
ABSTRACT
Neurocristopathies form a specific group of rare genetic diseases in which a defect in neural crest cell development is causal. Because of the large number of neural crest cell derivatives, distinct structures/cell types (isolated or in combination) are affected in each neurocristopathy. The most important issues in this research field is that the underlying genetic cause and associated pathogenic mechanism of most cases of neurocristopathy are poorly understood. This article describes how a relatively simple insertional mutagenesis approach in the mouse has proved useful for identifying new candidate genes and pathogenic mechanisms for diverse neurocristopathies.
A forward genetic screen for neurocristopathy-associated genes
Neural crest cells (NCC) are a multipotent embryonic cell population that migrates extensively from the dorsal region of the neural tube (the central nervous system primordium). Such characteristics allow NCC to colonize several regions of the developing embryo and generate a wide array of specialized cell types such as melanocytes (pigment cells), peripheral neurons and glia, adrenal chromaffin cells, cranio-facial chondrocytes and osteoblasts as well as specific cell subsets of the heart, inner ear and thymus.Citation1 Accordingly, abnormal NCC development results in congenital malformations (e.g. aganglionic megacolon, cleft palate and conotruncal heart malformations) and cancers (e.g., neuroblastoma, melanoma and multiple endocrine neoplasia) that may concomitantly affect several tissues as seen in Waardenburg and CHARGE syndromes. These diverse pathological entities (>40) are collectively regrouped under the name neurocristopathy.Citation2,3 Neurocristopathies have also in common that a large number of cases are currently unexplained at the genetic level.
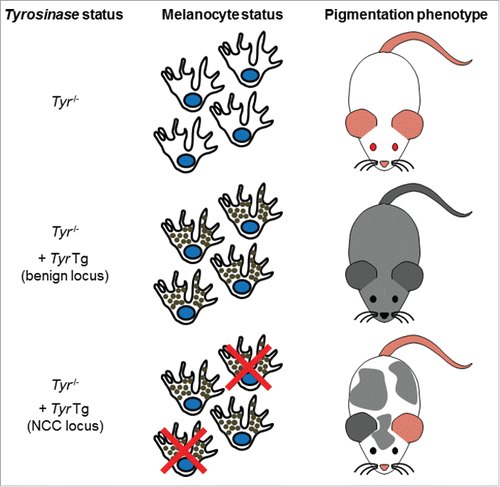
In two of our latest publications,Citation4,5 we show that the wide developmental potential of NCC can be used in screening efforts aimed at identifying new neurocristopathy-related genes. These articles clearly demonstrate the simplicity and usefulness of using pigmentation anomalies as a proxy for other NCC defects. The general idea of our approach is based on the facts that pigmentation is controlled via the secretion of melanin pigments by the NCC-derived melanocytes and that albino mice are devoid of pigmentation because of an inactivating mutation in the key melanin-producing enzyme Tyrosinase (Tyr).Citation6 In this regard, the development of a co-injection strategy involving the rescue of the albino mutation with a Tyr minigene has proved to be a clever way for facilitating identification of transgenic animals by simple visual inspection of coat color.Citation7,8 The description of the TashTCitation4 and HolsteinCitation5 mouse lines are now the proof of concept that the Tyr minigene can also be used as a potent screening tool for insertional mutagenesis-mediated identification of loci with key roles in NCC. Indeed, when the Tyr minigene is inserted in a locus important for NCC development, rescued pigmentation is non-uniform due to an insufficient number of melanocytes in the epidermis ().
Figure 1. Description of the pigmentation-based forward genetic screen in albino mice. Albino mice are unpigmented due to a G to C nucleotide change at position 308 of the Tyr gene, causing a cysteine to serine mutation at amino acid 103 of the Tyrosinase protein. Reintroduction of a functional Tyr gene via transgenesis can rescue the albino phenotype. Tyr transgene-induced pigmentation is uniform if inserted in a locus not important for NCC and not uniform if inserted in a locus important for NCC.
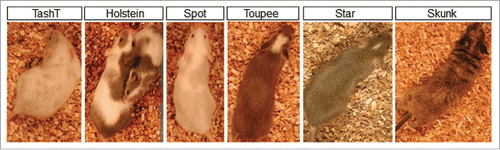
As of today, we have obtained five hypopigmented mutant lines out of a total of ˜200 lines generated. As shown in , these lines have been named after their distinctive pigmentation pattern observed in heterozygous animals. Importantly, when these heterozygotes are crossed to generate homozygotes, other NCC-related phenotypes show up. Among these, aganglionic megacolon is the most frequently observed, being present in three lines (TashT, Holstein and Spot). While this phenotype appears isolated – apart from the pigmentation defects – in the TashT and Holstein lines, it is observed in combination to inner ear defects in the Spot line. Consequently, this makes TashTCitation4 and HolsteinCitation5 new models for Hirshsprung's disease and Spot (to be described elsewhere) a new model for Waardenburg syndrome type 4. In the Toupee line (to be described elsewhere), homozygous animals display a complex phenotype that is reminiscent of CHARGE syndrome and which notably includes multiple congenital malformations affecting the face, palate, eyes, inner ears, heart, thymus and cranial nerves. Finally, in the Star line (to be described elsewhere), homozygosity appears to be embryonic lethal. Of note, a sixth line, named Skunk (to be described elsewhere) was also identified based on the presence of hyperpigmented patches.
Figure 2. Overview of the mutant lines obtained via pigmentation-based forward genetic screening. For each line, a characteristic pigmentation pattern can be observed in heterozygous animals.
Thanks to current high-throughput sequencing methods, both the site of transgene integration as well as the presence of associated small scale genomic rearrangements (insertions, duplications or deletions) have been precisely identified in four lines to date (TashT, Holstein, Spot and Toupee). Importantly, in all four cases, this resulted in the identification of a locus not previously associated with the modeled neurocristopathy. It is also noteworthy that, for three of these lines, the insertion has perturbed the function of nearby highly-conserved non-coding regulatory sequences. Considering that small-scale genomic rearrangements are more representative of the human disease state than single nucleotide polymorphisms,Citation9,10 this strongly suggests that a large number of neurocristopathy cases currently unexplained at the genetic level could be explained by similar outcomes. In this regard, although the ENCODE project has started to reveal the extent of the regulatory genome in both humans in mice,Citation11,12 the majority of it still awaits functional annotation. We are convinced that mouse models like ours will be instrumental for uncovering those regulatory elements that are relevant to human pathologies.
Important breakthroughs for Hirschsprung's disease
Hirschsprung's disease – the best known neurocristopathy – is a life-threatening congenital malformation of the enteric nervous system that affects 1 in 5000 newborns.Citation13 This severe condition is caused by the failure of NCC-derived enteric neural progenitors to reach the terminal regions of the developing intestines.Citation14 As these progenitors notably generate the peristalsis-controlling myenteric neural ganglia, the affected segment (aganglionic) remains tonically contracted, resulting in functional intestinal obstruction and accumulation of fecal material (megacolon). Hirschsprung's disease is clinically subdivided in short-segment (i.e. restricted to the rectosigmoid colon) and long-segment forms.Citation13 The short-segment form represents the vast majority of cases and is more common in males than females, with an overall ratio of ˜4:1. In addition to maleness, Down syndrome (trisomy 21) is another condition that predisposes to Hirschsprung's disease. Indeed, Down syndrome patients are estimated to be up to 100-fold more likely to develop Hirschprung's disease in comparison to the general population.Citation13
The vast majority of enteric neural progenitors are specifically derived from NCC of vagal origin – although a minor contingent is also provided by NCC of sacral origin.Citation15 The particular migration pattern of such “enteric” NCC (eNCC) is believed to be the main contributing factor to the pathogenesis of Hirschsprung's disease. In the mouse, eNCC of vagal origin initially reach the foregut mesenchyme around e9.5 and then migrate in the posterior direction to eventually reach the end of the hindgut by e14.5. Tight coordination of eNCC migration with cell proliferation, survival and differentiation mechanisms is critically required for completion of enteric nervous system formation.Citation16 Therefore, disruption of any of these processes can result in aganglionic segments of varying length.Citation17
At the genetic level, Hirschsprung's disease is considered a complex disease and current estimations indicate that mutations in known genes collectively explains less than one-third of the cases.Citation18 The main Hirschsprung's disease-associated genes encode members of GDNF/RET and EDN3/EDNRB signaling pathways, both of which having been shown to regulate virtually all cellular processes involved in the formation of the enteric nervous system. Indeed, the study of mouse models bearing mutation of these genes revealed important roles for both of these signaling pathways in the control of migration, proliferation, survival and differentiation of eNCC.Citation17,19 On the other hand, both the male sex bias and the strong association with Down syndrome are intriguing hallmarks of Hirschsprung's disease that were – before our work with the TashT and Holstein lines (see below) – very poorly understood at the molecular level.
The TashT line is the first mouse model to display a partially penetrant aganglionic megacolon phenotype in a strong male-biased manner.Citation4 In contrast to the megacolon phenotype, colonic aganglionosis is almost fully penetrant in homozygous TashT animals (TashTTg/Tg). The sex bias in megacolon expressivity can be explained by the fact that the MaleTashTTg/Tg enteric nervous system ends around a “tipping point” of minimal colonic ganglionosis (˜80% of colon length) while the FemaleTashTTg/Tg enteric nervous system ends just beyond it. Detailed analysis of embryonic intestines – notably via time-lapse imaging as well as via a novel quantitative method developed in our lab Citation20 – revealed that aganglionosis in TashTTg/Tg animals is due to slower migration of eNCC. The TashT insertional mutation is located in a Chr.10 gene desert containing multiple highly conserved elements that exhibit repressive activity in reporter assays. RNAseq analyses and 3C (chromosome conformation capture) assays revealed that these silencer elements normally repress Fam162b (at 3.6Mb from the transgenic insertion) and that the TashT insertion results, at least in part, in the NCC-specific relief of repression of this uncharacterized gene. We are currently testing the hypothesis that these silencer elements might regulate supplemental genes using circular chromosome conformation capture assays coupled to high-throughput sequencing (4C-seq). The transcriptional signature of eNCC from TashTTg/Tg embryos is also notably characterized by the deregulation of genes encoding members of the most important signaling pathways for enteric nervous system formation – Gdnf/Ret and Edn3/Ednrb. Very interestingly, cross-analysis of this WT-vs-TashTTg/Tg dataset (1243 genes differently expressed >2-fold at p<0.001) with a more recent MaleTashTTg/Tg-vs-FemaleTashTTg/Tg data set (18 genes differently expressed >2-fold at p<0.001; unpublished) revealed that only one gene is significantly deregulated. This Y-linked gene is specifically overexpressed in MaleTashTTg/Tg eNCC and its candidacy for explaining the male bias of the TashT megacolon phenotype is currently being tested with new mouse models.
In the Holstein line, all homozygotes (HolTg/Tg) die from aganglionic megacolon around weaning age. No sex bias is observed for this phenotype and, accordingly, the enteric nervous system of both male and female HolTg/Tg animals fails to reach the minimal level of colonic ganglionosis discovered with the TashT line. The Holstein transgene insertion site is located upstream of the Collagen-6α-4 (Col6a4) gene on Chr.9. We found that this insertion induces an eNCC-specific upregulation of Col6a4 gene expression in the developing intestines, most likely via disruption of CTCF-mediated insulation from a large genomic region specifically active in NCC. This ultimately leads to an increase in total collagen VI protein levels in the extracellular matrix surrounding both the developing and the postnatal enteric nervous system. Other data – notably from time-lapse imaging and heterotopic transplantation studies – indicated that such increased collagen VI levels during development mainly results in slower migration of HolTg/Tg eNCC. Further ex vivo analyses revealed that collagen VI is simply a poor substratum for supporting eNCC migration and can even interfere with the migration-promoting effects of fibronectin. Finally, in total agreement with our findings made with HolTg/Tg mice, we also observed that the enteric ganglia from the ganglionated region of a majority of human Hirschsprung's disease patients are specifically surrounded by abundant collagen VI microfibrils. Importantly, we further found that this outcome was accentuated in patients also suffering from Down syndrome. We believe that this latter finding can be explained by the facts that human COL6A1 and COL6A2 genes are jointly located on human Chr.21q and that the collagen VI α1 and α2 chains encoded by these genes are critically required for the assembly of every collagen VI microfibril – together with any one of the structurally related α3, α4, α5, or α6 chains.Citation21,22 Therefore, this work not only allowed us to identify a new clinically-relevant pathogenic mechanism for Hirschsprung's disease but also to offer a plausible explanation to the predisposition of Down syndrome patients to Hirschsprung's disease.
Conclusion
The work mentioned here clearly demonstrates that our pigmentation-based forward genetic screen approach can allow identifying candidate loci as well as pathogenic mechanisms for neurocristopathies. Through this work, we expect to significantly improve the way children suffering from such life-threatening diseases – as well as their family – are currently being taken in charge.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The author thanks David W. Silversides in whose laboratory the transgenic mice were generated.
Funding
The Pilon laboratory is funded by grants from the Canadian Institute of Health Research (CIHR), the Natural Science and Engineering Research Council of Canada (NSERC), the Fonds de la recherche du Québec – Santé (FRQS) and the Fondation du grand défi Pierre Lavoie.
References
- Bronner ME, LeDouarin NM. Development and evolution of the neural crest: an overview. Dev Biol 2012; 366:2-9; PMID:22230617; http://dx.doi.org/10.1016/j.ydbio.2011.12.042
- Bolande RP. Neurocristopathy: its growth and development in 20 years. Pediatr Pathol Lab Med 1997; 17:1-25; PMID:9050057; http://dx.doi.org/10.1080/15513819709168343
- Etchevers HC, Amiel J, Lyonnet S. Molecular bases of human neurocristopathies. Adv Exp Med Biol 2006; 589:213-34; PMID:17076285; http://dx.doi.org/10.1007/978-0-387-46954-6_14
- Bergeron KF, Cardinal T, Toure AM, Beland M, Raiwet DL, Silversides DW, Pilon N. Male-biased aganglionic megacolon in the TashT mouse line due to perturbation of silencer elements in a large gene desert of chromosome 10. PLoS Genet 2015; 11:e1005093; PMID:25786024; http://dx.doi.org/10.1371/journal.pgen.1005093
- Soret R, Mennetrey M, Bergeron KF, Dariel A, Neunlist M, Grunder F, Faure C, Silversides DW, Pilon N. A collagen VI-dependent pathogenic mechanism for Hirschsprung's disease. J Clin Invest 2015; 125:4483-96; PMID:26571399; http://dx.doi.org/10.1172/JCI83178
- Yokoyama T, Silversides DW, Waymire KG, Kwon BS, Takeuchi T, Overbeek PA. Conserved cysteine to serine mutation in tyrosinase is responsible for the classical albino mutation in laboratory mice. Nucleic Acids Res 1990; 18:7293-8; PMID:2124349; http://dx.doi.org/10.1093/nar/18.24.7293
- Methot D, Reudelhuber TL, Silversides DW. Evaluation of tyrosinase minigene co-injection as a marker for genetic manipulations in transgenic mice. Nucleic Acids Res 1995; 23:4551-6; PMID:8524641; http://dx.doi.org/10.1093/nar/23.22.4551
- Overbeek PA, Aguilar-Cordova E, Hanten G, Schaffner DL, Patel P, Lebovitz RM, Lieberman MW. Coinjection strategy for visual identification of transgenic mice. Transgenic Res 1991; 1:31-7; PMID:1844573; http://dx.doi.org/10.1007/BF02512994
- Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet 2007; 39:S43-7; PMID:17597781; http://dx.doi.org/10.1038/ng2084
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med 2010; 61:437-55; PMID:20059347; http://dx.doi.org/10.1146/annurev-med-100708-204735
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/10.1038/nature11247
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 2014; 515:355-64; PMID:25409824; http://dx.doi.org/10.1038/nature13992
- Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 2008; 45:1-14; PMID:17965226; http://dx.doi.org/10.1136/jmg.2007.053959
- Bergeron KF, Silversides DW, Pilon N. The developmental genetics of Hirschsprung's disease. Clin Genet 2013; 83:15-22; PMID:23043324; http://dx.doi.org/10.1111/cge.12032
- Goldstein AM, Hofstra RM, Burns AJ. Building a brain in the gut: development of the enteric nervous system. Clin Genet 2013; 83:307-16; PMID:23167617; http://dx.doi.org/10.1111/cge.12054
- Lake JI, Heuckeroth RO. Enteric nervous system development: migration, differentiation, and disease. Am J Physiol Gastrointestinal Liver Physiol 2013; 305:G1-24; http://dx.doi.org/10.1152/ajpgi.00452.2012
- Obermayr F, Hotta R, Enomoto H, Young HM. Development and developmental disorders of the enteric nervous system. Nat Rev Gastroenterol Hepatol 2013; 10:43-57; PMID:23229326; http://dx.doi.org/10.1038/nrgastro.2012.234
- Alves MM, Sribudiani Y, Brouwer RW, Amiel J, Antinolo G, Borrego S, Ceccherini I, Chakravarti A, Fernandez RM, Garcia-Barcelo MM, et al. Contribution of rare and common variants determine complex diseases-Hirschsprung disease as a model. Dev Biol 2013; 382:320-9; PMID:23707863; http://dx.doi.org/10.1016/j.ydbio.2013.05.019
- Barlow A, de Graaff E, Pachnis V. Enteric nervous system progenitors are coordinately controlled by the G protein-coupled receptor EDNRB and the receptor tyrosine kinase RET. Neuron 2003; 40:905-16; PMID:14659090; http://dx.doi.org/10.1016/S0896-6273(03)00730-X
- Bergeron KF, Cardinal T, Pilon N. A quantitative cell migration assay for murine enteric neural progenitors. J Visualized Exp 2013; 18(79):e50709
- Fitzgerald J, Holden P, Hansen U. The expanded collagen VI family: new chains and new questions. Connective Tissue Res 2013; 54:345-50; http://dx.doi.org/10.3109/03008207.2013.822865
- Gara SK, Grumati P, Urciuolo A, Bonaldo P, Kobbe B, Koch M, Paulsson M, Wagener R. Three novel collagen VI chains with high homology to the alpha3 chain. J Biol Chem 2008; 283:10658-70; PMID:18276594; http://dx.doi.org/10.1074/jbc.M709540200