier [Citation27,Citation28].
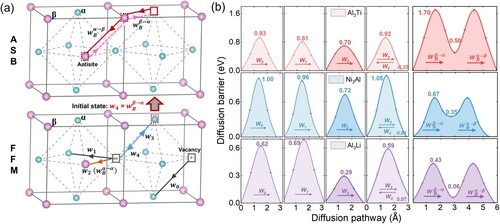
The atomic movement in ordered intermetallics, on the other hand, is not short ranged, since the direct exchange of atomic position of different sublattice will lead to the frustration of ordered structure. A common L12 A3B intermetallics, which is widely existed in superalloys, bimetallic catalysts, high-entropy alloys, etc. [Citation29–31], needs anti-site defects to fulfill the transportation of B atoms. Five-frequency model (FFM) and anti-site bridge model (ASB) are two well-documented diffusion models describing the diffusion pathways of anti-site B atoms in the L12 A3B intermetallics (refer to supplementary material for further details) [Citation32,Citation33]. B atoms can achieve effective migration in these models without breaking the structural integrity. As depicted in Figure (a), these two models are not independent due to a shared diffusion state. The initial state of is identical to the initial state of
marked by the red rectangle vacancy, leading to a blending of the two diffusion models [Citation29]. Consequently, when the vacancy ‘disassociates’ with the anti-site B atom along the
pathway to the initial state of
, it becomes uncertain whether the next jump will follow the FFM pathway (
) or the ASB pathway (
).
Figure 1. (a) Atomic illustration of FFM and ASB model for L12 intermetallics. (b) Calculated diffusion energy barriers of FFM and ASB in L12 Al3Ti, Ni3Al and Al3Li at 0 K.
In this work, a simple yet ingenious physical picture is constructed by considering the same route in these two diffusion models as a degenerate diffusion state. The bifurcation of this degeneracy state thus lead to quite distinct phenomenological Onsager transport coefficients and atomistic correlation relationship [Citation34]. In order to unravel the decomposer of this degenerate state, how it decouples this state and determines the final diffusion coefficient, 11 common L12 intermetallics are chosen and calculated using both FFM and ASB models through density function theory. Al3Ti, Ni3Al and Al3Li are discussed in detail to show how electronic wind force untangles this degeneracy.
2. Methods
The factors affecting the selection of B atom diffusion model are studied using first principles calculations using Vienna ab initio simulation package (VASP) [Citation35] with projector augmented wave (PAW) method [Citation36] and the Perdew−Burke−Ernzerhof (PBE) exchange−correlation functional [Citation37]. Monkhorst−Pack method [Citation38] was used to sample the 7 × 7 × 7 k−point mesh, and 2 × 2 × 2 supercell was adopted in L12 structure which has sufficient precision compared with the larger 3 × 3 × 3 supercell shown in Figure S1 and Table S1. A cutoff energy for the plane−wave basis was set to 500 eV. Convergence criteria for electronic and ionic relaxations were 0.001 meV and 0.01 eV/Å, respectively, whereas that for static, self−consistent calculations was 0.001 meV. The energy barriers of the atom migration were calculated using climb−image nudged elastic band (CI−NEB) [Citation39] calculations. Alloy Theoretic Automated Toolkit (ATAT) [Citation40] package was used to obtain the effective frequency and quasi−harmonic thermodynamic properties through phonon calculation based on the direct force constant supercell approach [Citation41]. The phonon dispersion results provide information about the atomic diffusion behaviors and the onset of the diffusion processes [Citation42–45], which are available in the supplementary material. All parameters have been tested in our previous work [Citation46].
3. Results and discussion
The diffusion energy barriers corresponding to the pathways in Figure (a) of the studied L12 systems are listed in Table . Al3Ti, Ni3Al and Al3Li, as typical examples, are illustrated in Figure (b). The calculated diffusion energy barrier data align well with other literature values (Table S1) [Citation47,Citation48]. It is observed that the diffusion energy barrier
is the lowest among FFM pathways, especially lower than the degenerated
. In ASB, the degenerated
is always larger than
, indicating that the first jump is harder than the second jump due to the introduction of two anti-site defects. Generally speaking, the relative energy barrier of
and
should be able to differentiate the atomic propensity toward to FFM or ASB. As shown in Figure (b), Al3Ti and Al3Li would choose FFM, while Ni3Al adopt ASB. Even though there are many works using the energy barriers to select the preferred diffusion pathways [Citation49,Citation50], it is widely acknowledged that energy barrier itself cannot solely determine the diffusion behaviors [Citation51].
Table 1. Calculated diffusion energy barriers , works
, pre-factor
, activation energy
, the values of
and
of all studied L12 intermetallics.
A more accurate description of atomic mechanisms on the diffusion coefficient is proposed by Bakker [Citation52], which is determined by two parts: the jump frequency of the target diffusion atom exchanging with its nearest neighbor (NN) vacancy (effective jump frequencies
in FFM and the
in ASB, see supplementary material for the definition), and the probability
that the vacancy is at the NN sites of the target diffusion atom. The diffusion coefficient is then obtained by:
(1)
(1) where
is a constant for the same L12 structure in FFM and ASB, determined by lattice constant and point defect concentrations.
The calculated results of in FFM and
in ASB are shown in Figure (a–c) and Figure S3. It can be seen that the relative values of
and
are consistent with the relative value of
and
. That is the lower the energy barrier, the higher the internal jump frequency. In this way, the preference of FFM or ASB is the same as that indicated by the energy barrier. As the stochastic methods use energy barrier or jump frequency to compute the diffusion coefficient, they may overweight the contribution of energy barrier and prejudice the lower one against the higher one [Citation27,Citation28]. For example, the ASB pathway proposed by Belova and Murch is first concepted by the direct movement of anti-site B atom in
-sublattice (major A sublattice) through a transition state of bridged two anti-site B states. The diffusion coefficient of Ni3X (X = Al, Ga, Ge) obtained from kinetic Monte Carlo calculations is closer to experimental values than those from the previous FFM mechanism [Citation33]. With almost a decade development, the ASB mechanism is constructed analytically like FFM. However, still many works adopted or found FFM over ASB [Citation32,Citation48,Citation50]. This may originate from the factor that there is a degenerate state between these two mechanisms.
Figure 2. Calculated results of ,
and
in Equation (1) of L12 Al3Ti, Ni3Al and Al3Li, with available data in the literature [Citation29,Citation53–59].
![Figure 2. Calculated results of P, w and D in Equation (1) of L12 Al3Ti, Ni3Al and Al3Li, with available data in the literature [Citation29,Citation53–59].](/cms/asset/9f9d215f-82c2-40b7-b69a-2fd65b44b619/tmrl_a_2367645_f0002_oc.jpg)
Figure (d–f) and Figure S8 show the calculated probabilities and
. The
value of the two models differ greatly in different diffusion systems, and do not have apparent relationship with energy barriers or jump frequency of effective jump for FFM or ASB. For instance,
is smaller than
for Al3Li, while the respective
and
show reversed trend. Similar phenomenon is also observed in other L12 intermetallic compounds like Al3Sc, Al3Mg and Co3Al as shown in Figure S8. This reversed relationship makes the diffusion mode deduced solely from energy barriers or jump frequency ambiguous. In Al3Ti and Ni3Al, the relative value of
follows the same rule with energy barriers or jump frequency, that
is larger than
. However, the slope trend does not resemble with each other, i.e. the slope is almost the same for
in Ni3Al, while that differs greatly for
.
Based on the and
values, the resulting diffusion coefficients are calculated analytically and shown in Figure (g–i) and Figure S8 together with available experimental values [Citation29,Citation53–59]. The diffusion coefficients of majority A in L12 A3B are also plotted for comparison. Compared with experimental values,
obtained from different models show quite distinct behaviors. For example,
of Al3Ti from FFM agree well with experimental ones, while those of Ni3Al and Al3Li from ASB are consistent with experiments. This behavior indicates the transportation of B atoms may prefer one model over another, resembling the rate-determined reaction in chemical science. Comparing the results of
,
and
, we find the relative values and variation trends of
and
are almost the same. This finding indicates that the probability of the vacancy remaining at the NN site of the target diffusion atom could be regarded the manipulator of the ‘rate-determined pathway’.
We now look into the internal connection between and the degenerate diffusion state. The meaning of
is how likely the vacancy provides new diffusion sites for B atoms to make the intended move (
or
) for FFM or ASB pathways. According to Einstein’s atomic mechanism [Citation1], the probability
could represent the correlation factor as it determines the effective jump in the diffusion process. The degenerate state
and
is the jump of vacancy to the NN of anti-site B. Thus, how many vacancies available for the effective jump of FFM or ASB is associated with how many jumps from degenerate
or
, respectively. As a result,
and
can be interpreted as the probability amplitude of degenerate diffusion state, while
and
can be used to determine the preferred diffusion pathway. As shown in Table , the diffusion energy barriers
and
(as they does not change with temperature like
and
) are compared with each other and the preferred diffusion model. When the B atom prefers FFM, the value of
, and when B atom prefers ASB,
.
As the atomic mechanism suggests the deciding role of degenerate or
, its phenomenological understanding deserves further attentions. According to Onsager relationship, the movement direction of the vacancy towards the diffusion atom can be predicted by the drag ratio
. The definition of
implies that the drag ratio complies with
, that how many vacancies are brought to the NN of anti-site B. Accordingly, the degenerate
or
also reflect the effect of drag ratio. Indeed, Trinkle et al. pointed out that during the jump of
in FFM, the barycenter of the solute-vacancy pair remains unchanged, so that
has less impact on the drag radio than
[Citation22].
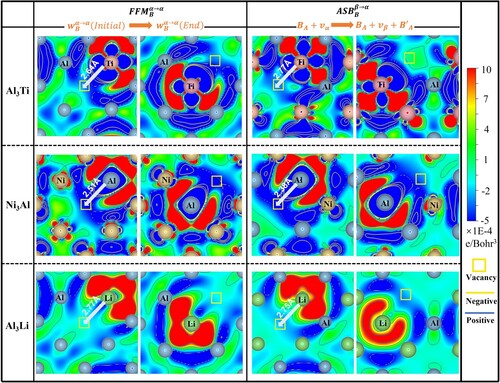
The intrinsic mechanism of the degenerate pathway and its role in the final (observable) diffusion coefficient is analyzed by the electronic density difference. Figure shows charge density difference contour plots on (010) plane intersecting the vacancy in sublattice of the initial and final state of
in FFM and the first jump
of ASB. Charge densities in column 1 and 3 represent the end of untanglement and those in column 2 and 4 are the end of effective movement of B atom. It can be seen that the introduction of vacancy near the anti-site B induces charge redistribution between the vacancy and anti-site B atom [Citation60,Citation61]. The partial covalent bonding feature of L12 intermetallics, as reflected by the red regions in Figure , makes the diffusion of anti-site B atoms directional. The eletrons are accumulated around the anti-site B, while electrons are depleted on the vacancy. However, the amount of electrons accumulated or depleted is different for FFM and ASB models. For example, in Al3Ti the vacancy has negative value of charge density for FFM model, while it is positive for ASB model. As the Ti atoms are surrounded by positive charge density, it will push Ti into vacancy easily in FFM model, while hard for ASB. On the contrary, in Al3Li the vacancy has positive charge density in the case of FFM, while slight negative charge density for ASB, leading to the movement of B atoms toward ASB. In Ni3Al, the charge density distribution, on the other hand, shows similar feature between FFM and ASB, but the distance from the vacancy to the anti-site B is different. Comparing the charge density distribution with
value, it could be deduced that they are in line with each other in decoupling the degenerate pathways.
Figure 3. The charge density difference contour plots of L12 Al3Ti, Ni3Al and Al3Li on the plane.
As mentioned before, the value is closely related to correlation in FFM and ASB. The electron density is also associated with correlation through the electron wind force [Citation62] proposed by Huntington (details are presented in the supplementary material). As shown in Figure , the accumulation and depletion of charge density build a naturally electrostatic potential
. The atoms carry electrons will move in that potential. Based on the Huntington and Onsager theory, the correlation factor
can be represented by
(2)
(2) where
is the diffusion coefficient considering the correlation effect,
is the random diffusion coefficient,
is the concentration of diffusion atom (the anti-site B atom in this work),
is the effective charge number of B atom,
is the absolute value of the electronic charge and
is the Boltzmann’s constant (The derivation of this equation is put in the supplementary material.). From above equation, the key factor in correlation is
, the potential gradient induced by charge density redistribution. Thus, the steeper the gradient, the larger the correlation factor. As discussed before, the larger charge density difference between the anti-site B and vacancy facilitates the atomic movement and decouples the degenerate pathways. Note that the energy barrier cannot be simply related to the charge density difference, as it is also influenced by volume, long-range interactions, lattice distortion, and so on.
Similar to the /
value (Figure (a)), we construct a quantitative description of electron wind force. Thus, the electron wind force induced by electrostatic potential
for the given atom is defined as:
(3)
(3) where
is the absolute value of the electronic charge,
is the effective charge number of B atom, and
is the electrostatic potential. On the basis of symmetry, we only consider the electric field generated by the NN atoms of the anti-site diffusion atom along the direction of the diffusion pathway (A atom shown in Figure (b)). The
is estimated through Bader charge analysis [Citation63]. Unfortunately, direct comparison of diffusion driving force may not be adequate since the diffusion is also related to the distance of atom movement. The diffusion distance, as noticed by Janotti et al., causes ‘Larger Atoms Can Move Faster’ in metals [Citation49].
Figure 4. Schematic illustration of (a) the decoupling direction and (b) the electronic work during the diffusion process. (c) Guiding diagram for the diffusion model selection.
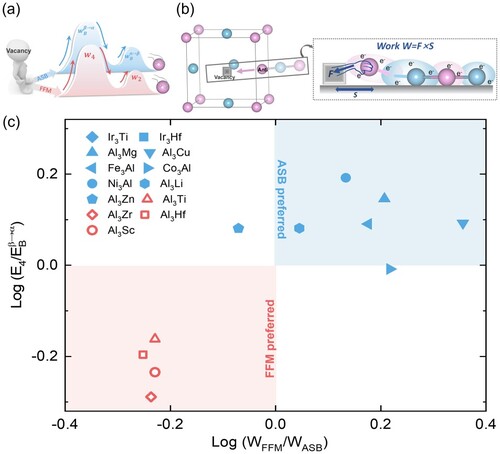
Accordingly, we use the work done in the process of atomic diffusion to quantitatively assess the impacts of the charge density differences and displacements in Figure , and to differentiate the role of electron wind force in untangling of degenerate pathways. The work is defined as:
(4)
(4) where
is the distance from the A atom,
is the Coulomb constant,
is the charge of the NN atom. It should be noted that the displacement (the white arrow in Figure ) during diffusion process is equal to
. See Table and Table S4 for the value of each parameter in Equation (4). In practice, the more work the diffusion atoms do during diffusion, the harder the diffusion atom to get rid of the surrounding A atoms to complete diffusion. With the work for diffusion, we can quantitatively decouple the degeneracy in diffusion pathways. Comparing
in FFM (
) with
in ASB (
), when
, it indicates that more energy is required for diffusion through FFM. As shown in Figure (c), the relative value of
and
are consistent with each other in decoupling the degenerate pathway into the most probable diffusion model, which reflect the correlation between the probability
and the charge density differences quantitatively. Among the L12 structure intermetallics we studied, Al3Ti, Al3Zr, Al3Hf and Al3Sc in the red region (
and
) choose FFM model for diffusion, while Ni3Al, Fe3Al, Al3Mg and others in the blue region (
and
) will choose ASB for diffusion.
4. Conclusion
In summary, the decisive role of the degenerate and
on the most probable diffusion model has been uncovered from the perspective of the probability
correlated to the Onsager relationship and the electronic work
done by the electron wind force. This decoupling rule establishes a framework for choosing the exact analytical model of diffusion coefficient in L12 intermetallics. Our study is pivotal for unraveling the intricacies of atomic diffusion in solids and addressed the limitations of current diffusion research methodologies. The physical picture provided in this work not only sheds light on the atomistic and phenomenological theory of current state-of-art diffusion models, but also would facilitate the development of long-ranged diffusion model and reliable stochastic methods.
Supplemental Material
Download PDF (1.2 MB)Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Einstein A. Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann Phys. 1905;322:549–560. doi:10.1002/andp.19053220806
- Leclaire AD, Lidiard AB. Correlation effects in diffusion in crystals. Philos Mag. 1956;1:518–527. doi:10.1080/14786435608238133
- Koiwa M. Diffusion in materials—history and recent developments. Mater Trans JIM. 1998;39:1169–1179. doi:10.2320/matertrans1989.39.1169
- Manning JR. Correlation effects in impurity diffusion. Phys Rev [Internet]. 1959;116:819–827. Available from: https://link.aps.org/doi/10.1103PhysRev.116.819
- Sangiovanni DG, Klarbring J, Smirnova D, et al. Superioniclike Diffusion in an elemental crystal: bcc titanium. Phys Rev Lett [Internet]. 2019;123:105501. Available from: https://link.aps.org/doi/10.1103PhysRevLett.123.105501
- Messina L, Nastar M, Garnier T, et al. Exact ab initio transport coefficients in bcc Fe − X (X = Cr, Cu, Mn, Ni, P, Si) dilute alloys. Phys Rev B [Internet]. 2014;90:104203. Available from: https://link.aps.org/doi/10.1103PhysRevB.90.104203
- Zhang W, Seo D-H, Chen T, et al. Kinetic pathways of ionic transport in fast-charging lithium titanate. Science (80-). 2020;367:1030–1034. doi:10.1126/science.aax3520
- Wolverton C. Solute–vacancy binding in aluminum. Acta Mater. 2007;55:5867–5872. doi:10.1016/j.actamat.2007.06.039
- Mantina M, Wang Y, Arroyave R, et al. First-principles calculation of self-diffusion coefficients. Phys Rev Lett. 2008;100:215901. doi:10.1103/PhysRevLett.100.215901
- Hargather CZ, Shang S-L, Liu Z-K. A comprehensive first-principles study of solute elements in dilute Ni alloys: diffusion coefficients and their implications to tailor creep rate. Acta Mater. 2018;157:126–141. doi:10.1016/j.actamat.2018.07.020
- Belova IV, Brown MJ, Allnatt AR, et al. Diffusion kinetics of isolated and paired species: a Monte Carlo study. Philos Mag [Internet]. 2005;85:2787–2798. Available from: http://www.tandfonline.com/doi/abs/10.108014786430500155296
- Allnatt AR, Belova IV, Murch GE. Diffusion kinetics in dilute binary alloys with the h.c.p. crystal structure. Philos Mag [Internet]. 2014;94:2487–2504. Available from: http://www.tandfonline.com/doi/abs/10.108014786435.2014.916426
- Belova IV, Ahmed T, Sarder U, et al. The Manning factor for direct exchange and ring diffusion mechanisms. Philos Mag. 2017;97:230–247. doi:10.1080/14786435.2016.1255368
- Sato H, Kikuchi R. Theory of many-body diffusion by the path-probability method: Conversion from ensemble averaging to time averaging. Phys Rev B [Internet]. 1983;28:648–664. Available from: https://link.aps.org/doi/10.1103PhysRevB.28.648
- Ishii T, Sato H, Kikuchi R. Theory of hopping conduction by the path-probability method. Phys Rev B [Internet]. 1986;34:8335–8344. Available from: https://link.aps.org/doi/10.1103PhysRevB.34.8335
- Huang L, Ma K, Belkacemi LT, et al. Impact of the local microstructure fluctuations on radiation-induced segregation in dilute Fe-Ni and Ni-Ti model alloys: a combined modeling and experimental analysis. Acta Mater [Internet]. 2022;225:117531. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359645421009095
- Huang L, Nastar M, Schuler T, et al. Multiscale modeling of the effects of temperature, radiation flux, and sink strength on point-defect and solute redistribution in dilute Fe-based alloys. Phys Rev Mater [Internet]. 2021;5:033605. Available from: https://link.aps.org/doi/10.1103PhysRevMaterials.5.033605
- Messina L, Schuler T, Nastar M, et al. Solute diffusion by self-interstitial defects and radiation-induced segregation in ferritic Fe–X (X = Cr, Cu, Mn, Ni, P, Si) dilute alloys. Acta Mater [Internet]. 2020;191:166–185. doi:10.1016/j.actamat.2020.03.038 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359645420302251
- Krzyżewski F, Załuska-Kotur MA. Coupling of orthogonal diffusion modes in two-dimensional nonhomogeneous systems. Phys Rev B [Internet]. 2008;78:235406. Available from: https://link.aps.org/doi/10.1103PhysRevB.78.235406
- Mińkowski M, Załuska-Kotur MA. Influence of metastable states on collective particle diffusion in the interacting lattice gas model. J Stat Mech Theory Exp [Internet]. 2013;2013:P05004. Available from: https://iopscience.iop.org/article/10.10881742-5468/2013/05/P05004
- Mińkowski M, Załuska–Kotur MA. The collective diffusion coefficient as a shape detector of the surface energy landscape. J Stat Mech Theory Exp [Internet]. 2018;2018:013204. Available from: https://iopscience.iop.org/article/10.10881742-5468/aa9da9
- Garnier T, Trinkle DR, Nastar M, et al. Quantitative modeling of solute drag by vacancies in face-centered-cubic alloys. Phys Rev B [Internet]. 2014;89:144202. Available from: https://link.aps.org/doi/10.1103PhysRevB.89.144202
- Agarwal R, Trinkle DR. Exact model of vacancy-mediated solute transport in magnesium. Phys Rev Lett. 2017;118:105901. doi:10.1103/PhysRevLett.118.105901
- Trinkle DR. Variational principle for mass transport. Phys Rev Lett [Internet]. 2018;121:235901. Available from: https://link.aps.org/doi/10.1103PhysRevLett.121.235901
- Kulitckii V, Schneider A, Lukianova O, et al. Atomic diffusion in bcc Fe–Mn alloys: theoretical analysis and experimental measurements across the Curie temperature. Acta Mater [Internet]. 2023;251:118883. doi:10.1016/j.actamat.2023.118883 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359645423002148
- Liu Z-K. Theory of cross phenomena and their coefficients beyond Onsager theorem. Mater Res Lett [Internet]. 2022;10:393–439. Available from: https://www.tandfonline.com/doi/full/10.108021663831.2022.2054668
- Schneider A, Fu C-C, Soisson F, et al. Atomic diffusion in α-iron across the Curie point: an efficient and transferable Ab initio–based modeling approach. Phys Rev Lett [Internet]. 2020;124:215901. Available from: https://link.aps.org/doi/10.1103PhysRevLett.124.215901
- Thomas SL, Patala S. Vacancy diffusion in multi-principal element alloys: the role of chemical disorder in the ordered lattice. Acta Mater [Internet]. 2020;196:144–153. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359645420304547
- Pelleg J. Diffusion in the Iron Group L12 and B2 intermetallic compounds [Internet]. Cham: Springer International Publishing; 2017. Available from: http://link.springer.com/10.1007978-3-319-39522-7
- Luo H, Liu W, Gong H, et al. First principles calculation of composition dependence tracer and interdiffusion with phase change in γ/γ′ superalloy: a case study of Ir/Ir3Nb. Calphad [Internet]. 2024;84:102659. doi:10.1016/j.calphad.2024.102659 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0364591624000014
- Luo H, Liu W, Ma Y, et al. Unraveling L12 Al3X (X = Ti, Zr, Hf) nano-precipitate evolution in aluminum alloys via multi-scale diffusion simulation. J Mater Res Technol [Internet]. 2024;30:7104–7114. doi:10.1016/j.jmrt.2024.05.050 Available from: https://linkinghub.elsevier.com/retrieve/pii/S2238785424010925
- Numakura H, Ikeda T, Nakajima H, et al. Diffusion in Ni3Al, Ni3 Ga and Ni3Ge. Mater Sci Eng A. 2001;312:109–117. doi:10.1016/S0921-5093(00)01864-5
- Belova IV, Murch GE. Analysis of diffusion mechanisms in Ni3Al, Ni3Ge and Ni3 Ga. Defect Diffus Forum. 2000;177–178:59–68. doi:10.4028/www.scientific.net/DDF.177-178.59
- Pikal MJ. Theory of the Onsager transport coefficients lij and Rij for electrolyte solutions. J Phys Chem [Internet]. 1971;75:3124–3134. Available from: https://pubs.acs.org/doi/abs/10.1021j100689a016
- Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54:11169–11186. doi:10.1103/PhysRevB.54.11169
- Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59:1758–1775. doi:10.1103/PhysRevB.59.1758
- Perdew JP, Chevary JA, Vosko SH, et al. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys Rev B. 1992;46:6671–6687. doi:10.1103/PhysRevB.46.6671
- Blöchl PE, Jepsen O, Andersen OK. Improved tetrahedron method for Brillouin-zone integrations. Phys Rev B. 1994;49:16223–16233. doi:10.1103/PhysRevB.49.16223
- Henkelman G, Jónsson H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J Chem Phys [Internet]. 2000;113:9978–9985. doi:10.1063/1.1323224 Available from: https://pubs.aip.org/jcp/article/113/22/9978/184858/Improved-tangent-estimate-in-the-nudged-elastic
- van de Walle A, Asta M, Ceder G. The alloy theoretic automated toolkit: a user guide. Calphad. 2002;26:539–553. doi:10.1016/S0364-5916(02)80006-2
- Wei S, Chou MY. Ab initio calculation of force constants and full phonon dispersions. Phys Rev Lett. 1992;69:2799–2802. doi:10.1103/PhysRevLett.69.2799
- Ren Q, Gupta MK, Jin M, et al. Extreme phonon anharmonicity underpins superionic diffusion and ultralow thermal conductivity in argyrodite Ag8SnSe6. Nat Mater [Internet]. 2023;22:999–1006. Available from: https://www.nature.com/articles/s41563-023-01560-x
- Gupta MK, Ding J, Bansal D, et al. Strongly anharmonic phonons and their role in superionic diffusion and ultralow thermal conductivity of Cu 7 PSe 6. Adv Energy Mater [Internet]. 2022:12. Available from: https://onlinelibrary.wiley.com/doi/10.1002aenm.202200596
- Gupta MK, Ding J, Osti NC, et al. Fast Na diffusion and anharmonic phonon dynamics in superionic Na3PS4. Energy Environ Sci [Internet]. 2021;14:6554–6563. Available from: https://xlink.rsc.org/?DOI=D1EE01509E
- Gupta MK, Singh B, Goel P, et al. Lithium diffusion in Li2X (X = O,S,and Se): Ab initio simulations and inelastic neutron scattering measurements. Phys Rev B [Internet]. 2019;99:224304. Available from: https://link.aps.org/doi/10.1103PhysRevB.99.224304
- Luo H, Tang S, Liang C, et al. Correlation governs the impurity (Ti, Zr, Hf) diffusion in face−centered cubic iridium through first−principles calculation. Calphad [Internet]. 2022;77:102433. doi:10.1016/j.calphad.2022.102433 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0364591622000402
- Gopal P, Srinivasan SG. First-principles study of self- and solute diffusion mechanisms inγ’-Ni3Al. Phys Rev B [Internet]. 2012;86:014112. Available from: https://link.aps.org/doi/10.1103PhysRevB.86.014112
- Zhang X, Deng H, Xiao S, et al. First-principles calculation of self-diffusion coefficients in Ni3Al. J Alloys Compd. 2014;612:361–364. doi:10.1016/j.jallcom.2014.05.218
- Janotti A, Krčmar M, Fu CL, et al. Solute diffusion in metals: larger atoms can move faster. Phys Rev Lett. 2004;92:085901. doi:10.1103/PhysRevLett.92.085901
- Ren Z, Xue Z, Zhang X, et al. Atomic diffusion mediated by vacancy defects in L12-Zr3Al: a first-principles study. J Alloys Compd. 2020;821:153223. doi:10.1016/j.jallcom.2019.153223
- Naghavi SS, Hegde VI, Wolverton C. Diffusion coefficients of transition metals in fcc cobalt. Acta Mater. 2017;132:467–478. doi:10.1016/j.actamat.2017.04.060
- Bakker H, Westerveld JPA. Expression for the diffusion coefficient in intermetallic compounds with cubic structures. Phys Status Solidi. 1988;145:409–417. doi:10.1002/pssb.2221450206
- Barlow IC, Jones H, Rainforth WM. Evolution of microstructure and hardening, and the role of Al3Ti coarsening, during extended thermal treatment in mechanically alloyed Al-Ti-O based materials. Acta Mater [Internet]. 2001;49:1209–1224. doi:10.1016/S1359-6454(01)00016-7 Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359645401000167
- Thiyaneshwaran N, Sivaprasad K, Ravisankar B. Nucleation and growth of TiAl3 intermetallic phase in diffusion bonded Ti/Al Metal Intermetallic Laminate. Sci Rep [Internet]. 2018;8:16797. Available from: https://www.nature.com/articles/s41598-018-35247-0
- Tardy J, Tu KN. Solute effect of Cu on interdiffusion in Al3Ti compound films. Phys Rev B [Internet]. 1985;32:2070–2081. Available from: https://link.aps.org/doi/10.1103PhysRevB.32.2070
- Ikeda T, Almazouzi A, Numakura H, et al. Single-phase interdiffusion in Ni3Al. Acta Mater [Internet]. 1998;46:5369–5376. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1359645498002092
- Rothova V, Cermak J. Bulk and grain boundary diffusion of 67 Ga in Ni3Al—influence of composition. Intermetallics [Internet]. 2005;13:113–120. doi:10.1016/j.intermet.2004.06.006 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0966979504001840
- Shi Y, Frohberg G, Wever H. Diffusion of63Ni and114mIn in the γ′-phase Ni3Al. Phys Status Solidi [Internet]. 1995;152:361–375. Available from: https://onlinelibrary.wiley.com/doi/10.1002pssa.2211520205
- Van der Ven A, Thomas JC, Xu Q, et al. Linking the electronic structure of solids to their thermodynamic and kinetic properties. Math Comput Simul [Internet]. 2010;80:1393–1410. doi:10.1016/j.matcom.2009.08.008 Available from: https://linkinghub.elsevier.com/retrieve/pii/S0378475409002456
- Mattsson TR, Mattsson AE. Calculating the vacancy formation energy in metals: Pt, Pd, and Mo. Phys Rev B [Internet]. 2002;66:214110. Available from: https://link.aps.org/doi/10.1103PhysRevB.66.214110
- Carling K, Wahnström G, Mattsson TR, et al. Vacancies in metals: from first-principles calculations to experimental data. Phys Rev Lett. 2000;85:3862–3865. doi:10.1103/PhysRevLett.85.3862
- Huntington HB. Electromigration in metals. Diffus Solids [Internet]. 1975;Elsevier:303–352. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780125226608500118
- Henkelman G, Arnaldsson A, Jónsson H. A fast and robust algorithm for Bader decomposition of charge density. Comput Mater Sci [Internet]. 2006;36:354–360. Available from: https://www.science.org/doi/10.1126science.252.5012.1566.b