
ABSTRACT
Millions of children worldwide are born with rare and debilitating developmental disorders each year. Although an increasing number of these conditions are being recognized at the molecular level, the characterization of the underlying pathophysiology remains a grand challenge. This is often due to the lack of appropriate patient material or relevant animal models. Dogs are coming to the rescue as physiologically relevant large animal models. Hundreds of spontaneous genetic conditions have been described in dogs, most with close counterparts to human rare disorders. Our recent examples include the canine models of human Caffey (SLC37A2), van den Ende-Gupta (SCARF2) and Raine (FAM20C) syndromes. These studies demonstrate the pathophysiological similarity of human and canine syndromes, and suggest that joint efforts to characterize both human and canine rare diseases could provide additional benefits to the advancement of the field of rare diseases. Besides revealing new candidate genes, canine models allow access to experimental resources such as cells, tissues and even live animals for research and intervention purposes.
Introduction
Dog, Canis lupus familiaris, is considered to be the first domesticated animal. Well-supported evidence suggests that all dog breeds originate from the Eurasian gray wolf (Canis lupus lupus), potentially from Southeast Asia,Citation1 although the exact geographical location and the timing of the process remains elusive.Citation2 The majority of the 400 current breeds, each defined by specific physical and behavioral characteristics, are less than 200 y old. The phenotypic variation seen in dogs today is mainly a result of recent strong artificial selection driven by dog breeders admiring distinctive features. The establishment of registering bodies (e.g. kennel clubs), together with breeding standards, has led to controlled and restricted breeding practices. Many breeds have arisen from a limited number of founder animals and the use of popular sires is a common practice. As a consequence, each breed represents an isolated breeding population with high levels of phenotypic homogeneity, reduced genetic diversity and enrichment of breed-specific disorders.
The sequencing of the canine genome and subsequent development of powerful gene discovery tools has remarkably facilitated gene discoveries in disease, morphology and behavior.Citation3-5 Dogs possess a unique genetic architecture, consisting of an ancient short and recent long-range linkage disequilibrium (LD).Citation3,6 This genetic architecture facilitates gene discovery by providing a powerful mapping approach within a breed while an across-breed mapping may significantly narrow down the associated region in order to expedite the identification of the causal risk variant as efficiently exemplified in a study of canine ectodermal dysplasia.Citation7 The dog genome is less divergent from the human genome than from the mouse genome and, therefore, more of the human genomic sequence can be syntenically aligned to the genome of the dog than that of the mouse.Citation3 Dogs have approximately the same number of genes as humans, most of them being close orthologues.
Dog is a large animal model and physiologically and clinically more similar to human than mouse. Importantly, pet dogs share also the environmental conditions of their owners and are thus not only affected by genetic traits but also by “life style.” Hundreds of spontaneously occurring common canine conditions are analogous to human diseases such as diabetes, cancers, epilepsies, eye diseases and autoimmune diseases, not to mention the high numbers of rare monogenic diseases. Canine heritable monogenic diseases, with the current list of 201 known causative genetic variants, are recorded in the public database Online Mendelian Inheritance in Animals (OMIA) (http://omia.angis.org.au). Most of the genes underlying canine diseases are orthologues of the genes in the corresponding human rare conditions.
Well-recorded genealogical data is also available, in many places, for dogs, which can assist in the design of genetic studies. Purebred dog registries are maintained by kennel clubs or other equivalent organizations. For example, the Finnish Kennel Club maintains an open dog registry in Finland (http://jalostus.kennelliitto.fi) and contains useful information about each registered Finnish dog, e.g., pedigrees and some results of health examinations. Dogs also have good-quality veterinary medical care available and professional veterinary records are essential to reliable and successful research.
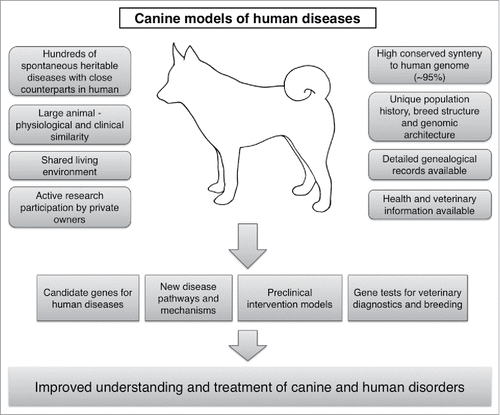
To utilize the canine models of human hereditary disorders, we have established a comparative genetic program in Finland with a large canine biobank, including ∼60 000 samples from over 300 breeds of dogs. Leveraging this biobank, the unique canine genetic architecture and genomic tools, the presence of hundreds of breed-specific conditions, and active research participation of the canine and veterinary communities, we have reported numerous new gene discoveries in dogs across disease groups,Citation7-13 including several developmental disorders that model human pediatric disorders. The identification of the genetic causes establishes the affected breeds as preclinical models, including examples such as the recombinant protein replacement for X-linked hypohidrotic ectodermal dysplasia (XLHED),Citation14 and gene therapies for hemophilia ACitation15 and retinal degeneration.Citation16 Given the accelerating pace of gene discoveries, new opportunities exist for intervention trials. A summary of the special features that facilitate the use of the dog as a complementary model to human disorders is shown in .
Figure 1. Various clinical, genetic, physiological and environmental characteristics in dogs make them excellent comparative models for facilitating the understanding and treatment of human disorders.
Our most recent study revealed 3 new developmental disorders and causative genes in dogs.Citation17 We identified SLC37A2 as novel candidate gene for human infantile cortical hyperostosis (Caffey disease) and implicated SCARF2 and FAM20C variants in the canine forms of van den Ende-Gupta and Raine syndromes, respectively. The study demonstrates the power of next-generation sequencing approaches in identifying the causative genes, the molecular diagnoses and the names of illnesses in canine syndromes with high relevance to rare human diseases. All 3 spontaneous canine conditions closely resemble the human conditions and provide physiologically relevant models to better understand poorly characterized gene functions and molecular pathologies toward new therapeutic innovations.
A novel sugar-phosphate transporter candidate gene for infantile hyperostosis
Craniomandibular osteopathy (CMO) is a self-limiting hyperostosis with a painful swelling of the jaw, leading to dysphagia and difficulty in opening the mouth in young dogs. It is equivalent to human infantile cortical hyperostosis.Citation18,19 We discovered a splicing defect in the SLC37A2 gene in CMO and propose it as an excellent candidate for mutation screening in Caffey patients. Our discovery is important since the molecular etiology remains unknown in many cases and early molecular diagnosis would avoid invasive procedures. Furthermore, it is still mechanistically unclear how the previously-discovered missense variant in COL1A1 could lead to self-limiting hyperostosis.Citation20,21 SLC37A2 may provide new insights to disease etiology.
SLC37A2 belongs to the SLC37 family of 4 ER-associated glucose-phosphate transporters.Citation22 It is abundantly expressed in disease-relevant tissues such as bone marrow and haematopoietic cell linages such as osteoclasts and macrophages,Citation23,24 and may play a central role in glucose homeostasis in osteogenesis. We hypothesize that an impaired function of SLC37A2 disturbs proper glucose supply in the osteoclasts, decreasing their overall activity, which in turn results in an imbalance between osteoblastic and osteoclastic functions in the developing bones leading to hyperostosis. Interestingly, SLC37A2 has been found also as a primary target for vitamin D,Citation25 which could be a potent therapeutic booster to alleviate clinical signs in affected dogs, which are due to diminished expression of wild type expression of SLC37A2.
Our canine study not only discovered a novel Caffey disease gene but also unraveled a physiological function of SLC37A2, providing new insights and hypotheses into infantile swelling diseases, which may be related to impaired glucose homeostasis during the bone development. The affected dogs now provide unique resources to address these hypotheses and other SLC37A2-related mechanisms in osteogenesis biology.
A canine model of van den Ende-Gupta syndrome
Our second example highlights the importance of the gene discovery to naming the illness and establishing a model for the human syndrome. We found a severe truncating mutation in SCARF2 in an unknown developmental syndrome in Wire Fox Terriers, characterized by severe mandibular prognathia and other skeletal abnormalities. The SCARF2 mutations have been found in less than 30 reported patients of van den Ende-Gupta syndrome (VDEGS). These patients are characterized by craniofacial and skeletal abnormalities including blepharophimosis, a flat and wide nasal bridge, narrow and beaked nose, hypoplastic maxilla with or without cleft palate and everted lower lip, prominent deformed ears, down-slanting eyes, arachnodactyly and camptodactyly.Citation26
Human and canine VDEGS patients share many similarities, including hypoplastic maxilla, dislocated radial head, patellar dislocation, deviated nasal septum and small eyes.Citation17 Therefore, the SCARF2 discovery established the canine diagnosis and a model for human VDEGS. This is important since the lack of a transgenic mouse model and scarcity of human patients highlight the role of affected dogs as a novel resource for understanding SCARF2 functions and molecular pathology. In addition, given that some of the VDEGS dogs survive past 10 years, they could potentially serve as preclinical models.
SCARF2 is a very poorly-characterized member of the scavenger receptor type F family.Citation27 It is a single-pass transmembrane protein with homology to calmodulin (CaM)-like Ca2+-binding proteins. The extracellular domain contains several putative epidermal growth factor-like (EGF) domains, and it has a number of positively-charged residues within the intracellular domain. Scarf2 is expressed in disease-relevant tissues such as epidermis, branchial arches, mandible, maxilla and urogenital ridge tissue of developing mouse embryos.Citation28,29 However, nothing is known about its function.
A detailed histopathological characterization of the canine VDEGS should be performed to learn about SCARF2 functions, regulation and the related cellular pathway, in order to improve our understanding of the disease mechanisms, which in turn could lead to therapeutic hypotheses. The established canine model provides powerful new resources toward these aims.
A canine model of Raine syndrome
FAM20C mutations have been described in human autosomal recessive osteosclerotic bone dysplasia (Raine syndrome). Less than 40 cases with variable severity and clinical heterogeneity have been reported including those which survive into childhood.Citation30-33 Typical characteristics include variable extent of hypophosphatemia, abnormal and hypomineralized teeth, craniofacial anomalies, such as exophthalmos, midface hypoplasia, microcephaly and cleft palate, as well as gingival hyperplasia, generalized osteosclerosis and intracerebral calcifications.
We found a recessive missense variant in the kinase domain of the FAM20C gene in a family of several affected dogs that suffered from severe dental wear and loss of teeth. Clinical findings in dogs were limited to severe hypomineralization of teeth, leading to extensive wear and inflammation as prominent features. We did not observe some of the typical gross changes described in Raine patients such as hypophosphatemia and craniofacial anomalies. Our canine study raises the question of whether a more limited human phenotype exists and should be targeted for mutation screening. However, there is a significant clinical heterogeneity in the symptoms between human patients and more detailed radiographic analyses should be performed in dogs to observe potential mild changes outside the dental phenotype.
Different species may expand our understanding of the heterogeneity of the clinical picture and provide important information about genotype-phenotype correlations. Fam20c-deficient mice have a prominent dental phenotype.Citation34 Our FAM20C-deficient dogs have a dental phenotype similar to that of mice and humans. Considering that, unlike rodents, dogs have a dental physiology similar to that of humans, having both deciduous and permanent dentitions, they provide a relevant preclinical model for this rare human bone disease.
Conclusion
We and others have demonstrated that dogs carry numerous clinically and genetically close counterparts of human rare diseases. Better characterization and utilization of the canine models could be highly useful, not only for the development of new therapies for rare human diseases, but also for the better understanding of the molecular pathophysiology of the conditions in general. A rapidly-growing number of new gene discoveries in dogs will establish interesting new models for human disease. Direct encouragement of private pet owners to participate in preclinical trials could provide a straightforward route to facilitate the validation of human treatment approaches while also benefitting canine health and welfare. The full preclinical potential of canine models has not been realized yet.
Disclosure of potential conflicts of interest
HL is an owner of Genoscoper Ltd., a DNA diagnostics company that provides genetic testing for dogs.
Funding
This work was supported by the Academy of Finland (1268091), the Sigrid Juselius Foundation, the Jane and Aatos Erkko Foundation, ERCStG (260997), Biocentrum Helsinki, and the Morris Animal Foundation (D13CA-403).
References
- Wang GD, Zhai W, Yang HC, Wang L, Zhong L, Liu YH, Fan RX, Yin TT, Zhu CL, Poyarkov AD, et al. Out of southern East Asia: the natural history of domestic dogs across the world. Cell Res 2016; 26:21-33; PMID:26667385; http://dx.doi.org/10.1038/cr.2015.147
- Frantz LA, Mullin VE, Pionnier-Capitan M, Lebrasseur O, Ollivier M, Perri A, Linderholm A, Mattiangeli V, Teasdale MD, Dimopoulos EA, et al. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science 2016; 352:1228-31; PMID:27257259; http://dx.doi.org/10.1126/science.aaf3161
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, 3rd, Zody MC, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005; 438:803-19; PMID:16341006; http://dx.doi.org/10.1038/nature04338
- Karlsson EK, Baranowska I, Wade CM, Salmon Hillbertz NH, Zody MC, Anderson N, Biagi TM, Patterson N, Pielberg GR, Kulbokas EJ, 3rd, et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat Genet 2007; 39:1321-8; PMID:17906626; http://dx.doi.org/10.1038/ng.2007.10
- van Steenbeek FG, Hytönen MK, Leegwater PA, Lohi H. The canine era: the rise of a biomedical model. Anim Genet 2016; 47(5):519-27; PMID:27324307
- Sutter NB, Eberle MA, Parker HG, Pullar BJ, Kirkness EF, Kruglyak L, Ostrander EA. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res 2004; 14:2388-96; PMID:15545498; http://dx.doi.org/10.1101/gr.3147604
- Drögemüller C, Karlsson EK, Hytönen MK, Perloski M, Dolf G, Sainio K, Lohi H, Lindblad-Toh K, Leeb T. A mutation in hairless dogs implicates FOXI3 in ectodermal development. Science 2008; 321:1462; PMID:18787161; http://dx.doi.org/10.1126/science.1162525
- Hytönen MK, Arumilli M, Lappalainen AK, Kallio H, Snellman M, Sainio K, Lohi H. A novel GUSB mutation in Brazilian terriers with severe skeletal abnormalities defines the disease as mucopolysaccharidosis VII. PLoS One 2012; 7:e40281; PMID:22815736; http://dx.doi.org/10.1371/journal.pone.0040281
- Seppälä EH, Reuser AJ, Lohi H. A nonsense mutation in the acid α-glucosidase gene causes Pompe disease in Finnish and Swedish Lapphunds. PLoS One 2013; 8:e56825; http://dx.doi.org/10.1371/journal.pone.0056825
- Kyöstila K, Lappalainen AK, Lohi H. Canine chondrodysplasia caused by a truncating mutation in collagen-binding integrin α subunit 10. PLoS One 2013; 8:e75621; PMID:24086591; http://dx.doi.org/10.1371/journal.pone.0075621
- Kyöstila K, Cizinauskas S, Seppälä EH, Suhonen E, Jeserevics J, Sukura A, Syrjä P, Lohi H. A SEL1L mutation links a canine progressive early-onset cerebellar ataxia to the endoplasmic reticulum-associated protein degradation (ERAD) machinery. PLoS Genet 2012; 8:e1002759; PMID:22719266; http://dx.doi.org/10.1371/journal.pgen.1002759
- Kyöstila K, Syrjä P, Jagannathan V, Chandrasekar G, Jokinen TS, Seppälä EH, Becker D, Drögemüller M, Dietschi E, Drögemüller C, et al. A missense change in the ATG4D gene links aberrant autophagy to a neurodegenerative vacuolar storage disease. PLoS Genet 2015; 11:e1005169; PMID:25875846; http://dx.doi.org/10.1371/journal.pgen.1005169
- Seppälä EH, Jokinen TS, Fukata M, Fukata Y, Webster MT, Karlsson EK, Kilpinen SK, Steffen F, Dietschi E, Leeb T, et al. LGI2 truncation causes a remitting focal epilepsy in dogs. PLoS Genet 2011; 7:e1002194; http://dx.doi.org/10.1371/journal.pgen.1002194
- Casal ML, Lewis JR, Mauldin EA, Tardivel A, Ingold K, Favre M, Paradies F, Demotz S, Gaide O, Schneider P. Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am J Hum Genet 2007; 81:1050-6; PMID:17924345; http://dx.doi.org/10.1086/521988
- Callan MB, Haskins ME, Wang P, Zhou S, High KA, Arruda VR. Successful phenotype improvement following gene therapy for severe Hemophilia A in privately owned dogs. PLoS One 2016; 11:e0151800; PMID:27011017; http://dx.doi.org/10.1371/journal.pone.0151800
- Beltran WA, Cideciyan AV, Iwabe S, Swider M, Kosyk MS, McDaid K, Martynyuk I, Ying GS, Shaffer J, Deng WT, et al. Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc Natl Acad Sci U S A 2015; 112:E5844-53; PMID:26460017; http://dx.doi.org/10.1073/pnas.1509914112
- Hytönen MK, Arumilli M, Lappalainen AK, Owczarek-Lipska M, Jagannathan V, Hundi S, Salmela E, Venta P, Sarkiala E, Jokinen T, et al. Molecular characterization of three canine models of human rare bone diseases: Caffey, van den Ende-Gupta, and Raine Syndromes. PLoS Genet 2016; 12:e1006037; PMID:27187611; http://dx.doi.org/10.1371/journal.pgen.1006037
- Thornburg LP. Infantile cortical hyperostosis (Caffey-Silverman syndrome). Animal model: craniomandibular osteopathy in the canine. Am J Pathol 1979; 95:575-8; PMID:377993
- Nistala H, Mäkitie O, Juppner H. Caffey disease: new perspectives on old questions. Bone 2014; 60:246-51; PMID:24389367; http://dx.doi.org/10.1016/j.bone.2013.12.030
- Gensure RC, Makitie O, Barclay C, Chan C, Depalma SR, Bastepe M, Abuzahra H, Couper R, Mundlos S, Sillence D, et al. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest 2005; 115:1250-7; PMID:15864348; http://dx.doi.org/10.1172/JCI22760
- Kitaoka T, Miyoshi Y, Namba N, Miura K, Kubota T, Ohata Y, Fujiwara M, Takagi M, Hasegawa T, Juppner H, et al. Two Japanese familial cases of Caffey disease with and without the common COL1A1 mutation and normal bone density, and review of the literature. Eur J Pediatr 2014; 173:799-804; PMID:24390061; http://dx.doi.org/10.1007/s00431-013-2252-8
- Chou JY, Mansfield BC. The SLC37 family of sugar-phosphate/phosphate exchangers. Curr Top Membr 2014; 73:357-82; PMID:24745989; http://dx.doi.org/10.1016/B978-0-12-800223-0.00010-4
- Kim JY, Tillison K, Zhou S, Wu Y, Smas CM. The major facilitator superfamily member Slc37a2 is a novel macrophage- specific gene selectively expressed in obese white adipose tissue. Am J Physiol Endocrinol Metab 2007; 293:E110-20; PMID:17356011; http://dx.doi.org/10.1152/ajpendo.00404.2006
- Ha BG, Hong JM, Park JY, Ha MH, Kim TH, Cho JY, Ryoo HM, Choi JY, Shin HI, Chun SY, et al. Proteomic profile of osteoclast membrane proteins: identification of Na+/H+ exchanger domain containing 2 and its role in osteoclast fusion. Proteomics 2008; 8:2625-39; PMID:18600791; http://dx.doi.org/10.1002/pmic.200701192
- Saksa N, Neme A, Ryynänen J, Uusitupa M, de Mello VD, Voutilainen S, Nurmi T, Virtanen JK, Tuomainen TP, Carlberg C. Dissecting high from low responders in a vitamin D3 intervention study. J Steroid Biochem Mol Biol 2015; 148:275-82; PMID:25448738; http://dx.doi.org/10.1016/j.jsbmb.2014.11.012
- Anastasio N, Ben-Omran T, Teebi A, Ha KC, Lalonde E, Ali R, Almureikhi M, Der Kaloustian VM, Liu J, Rosenblatt DS, et al. Mutations in SCARF2 are responsible for Van Den Ende-Gupta syndrome. Am J Hum Genet 2010; 87:553-9; PMID:20887961; http://dx.doi.org/10.1016/j.ajhg.2010.09.005
- Ishii J, Adachi H, Aoki J, Koizumi H, Tomita S, Suzuki T, Tsujimoto M, Inoue K, Arai H. SREC-II, a new member of the scavenger receptor type F family, trans-interacts with SREC-I through its extracellular domain. J Biol Chem 2002; 277:39696-702; PMID:12154095; http://dx.doi.org/10.1074/jbc.M206140200
- Hwang M, Morasso MI. The novel murine Ca2+-binding protein, Scarf, is differentially expressed during epidermal differentiation. J Biol Chem 2003; 278:47827-33; PMID:12970338; http://dx.doi.org/10.1074/jbc.M306561200
- Hwang M, Kalinin A, Morasso MI. The temporal and spatial expression of the novel Ca2+-binding proteins, Scarf and Scarf2, during development and epidermal differentiation. Gene Expr Patterns 2005; 5:801-8; PMID:15922673; http://dx.doi.org/10.1016/j.modgep.2005.03.010
- Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T, Hasegawa K, Kitaoka T, Kubota T, Imanishi Y, et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone 2014; 67:56-62; PMID:24982027; http://dx.doi.org/10.1016/j.bone.2014.06.026
- Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, Bjerknes R. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res 2013; 28:1378-85; PMID:23325605; http://dx.doi.org/10.1002/jbmr.1850
- Simpson MA, Hsu R, Keir LS, Hao J, Sivapalan G, Ernst LM, Zackai EH, Al-Gazali LI, Hulskamp G, Kingston HM, et al. Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 2007; 81:906-12; PMID:17924334; http://dx.doi.org/10.1086/522240
- Simpson MA, Scheuerle A, Hurst J, Patton MA, Stewart H, Crosby AH. Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet 2009; 75:271-6; PMID:19250384; http://dx.doi.org/10.1111/j.1399-0004.2008.01118.x
- Wang X, Wang S, Lu Y, Gibson MP, Liu Y, Yuan B, Feng JQ, Qin C. FAM20C plays an essential role in the formation of murine teeth. J Biol Chem 2012; 287:35934-42; PMID:22936805; http://dx.doi.org/10.1074/jbc.M112.386862