
Abstract
Human mutations in MTHFD1 have recently been identified in patients with severe combined immunodeficiency (SCID). SCID results from inborn errors of metabolism that cause impaired T- and B-cell proliferation and function. One of the most common causes of SCID is adenosine deaminase (ADA) deficiency, which ultimately inhibits DNA synthesis and cell division. MTHFD1 has been shown to translocate to the nucleus during S-phase of the cell cycle; this localization is critical for synthesis of thymidyate (dTMP or the “T” base in DNA) and subsequent progression through the cell cycle and cell proliferation. Identification of MTHFD1 mutations that are associated with SCID highlights the potential importance of adequate dTMP synthesis in the etiology of SCID.
Introduction
Primary immunodeficiencies (PIDs) can be a consequence of rare inborn errors of metabolism resulting in immunological insufficiencies. Severe combined immunodeficiency (SCID), generally considered the most severe type of PID, is defined by impaired adaptive immunity and T- and B-cell function. PIDs and SCID can result from defects in nucleotide, carbohydrate, amino acid, or lipid metabolism as well as from mitochondrial disorders.Citation1 Recently, expanded newborn screening has revealed that the prevalence of SCID is about twice as high as previously estimated.Citation1 Agnostic genome-wide DNA discovery approaches, such as whole-exome sequencing (WES), have also revealed previously unidentified genetic causes of SCID, which have implicated new metabolic pathways and associated nutritional exposures that may interact with and modify genetically-sensitized immunological phenotypes. In 2011, a girl presented with SCID combined with several features of impaired folate/vitamin B12-dependent one-carbon metabolism, including megaloblastic anemia, hyperhomocystenemia, and methylmalonic aciduria. Exome sequencing revealed 2 deleterious mutations in the MTHFD1 gene.Citation2 More recently, 4 additional MTHFD1-deficient patients have been identified using similar approaches.Citation3 Subsequent studies in fibroblasts from the original patient revealed that impaired thymidylate (dTMP) synthesis, leading to genome instability, was the primary metabolic defect causally related to SCID.Citation4 While inborn errors in the adenosine deaminase (ADA) gene and other purine synthesis genes are classically associated with SCID, this is the first study to provide evidence that impaired folate-dependent de novo thymidylate (dTMP) synthesis could contribute to the etiology of SCID, and suggests the potential for nutrition-based regimens for management and prevention of SCID.
Causes of SCID
All cases of SCID involve diminished T-cell proliferation and function, and most cases also involve insufficient B-cell proliferation.Citation1 Therefore, mutations that influence any basic cellular process that impair T-cell and/or B-cell survival and/or proliferation may present as SCID.Citation1,5 Infants with SCID are susceptible to life-threatening recurrent infections, especially respiratory and gut infections.Citation1,5 Many cases of SCID are treatable and can be managed with stem cell transplantation or enzyme replacement therapy, highlighting the importance of early detection and diagnosis.Citation1
SCID does not have a single etiology. It may result from genetic mutations in pathways involved in cytokine production, immuloglobin formation (i.e. VDJ recombination) and synthesis of T-cell antigen receptors.Citation6 Processes involved in DNA synthesis, DNA repair, and cell division have also been implicated, including impaired function of cell cycle checkpoint proteins and components of the replication machinery such as helicases and repair proteins.Citation6 However, the etiology of immunodeficiencies includes inborn errors of metabolism, including impairments in micronutrient and macronutrient metabolism.Citation7 Adenosine deaminase (ADA) deficiency is the classical metabolic cause of SCID, and accounts for 15–20% of all SCID cases.Citation8 ADA functions in purine catabolism, and its deficiency results in the accumulation of the toxic metabolites deoxyadenosine (dAdo) and deoxyadenosine triphosphate (dATP). These metabolites inhibit ribonucleotide reductase (RNR), a key enzyme in the conversion of deoxyribonucleotide triphosphates (NTPs) to dNTPs. Under normal physiological conditions, dATP regulates RNR activity through feedback inhibition. The accumulation of dATP in ADA deficiency leads to impaired synthesis of other DNA precursor dNTPs, resulting in impaired DNA replication and repair, DNA damage, and apoptosis. Although dAdo and dATP accumulation may cause SCID through multiple mechanisms,Citation9 other genes that encode enzymes that function in purine metabolism, including purine nucleotide phosphorylase (PNP), are associated with less severe combined immunodeficiencies. PNP deficiency causes accumulation of deoxyguanosine triphosphate (dGTP), which also inhibits RNR, perturbs dTNP pools and decreases rates of DNA synthesis.Citation7 The identification of MTHFD1 mutations as a potential contributor to the etiology of SCID emphasizes the importance of nucleotide biosynthesis to SCID phenotypes and expands the causal pathways to include de novo nucleotide biosynthesis, and also raises the possibility for the involvement of nutritional factors, including folate, in the etiology of SCID.
Folate and One-Carbon Metabolism
Impairments in folate-mediated one-carbon metabolism (FOCM) have been shown to be associated with risk for congenital anomalies, certain cancers, neurodegeneration and vascular disease and cognitive decline.Citation10-14 Folate, also known as vitamin B9, is a water-soluble vitamin found in fresh fruits and vegetables or as an oxidized, synthetic pro-vitamin known as folic acid. Folic acid is available as a dietary supplement and is added to fortified foods and converted to natural folate in cells to serve as a cofactor. FOCM refers to a highly interconnected network of intracellular reactions that utilize folate and are necessary for synthesis of 3 of the 4 nucleotide bases in DNA (purines adenosine (A) and guanosine (G) and thymidylate (dTMP)) and for the remethylation of homocysteine to methionine ().Citation15
Figure 1. One-carbon metabolism in the cytoplasm and nucleus. The products of one-carbon metabolism, purines, thymidylate (dTMP) and methionine are shown in red. Sources of one-carbon units are shown in green. Formate generated in the mitochondria serves as the major source of one-carbon units for cytoplasmic and nuclear one-carbon metabolism. Serine is also a source of one-carbon units. Enyzmes that translocate to the nucleus during S-phase of the cell cycle are surrounded by dashed-line boxes. THF, tetrahydrofolate; dTMP, thymidylate; MTHFD1, methylenetetrahydrofolate dehydrogenase 1, (S) synthetase activity, (C) cyclohydrolase activity, (D) dehydrogenase activity; SHMT1, cytoplasmic serine hydroxymethyltransferase; TYMS, dTMP synthase; MTR, methionine synthase; AdoMet, S-adenosylmethionine; AdoHcy, S-adenosylhomocysteine; MTHFR, methylenetetrahdryfolate reductase.
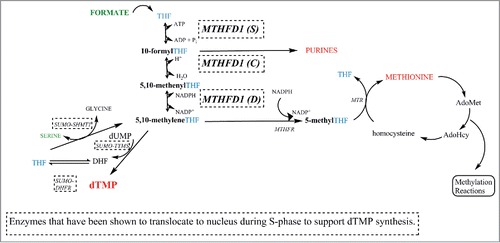
Functional biomarkers of FOCM capacity are commonly used to assess the function of de novo dTMP synthesis and homocysteine remethylation, but are lacking for de novo purine biosynthesis. Impaired dTMP synthesis leads to increased uracil (dU) misincorporation into DNA, DNA damage, impaired cell division, and megaloblastic anemia. Biomarkers of impaired homocysteine remethylation and cellular methylation capacity include depressed plasma methionine and S-adenosylmethionine (SAM, the principal methyl donor co-factor in cellular metabolism), elevated plasma homocysteine and S-adenosylhomocysteine, chromatin hypomethylation, and altered gene expression.
Inherited disorders of folate- and vitamin B12-dependent one-carbon metabolism are often characterized by both elevated homocysteine/impaired methionine synthesis and impaired de novo thymidylate biosynthesis leading to megaloblastic anemia. Elevated homocysteine is a biomarker for cardiovascular and cerebrovascular disease,Citation16 although lowering homocysteine levels does not improve outcomes.Citation17-19 Homocysteine levels are regulated through the activity of methionine synthase (MTR), betaine homocysteine methyltransferase (BHMT), and cystathionine β synthase (CBS). While folate deficiency can impair both de novo thymidylate synthesis and homocysteine remethylation, mutations that impact homocysteine metabolism and result in elevated homocysteine in the absence of impairments of de novo thymidylate biosynthesis (such as severe MTHFR deficiency) do not result in megaloblastic anemia, which is caused by impaired de novo thymidylate biosynthesis. The MTHFR 677C>T polymorphism, which has an allelic frequency as high as 70% in some populations,Citation20-22 is associated with decreased 5-methylTHF production, lower rates of methionine synthesis, elevated homocysteine, and a low folate status but improved de novo thymidylate biosynthesis.Citation23
Folate-dependent pathways and folate-utilizing enzymes within the cell compete for a limited amount of available folate cofactors, and this competition is most pronounced for binding the cofactor 5–10-methylenetetrahydrofolate (5,10-methyleneTHF). 5, 10-methyleneTHF exists at a branch point in FOCM, and is either used directly for dTMP synthesis or irreversibly methylated to form 5-methylTHF for methionine synthesis.Citation24 MTHFD1 is the primary source of folate-activated one-carbon units for de novo dTMP biosynthesis and homocysteine remethylation, and plays a key role in regulating the partitioning of 5,10-methyleneTHF to either homocysteine remethylation or de novo dTMP synthesis. MTHFD1 establishes this metabolic priority through its nuclear localization.Citation4,25 In the cytosol and nucleus, MTHFD1 is a trifunctional enzyme that serves as a primary entry point of chemically-activated single-carbon units into one carbon metabolism, by converting formate and THF to 10-formylTHF through its synthase domain, and by catalyzing the NADPH-dependent interconversion of the one-carbon substituted folate cofactors 10-formylTHF and 5,10-methyleneTHF through the cyclohydrolase/dehydrogenase (C/D) activity ().Citation25 Serine hydroxymethyltransferase (SHMT) can also generate 5,10-methyleneTHF using serine as a one-carbon source for dTMP synthesis and homocysteine remethylation in the nucleus and cytoplasm, but it makes relatively minor contributions to 5,10-methyleneTHF pools ().Citation24
Recent studies have demonstrated that nuclear localization of MTHFD1 regulates the partitioning of 5,10-methyleneTHF between homocysteine remethylation and de novo thymidylate biosynthesis. The folate-dependent enzymes of the de novo thymidylate cycle, including thymidylate synthase (TYMS), dihydrofolate reductase (DHFR), MTHFD1, and SHMT1/SHMT2α, translocate to the nucleus during the S-phase of the cell cycle and in response to UV radiation in mammalian cells.Citation25,26 These enzymes form a complex with the DNA replication complex to synthesize dTMP at sites of DNA synthesis.Citation27 Recently, we have shown that MTHFD1, which is the primary source of one-carbon units for 5,10-methyleneTHF and dTMP synthesis, translocates to the nucleus in response to folate deficiency and that it colocalizes with SHMT1, which serves as a scaffold for the dTMP synthesis enzyme complex.Citation25,27 Increased MTHFD1 nuclear enrichment during folate deficiency shifts the flux of one-carbon units in the form of 5,10-methyleneTHF from methionine synthesis, which occurs in the cytoplasm, to nuclear dTMP synthesis.
Vitamin B12 deficiency has also been linked to impaired de novo dTMP biosynthesis. It is required for the methionine synthase-dependent conversion of homocysteine to methionine, which regenerates THF from 5-methylTHF in the cytoplasm. In vitamin B12 deficiency, THF cannot be regenerated, causing folate co-factors to accumulate as 5-methylTHF. This comes at the expense of the availability of THF for other folate-dependent reactions such as purine and dTMP synthesis, thus creating a functional folate deficiency. This “methyl trap” then leads to impaired DNA synthesis and megaloblastic anemia.Citation28
One-Carbon Metabolism Pathways and SCID
Inborn errors in two FOCM genes have been implicated in SCID, among other adverse pathologies.Citation6 Hereditary folate malabsorption (HFM) is caused by mutations in the proton-coupled folate transporter (PCFT), which is responsible for intestinal folate uptake.Citation29,30 HFM patients exhibit severe folate deficiency, often with megaloblastic anemia and SCID, and can develop neurological symptoms in the absence of parenteral folate treatment.Citation30 Similarly, patients with deleterious mutations in transcobalamin (TC, product of the TCN2 gene), which is responsible for transporting vitamin B12 to peripheral tissues after intestinal absorption, also exhibit megaloblastic anemia, SCID, and numerous neurological impairments.Citation31–33 The lack of vitamin B12 within the cell leads to accumulation of intracellular folate as 5-methylTHF and a functional deficiency of other folate cofactors (by forming a “methyl trap” as described above). Impaired vitamin B12 metabolism and vitamin B12 deficiency have also been associated with impaired bone marrow function and immunological complications,Citation34 sometimes in conjunction with megaloblastic anemia. However megaloblastic anemia due to vitamin B12 deficiency does not always present with an impaired immune response.
Inborn errors of metabolism can result in metabolic impairments that may be managed and/or ameliorated with nutritional interventions that include dietary restrictions or nutrient supplementation. For example, the low tissue folate and abnormal hematological measures that characterize HFM can often be reversed with high-dose reduced folate treatment.Citation35,36 Similarly, patients with TC deficiency also exhibit clinical responses to vitamin B12 therapy.Citation37 However, there can be significant inter-individual variation in responses to these nutrient-based therapies,Citation37,38 likely due to the complexity of gene-nutrient interactions that underlie the biochemistry and physiology of human metabolism.Citation39
The Role of MTHFD1 Mutations in Folate Pathways and SCID
Mthfd1 has been shown to be an essential gene in mice, and human MTHFD1 single-nucleotide polymorphisms are associated with adverse pregnancy outcomes, including neural tube defects and congenital heart defects.Citation40–42 The recently reported patient with severe MTHFD1 deficiency presented with SCID and other classical signs of impaired vitamin B12 metabolism including elevated plasma homocysteine, decreased plasma methionine and megoblastic anemia. The proband was shown to have inherited deleterious mutations in both MTHFD1 alleles.Citation2 One allele resulted in an early stop codon in the C/D domain and the other resulted in a R173C mutation in the NADP binding site. In vivo studies comparing proband and matched-control fibroblasts revealed that patient fibroblasts expressed 50% less full-length MTHFD1 protein and displayed compromised methionine and dTMP synthesis; de novo purine synthesis was not affected. MTHFD1-catalyzed incorporation of formate-derived one-carbon units into methionine (a cytoplasmic process) was reduced by 90% in the patient's fibroblasts, while formate incorporation into dTMP (which occurs in the nucleus) was reduced by only 50%.Citation4 The decrease in de novo dTMP synthesis was associated with a 15% increase in uracil misincorporation into DNA and severe DNA damage. Further studies showed that the MTHFD1 protein was preferentially shuttled to the nucleus in patient fibroblasts. This increased nuclear translocation helped to protect nuclear dTMP synthesis at the expense of cytoplasmic methionine synthesis, which limited uracil misincorporation into DNA. The mechanisms by which MTHFD1 translocates to the nucleus and “responds” to impaired dTMP synthesis are currently under investigation.
It is likely that the impairment in dTMP synthesis, and not homocysteine remethylation, is responsible for the patient's SCID phenotype. There are numerous inborn errors in homocysteine remethylation and transsulfuration that are associated with high plasma homocysteine levels, but those that do not result in decreased dTMP synthesis (CBS deficiency, severe MTHFR deficiency) are not associated with hematological impairments including megaloblastic anemia or SCID.Citation16 These findings highlight the importance of de novo dTMP synthesis in maintaining genome stability, and by extension cell viability. The lack of association between elevated homocysteine and SCID in other inborn errors of metabolism suggests that the presentation of SCID in the MTHFD1-deficient proband is likely caused by impaired dTMP synthesis. MTHFD1-deficient patient fibroblasts also exhibited increased nuclear γH2AX levels, which serve as a marker of DNA double-strand breaks, relative to control fibroblasts. Interestingly, fibroblasts from SCID patients with ADA deficiency did not exhibit impaired de novo dTMP synthesis or increased nuclear γH2AX levels relative to control fibroblasts,Citation4 indicating that MTHFD1 and ADA deficiency cause SCID by distinct mechanisms that are independent of DNA damage.
Conclusion
Impaired purine nucleotide metabolism (as in ADA deficiency) and DNA synthesis have been shown to be causal in several types of SCID and other less severe PIDs.Citation6 As mentioned above, next-generation sequencing has revealed that the prevalence of SCID is much higher than previously appreciated and that it is associated with previously undiagnosed inborn errors of metabolism.Citation1 The folate and vitamin B12 metabolic pathways are tightly interconnected and the binding capacity of intracellular folate-utilizing enzymes exceeds the amount of available cofactor.Citation15 Therefore, the flow of one-carbon units to either nucleotide synthesis or methionine synthesis is strongly regulated by enzyme compartmentalization, multi-protein complex formation, and substrate channeling.Citation15 While numerous inborn errors of metabolism lead to impaired homocysteine remethylation and/or megaloblastic anemia, only severe disruption of folate absorption or vitamin B12 delivery (PCFT and TC mutations, respectively) have previously been associated with SCID. The association of MTHFD1 deficiency with SCID highlights the potential importance of de novo dTMP synthesis, as opposed to impaired purine synthesis (as observed in ADA deficiency), for lymphocyte proliferation and effective immune response. Although vitamin therapies, such as folic acid supplementation, may not be sufficient to overcome severe MTHFD1 mutations that lead to SCID, nutritional therapies that provide the cell with dTMP for DNA synthesis, including nucleoside supplementation, may prove to be beneficial, as has been shown for neural tube defect prevention in animal models.Citation43
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Funding for this study was provided by the National Institutes of Health R37DK58144 to PJS.
References
- Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, Baker M, Ballow M, Bartoshesky LE, Bonilla FA, et al. Newborn Screening for Severe Combined Immunodeficiency in 11 Screening Programs in the United States. Jama-J Am Med Assoc 2014; 312(7):729-38; http://dx.doi.org/10.1001/jama.2014.9132
- Watkins D, Schwartzentruber JA, Ganesh J, Orange JS, Kaplan BS, Nunez LD, Majewski J, Rosenblatt DS. Novel inborn error of folate metabolism: identification by exome capture and sequencing of mutations in the MTHFD1 gene in a single proband. J Med Genet 2011; 48(9):590-2; http://dx.doi.org/10.1136/jmedgenet-2011-100286
- Burda P, Kuster A, Hjalmarson O, Suormala T, Bürer C, Lutz S, Roussey G, Christa L, Asin-Cayuela J, Kollberg G, et al. Characterization and review of MTHFD1 deficiency: four new patients, cellular delineation and response to folic and folinic acid treatment. J Inherit Metab Dis 2015; 38(5):863-72.
- Field MS, Kamynina E, Watkins D, Rosenblatt DS, Stover PJ. Human mutations in methylenetetrahydrofolate dehydrogenase 1 impair nuclear de novo thymidylate biosynthesis. Proc Natl Acad Sci USA 2015; 112(2):400-5; http://dx.doi.org/10.1073/pnas.1414555112
- Fischer A. Severe combined immunodeficiencies (SCID). Clin Exp Immunol 2000; 122(2):143-9; http://dx.doi.org/10.1046/j.1365-2249.2000.01359.x
- Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Franco JL, Gaspar HB, Holland SM, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol 2014; 5:162
- Parvaneh N, Quartier P, Rostami P, Casanova JL, de Lonlay P. Inborn errors of metabolism underlying primary immunodeficiencies. J Clin Immunol 2014; 34(7):753-71; http://dx.doi.org/10.1007/s10875-014-0076-6
- Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Exp Opin Biol Ther 2014; 14(6):789-98; http://dx.doi.org/10.1517/14712598.2014.895811
- Cassani B, Mirolo M, Cattaneo F, Benninghoff U, Hershfield M, Carlucci F, Tabucchi A, Bordignon C, Roncarolo MG, Aiuti A. Altered intracellular and extracellular signaling leads to impaired T-cell functions in ADA-SCID patients. Blood 2008; 111(8):4209-19; http://dx.doi.org/10.1182/blood-2007-05-092429
- Wolff T, Witkop CT, Miller T, Syed SB, Force USPST. Folic acid supplementation for the prevention of neural tube defects: an update of the evidence for the US. Preventive Services Task Force. Annals Int Med 2009; 150(9):632-9; http://dx.doi.org/10.7326/0003-4819-150-9-200905050-00010
- Kronenberg G, Colla M, Endres M. Folic acid, neurodegenerative and neuropsychiatric disease. Curr Mol Med 2009; 9(3):315-23; http://dx.doi.org/10.2174/156652409787847146
- Ames BN. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutation Res 2001; 475(1-2):7-20; http://dx.doi.org/10.1016/S0027-5107(01)00070-7
- Choi SW, Mason JB. Folate and carcinogenesis: an integrated scheme. J Nutri 2000; 130(2):129-32
- Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci USA 1997; 94(7):3290-5
- Stover PJ, Field MS. Trafficking of intracellular folates. Adv Nutri 2011; 2(4):325-31; http://dx.doi.org/10.3945/an.111.000596
- Iacobazzi V, Infantino V, Castegna A, Andria G. Hyperhomocysteinemia: related genetic diseases and congenital defects, abnormal DNA methylation and newborn screening issues. Mol Genet Metabol 2014; 113(1-2):27-33; http://dx.doi.org/10.1016/j.ymgme.2014.07.016
- Clarke R, Halsey J, Lewington S, Lonn E, Armitage J, Manson JE, Bønaa KH, Spence JD, Nygård O, Jamison R, et al. Effects of lowering homocysteine levels with B vitamins on cardiovascular disease, cancer, and cause-specific mortality: Meta-analysis of 8 randomized trials involving 37 485 individuals. Arch Internal Med 2010; 170(18):1622-31; http://dx.doi.org/10.1001/archinternmed.2010.348
- Zhou YH, Tang JY, Wu MJ, Lu J, Wei X, Qin YY, Wang C, Xu JF, He J. Effect of folic acid supplementation on cardiovascular outcomes: a systematic review and meta-analysis. PloS One 2011; 6(9):e25142; http://dx.doi.org/10.1371/journal.pone.0025142
- Pan Y, Guo LL, Cai LL, Zhu XJ, Shu JL, Liu XL, Jin HM. Homocysteine-lowering therapy does not lead to reduction in cardiovascular outcomes in chronic kidney disease patients: a meta-analysis of randomised, controlled trials. Br J Nutri 2012; 108(3):400-7; http://dx.doi.org/10.1017/S0007114511007033
- Antonio-Vejar V, Del Moral-Hernández O, Alarcón-Romero LC, Flores-Alfaro E, Leyva-Vázquez MA, Hernández-Sotelo D, Illades-Aguiar B. Ethnic variation of the C677T and A1298C polymorphisms in the methylenetetrahydrofolate-reductase (MTHFR) gene in southwestern Mexico. Genetics Mol Res 2014; 13(3):7950-7; http://dx.doi.org/10.4238/2014.September.29.8
- Binia A, Contreras AV, Canizales-Quinteros S, Alonzo VA, Tejero ME, Silva-Zolezzi I. Geographical and ethnic distribution of single nucleotide polymorphisms within genes of the folatehomocysteine pathway metabolism. Genes Nutri 2014; 9(5):421; http://dx.doi.org/10.1007/s12263-014-0421-7
- Wilcken B, Bamforth F, Li Z, Zhu H, Ritvanen A, Renlund M, Stoll C, Alembik Y, Dott B, Czeizel AE, et al. Geographical and ethnic variation of the 677C>T allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): findings from over 7000 newborns from 16 areas world wide. J Med Genet 2003; 40(8):619-25; http://dx.doi.org/10.1136/jmg.40.8.619
- Tsang BL, Devine OJ, Cordero AM, Marchetta CM, Mulinare J, Mersereau P, Guo J, Qi YP, Berry RJ, Rosenthal J, Crider KS, et al. Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: a systematic review and meta-analysis of trials and observational studies. Am J Clin Nutrt 2015; 101(6):1286-94
- Herbig K, Chiang EP, Lee LR, Hills J, Shane B, Stover PJ. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J Biol Chem 2002; 277(41):38381-9; http://dx.doi.org/10.1074/jbc.M205000200
- Field MS, Kamynina E, Agunloye OC, Liebenthal RP, Lamarre SG, Brosnan ME, Brosnan JT, Stover PJ. Nuclear enrichment of folate cofactors and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) protect de novo thymidylate biosynthesis during folate deficiency. J Biol Chem 2014; 289(43):29642-50; http://dx.doi.org/10.1074/jbc.M114.599589
- Fox JT, Shin WK, Caudill MA, Stover PJ. A UV-responsive internal ribosome entry site enhances serine hydroxymethyltransferase 1 expression for DNA damage repair. J Biol Chem 2009; 284(45):31097-108; http://dx.doi.org/10.1074/jbc.M109.015800
- Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J Biol Chem 2012; 287(10):7051-62; http://dx.doi.org/10.1074/jbc.M111.333120
- Hoffbrand AV, Herbert V. Nutritional anemias. Seminars Hematol 1999; 36(4 Suppl 7):13-23
- Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006; 127(5):917-28; http://dx.doi.org/10.1016/j.cell.2006.09.041
- Kishimoto K, Kobayashi R, Sano H, Suzuki D, Maruoka H, Yasuda K, Chida N, Yamada M, Kobayashi K. Impact of folate therapy on combined immunodeficiency secondary to hereditary folate malabsorption. Clin Immunol 2014; 153(1):17-22; http://dx.doi.org/10.1016/j.clim.2014.03.014
- Teplitsky V, Huminer D, Zoldan J, Pitlik S, Shohat M, Mittelman M. Hereditary partial transcobalamin II deficiency with neurologic, mental and hematologic abnormalities in children and adults. Isr Med Assoc J 2003; 5(12):868-72
- Watkins D, Rosenblatt DS. Inborn errors of cobalamin absorption and metabolism. Am J Med Genet Seminars Med Genet 2011; 157C(1):33-44; http://dx.doi.org/10.1002/ajmg.c.30288
- Trakadis YJ, Alfares A, Bodamer OA, Buyukavci M, Christodoulou J, Connor P, Glamuzina E, Gonzalez-Fernandez F, Bibi H, Echenne B, et al. Update on transcobalamin deficiency: clinical presentation, treatment and outcome. J Inherited Metab Dis 2014; 37(3):461-73; http://dx.doi.org/10.1007/s10545-013-9664-5
- Solomon LR. Disorders of cobalamin (vitamin B12) metabolism: emerging concepts in pathophysiology, diagnosis and treatment. Blood Rev 2007; 21(3):113-30; http://dx.doi.org/10.1016/j.blre.2006.05.001
- Zhao R, Min SH, Qiu A, Sakaris A, Goldberg GL, Sandoval C, Malatack JJ, Rosenblatt DS, Goldman ID. The spectrum of mutations in the PCFT gene, coding for an intestinal folate transporter, that are the basis for hereditary folate malabsorption. Blood 2007; 110(4):1147-52; http://dx.doi.org/10.1182/blood-2007-02-077099
- Geller J, Kronn D, Jayabose S, Sandoval C. Hereditary folate malabsorption: family report and review of the literature. Medicine 2002; 81(1):51-68; http://dx.doi.org/10.1097/00005792-200201000-00004
- Haberle J, Pauli S, Berning C, Koch HG, Linnebank M. TC II deficiency: avoidance of false-negative molecular genetics by RNA-based investigations. J Hum Genet 2009; 54(6):331-4; http://dx.doi.org/10.1038/jhg.2009.34
- Thomas PK, Hoffbrand AV, Smith IS. Neurological involvement in hereditary transcobalamin II deficiency. J Neurol Neurosurg Psychiatry 1982; 45(1):74-77; http://dx.doi.org/10.1136/jnnp.45.1.74
- Ohlhorst SD, Russell R, Bier D, Klurfeld DM, Li Z, Mein JR, Milner J, Ross AC, Stover P, Konopka E. Nutrition research to affect food and a healthy life span. Am J Clin Nutri 2013; 98(2):620-5; http://dx.doi.org/10.3945/ajcn.113.067744
- Brody LC, Conley M, Cox C, Kirke PN, McKeever MP, Mills JL, Molloy AM, O'Leary VB, Parle-McDermott A, Scott JM, et al. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenasemethenyltetrahydrofolate cyclohydrolaseformyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: report of the Birth Defects Research Group. Am J Hum Genet 2002; 71(5):1207-15; PMID:12384833; http://dx.doi.org/10.1086/344213
- Parle-McDermott A, Kirke PN, Mills JL, Molloy AM, Cox C, O'Leary VB, Pangilinan F, Conley M, Cleary L, Brody LC, et al. Confirmation of the R653Q polymorphism of the trifunctional C1-synthase enzyme as a maternal risk for neural tube defects in the Irish population. Eur J Hum Genet 2006; 14(6):768-72; http://dx.doi.org/10.1038/sj.ejhg.5201603
- Parle-McDermott A, Pangilinan F, Mills JL, Signore CC, Molloy AM, Cotter A, Conley M, Cox C, Kirke PN, Scott JM, et al. A polymorphism in the MTHFD1 gene increases a mother's risk of having an unexplained second trimester pregnancy loss. Mol Hum Reprod 2005; 11(7):477-80; http://dx.doi.org/10.1093/molehr/gah204
- Martiniova L, Field MS, Finkelstein JL, Perry CA, Stover PJ. Maternal dietary uridine causes, and deoxyuridine prevents, neural tube closure defects in a mouse model of folate-responsive neural tube defects. Am J Clin Nutri 2015; 101(4):860-9; http://dx.doi.org/10.3945/ajcn.114.097279