
Abstract
Objective: As declining respiratory muscle function commonly leads to disability and death in amyotrophic lateral sclerosis (ALS), respiratory measurements such as slow vital capacity (SVC) may predict disease progression. This study evaluated the relationship between SVC and symptoms measured by the revised ALS Functional Rating Scale (ALSFRS-R).
Methods: About 453 ALS placebo-treated patients from the EMPOWER trial (NCT01281189) were evaluated. Correlations between %predicted SVC and individual respiratory ALSFRS-R subdomain items, respiratory subdomain score (maximum score of 12), and total ALSFRS-R score (maximum score of 48) were evaluated using the Pearson correlation coefficient. Pearson's chi-squared test was used to evaluate changes from baseline to week 48 in ALSFRS-R respiratory symptom and respiratory subdomain scores in patients with baseline %predicted SVC above/below the median at baseline and with more slowly/more rapidly decreasing %predicted SVC.
Results: The %predicted SVC showed significant correlations with dyspnea, orthopnoea, respiratory insufficiency, respiratory subdomain score, and total ALSFRS-R score (all p < 0.0001). Patients with baseline SVC values < median were significantly more likely than those with baseline SVC ≥ median to have a change in total ALSFRS-R respiratory subdomain score from 12 to <12 (40.9% vs. 30.2%, p = 0.0358) and from ≥10 to <10 (41.6% vs. 24.4%, p = 0.0005). Additionally, patients with smaller declines in SVC over time were significantly more likely to have smaller decreases in their respiratory subdomain scores (p < 0.0001).
Conclusions: The higher correlation between %predicted SVC and specific ALSFRS-R symptom scores in patients with rapidly versus more slowly progressing disease reinforces the importance of continually monitoring respiratory function throughout the disease course.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disease that causes irreversible weakness of all skeletal muscles, including those involved with respiration (Citation1–3). Weakness in respiratory muscles directly impacts respiratory function and is the most common cause of death in ALS (Citation4,Citation5); for that reason, regular monitoring of pulmonary function is essential. Respiratory distress is an uncommon presenting symptom in ALS (Citation6); even patients with significant respiratory deficits may not complain of symptoms because of an inability to exercise based on extremity weakness (Citation7). Vital capacity (VC) has been shown to be a clinically meaningful predictor of ALS disease progression and survival (Citation8,Citation9) that can be used to guide treatment decisions, including noninvasive ventilation (NIV) initiation, percutaneous endoscopic gastrostomy, and tracheostomy (Citation1,Citation8).
VC can be measured as (i) forced vital capacity (FVC), when the subject exhales rapidly with maximum effort (typically in 6 s), or as (ii) slow vital capacity (SVC), when the subject exhales slowly (Citation10). A recent study demonstrated that, in patients with ALS, FVC and SVC are highly correlated and can be used interchangeably to measure respiratory function as well as disease progression (Citation11). However, FVC may underestimate VC in patients with concomitant obstructive lung disease (Citation12), and SVC may be less demanding to perform (Citation11), especially for patients with bulbar or advanced disease (Citation13), resulting in a recent trend toward the use of SVC rather than FVC in clinical trials of patients with ALS (Citation10).
The purpose of this retrospective study was to evaluate the relationship between SVC and symptoms of disease and disease progression in patients with ALS. Specifically, the objectives of this study were to determine (i) the correlation between percent predicted SVC and respiratory symptoms measured with the revised ALS Functional Rating Scale (ALSFRS-R), a tool used to measure disease progression that has been shown to correlate with survival (Citation14,Citation15), and (ii) whether baseline SVC predicts disease progression as measured by the ALSFRS-R.
Methods
Subjects and assessments
This was a retrospective study of subjects who received placebo in the EMPOWER trial (Clinicaltrials.gov identifier, NCT01281189) (Citation16). EMPOWER was a large, randomized, double-blind, phase 3 trial designed to evaluate the safety and efficacy of dexpramipexole versus placebo in adults with possible, laboratory-supported probable, probable, or definite ALS according to the revised El Escorial criteria in the United States, Canada, Australia, and Europe. Subjects had onset of symptoms ≤24 months before screening and an upright SVC ≥65% of the predicted value for age, height, and sex and were assessed for ≥1 year, with maximum follow-up of 1.5 years.
Assessments in the EMPOWER study included SVC, ALSFRS-R, sniff nasal inspiratory pressure (Citation17), time to respiratory failure (defined as tracheostomy with permanent assisted ventilation or use of noninvasive ventilation for ≥22 hours per day for ≥10 consecutive days), and survival. The placebo-treated patients in the EMPOWER study had a mean age of 57.3 years and a mean duration of symptoms of 15.5 months at randomization (Citation16). Sixty-four percent were male and 24% had bulbar onset. In this group, mean time from diagnosis to baseline was 7.6 months, mean ALSFRS-R score at baseline was 37.9, and predicted upright SVC at baseline was 89.1.
Of the 467 patients randomized to the placebo group who had at least 1 post-baseline measurement of SVC, 453 had had paired ALSFRS-R and SVC measurements and were included in this analysis.
The SVC measurements were obtained using a Vitalograph 6800 spirometer. Calibrations were performed at each visit and Knudson 88 was used for reference values. The upright SVC was determined by performing 3–5 measures in accordance with criteria established by the American Thoracic Society (ATS) and the European Respiratory Society (ERS). For the SVC procedure, patients were instructed to perform an exhalation followed by 3–5 stable tidal breaths to average the baseline. Patients then were asked to take a slow and full inspiration within 4 s followed by a slow and complete expiration. A complete inspiration was then performed in order to complete the flow volume loop. Spirometry software (Biomedical Systems CardioPulmonary Suite) was used to qualify each effort and identify if it met the ATS/ERS Quality recommendations.
Statistical analysis
The Pearson moment correlation (r) test was used to evaluate the correlation between percent predicted SVC and the respiratory symptoms measured by the ALSFRS-R, such as dyspnea, orthopnoea, respiratory insufficiency (questions 10, 11, 12 of the scale, ranging from 0 to 4), the respiratory subdomain score (questions 10 + 11 + 12 of the scale, ranging from 0 to 12), and the total score for ALSFRS-R (ranging from 0 to 48). Pearson’s chi-squared test was used to compare scores of respiratory symptoms measured by the ALSFRS-R. Comparisons from baseline to 48 weeks included: reduction to less than 4 versus no reduction for dyspnea, orthopnoea, and respiratory insufficiency and reduction to less than 12 and less than 10 versus no reduction for ALSFRS-R respiratory subdomain score between patients with baseline percent predicted SVC below the median of the entire sample population and those with baseline percent predicted SVC at or above the median. Similarly, Pearson’s chi-squared test was used to compare the change from baseline to week 48 in the ALSFRS-R respiratory subdomain score (a decline of ≤2 points vs. ≥3 points) between patients who had a decline greater than 18 percentage points in percent predicted SVC (median change from baseline to week 48) and those with a decline of at most 18 percentage points over the 48 weeks. An analysis of variance model was used to evaluate the difference in percent predicted SVC at the first drop of ALSFRS-R dyspnea and orthopnoea scores between patients with slow decline and those with rapid decline in SVC. All of the hypothesis tests performed were 2-sided with a significance level of 0.05.
Results
Lower baseline SVC values and prediction of respiratory symptom progression
Lower baseline SVC values were associated with an increased likelihood of worsening symptoms as measured by respiratory symptom scores on the ALSFRS-R (). Specifically, a greater percentage of subjects with baseline percent predicted SVC values below median (88%) had significantly worsening orthopnoea or respiratory insufficiency and a trend toward worsening dyspnea, as measured on the ALSFRS-R (defined as having a score of 0–3) at week 48, than subjects with baseline percent predicted SVC values at or above median. For example, of the subjects with a baseline percent predicted SVC below median (<88% at baseline; n = 154), 39.0% developed orthopnoea and 36.4% developed respiratory insufficiency compared with only 25.9% (p = 0.0081) and 17.1% (p < 0.0001) of subjects with baseline percent predicted SVC values at or above median (n = 205) at week 48. A total of 35.7% of subjects with a baseline percent predicted SVC below median developed dyspnea compared with only 27.3% in the subjects with baseline percent predicted SVC values above median, although these results were not significant.
Figure 1. Scores of dyspnea (A), orthopnoea (B), and respiratory insufficiency (C) above/at median versus below median percent predicted SVC.
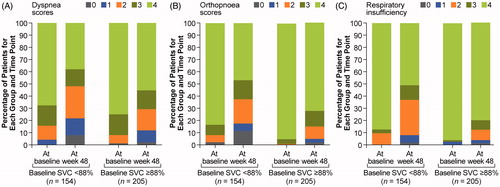
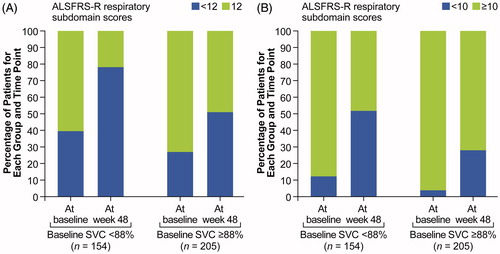
At week 48, subjects with baseline percent predicted SVC values below median (<88% predicted) were also significantly more likely to have a change in ALSFRS-R respiratory subdomain score from 12 to below 12 (40.9% vs. 30.2%; p = 0.0358) and a change in ALSFRS-R respiratory subdomain score from 10 or greater to below 10 (41.6% vs. 24.4%; p = 0.0005) compared with subjects with baseline percent predicted SVC values at or above the median ().
Figure 2. Respiratory insufficiency score 12 (A) or 10 (B): above/at median versus below median percent predicted SVC.
SVC and the ALSFRS-R
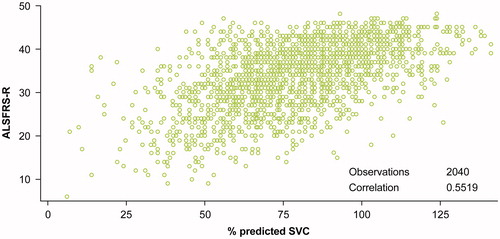
Percent predicted SVC was found to be significantly correlated with dyspnea (r = 0.3611; p < 0.0001), orthopnoea (r = 0.3230; p < 0.0001), respiratory insufficiency (r = 0.3496; p < 0.0001), and the respiratory subdomain score (r = 0.4262; p < 0.0001). The correlation between total ALSFRS-R score and percent predicted SVC was the highest (r = 0.5519; p < 0.0001) (). The proportion of SVC variance explained by dyspnea, orthopnoea, respiratory insufficiency, and the entire respiratory subdomain was 13%, 10%, 12%, and 18%, respectively. Thirty percent of the SVC variance was explained by the total ALSFRS-R score.
Figure 3. Correlation between percent predicted SVC and total ALSFRS-R score.
Similar results were found between the changes from baseline of percent predicted SVC and the individual respiratory symptom scores. Change from baseline of percent predicted SVC was found to be significantly correlated (p < 0.0001) with changes in dyspnea (r = 0.2158), orthopnoea (r = 0.2448), respiratory insufficiency (r = 0.2558), and the respiratory subdomain score (r = 0.3163). Moreover, the correlation between change in total ALSFRS-R score and change from baseline of percent predicted SVC was higher (r = 0.5261; p < 0.0001).
Decline in SVC over time and disease progression
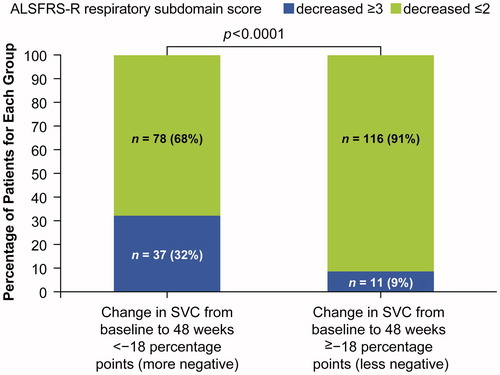
Decline in SVC over time was also found to be associated with worsening symptoms. For example, when subjects were classified by a change in percent predicted SVC from baseline to week 48 (< −18% percentage [more negative] vs. ≥ −18 percentage points [less negative]), subjects with the smaller (less negative) change were significantly more likely to have a smaller decrease in their respiratory subdomain score (≤2 points vs. ≥3 points) than were those with a greater (more negative) decline (p < 0.0001). For example, of those with a smaller decline in percent predicted SVC, 9.0% had a decrease in ALSFRS-R respiratory subdomain score of ≥3 points compared with 32.0% of those with a steeper decline in SVC ().
Figure 4. Relationship between change from baseline in SVC and changes in ALSFRS-R respiratory sub-score.
When rate of disease progression was considered, subjects classified as having rapidly progressing ALS (i.e. having a decline in SVC that is steeper than the population mean) reported symptoms that were better matched with the SVC values compared with those categorized as having slowly progressing disease. For example, at the first drop in orthopnoea score (to a 3, 2, 1, or 0), the least squares mean of percent predicted SVC is significantly different for those with rapidly progressing ALS compared with those with slowly progressing disease (56.0% vs. 74.3%; p = 0.0336) ().
Table 1. Difference in percent predicted SVC at the first drop of ALSFRS-R dyspnea/orthopnoea score between patients with slow decline and rapid decline in SVC.
Discussion
This analysis of placebo-treated subjects from the EMPOWER study revealed that baseline percent predicted SVC and change from baseline percent predicted SVC were weakly, yet significantly, correlated with specific respiratory symptoms as measured by the ALSFRS-R (i.e. dyspnea, orthopnoea, respiratory insufficiency [questions 10, 11, and 12 of the scale]) and the respiratory subdomain score (questions 10 + 11 + 12 of the scale), although the strongest correlation was found to be with total ALSFRS-R score. Baseline SVC also predicted the probability of developing respiratory symptoms in patients with ALS; that is, patients with percent predicted SVC values above the median at baseline were less likely to develop certain respiratory symptoms than those with a baseline percent predicted SVC below the median. Finally, larger decreases from baseline in SVC were associated with larger increases in respiratory symptoms as measured by the ALSFRS-R.
Previous studies have also shown correlations between VC and ALSFRS-R (Citation11,Citation18). For example, in a study of 469 ALS patients (2000–2014) in whom respiratory tests were performed at baseline and 4 months later, both SVC and FVC were found to be moderately correlated with ALSFRS-R (r = 0.506 and 0.498, respectively) and the respiratory subdomain of the ALSFRS-R (r = 0.453 and 0.435, p < 0.001, respectively) (Citation18). However, a previous study by the same group comparing the association between VC and ALSFRS-R (total and respiratory subdomain) in patients with spinal-onset ALS and those with bulbar-onset ALS found a weaker correlation between the respiratory subdomain of the ALSFRS-R and both FVC (r = 0.366; p < 0.001) and SVC (r = 0.385; p < 0.001). These findings suggest that the respiratory subscale is less sensitive to the decline in respiratory function (Citation11), particularly in different subsets of patients.
Monitoring respiratory function with direct objective measurements such as VC, therefore, is critical because decisions to implement interventions such as NIV, which has been shown to improve quality of life (QoL) (Citation19–21) and prolong survival (Citation19,Citation22–24), are often driven by specific values for different tests (Citation1,Citation8). However, these scores are less meaningful to patients and caregivers, who focus on symptoms and disability milestones when thinking about and planning for disease progression (Citation25). This focus, along with its high reliability (Citation26) and acceptance as a clinical trial outcome measure (Citation27), may be contributing to the widespread use of the ALSFRS-R as an instrument to measure disease progression and degree of functional impairment in ALS (Citation28). It is considered the “gold standard” for the functional evaluation of ALS (Citation29) even though it has been shown to be less sensitive to change than other measures, especially in patients with advanced disease (Citation30,Citation31). However, despite a consensus among 42 members of the Northeast ALS Consortium that a ≥20% decline in ALSFRS-R score represents a clinically meaningful change (Citation32), the total score, which is what is typically used in clinical trials, may not be the best result to report (Citation33), in part because it does not specify the symptoms that the patient is experiencing.
Our consideration of the patient population as consisting of 2 cohorts related to the rate of disease progression underscores the importance of continual monitoring with direct objective measures and patient-reported outcomes, and is supported by previous studies (Citation34). For example, a previous retrospective review of the medical records of 153 patients with ALS cared for at a single institution (The Penn State Milton S. Hershey Medical Center) over a 50-month period evaluated data from 139 patients with serial measurements of FVC who were stratified into two distinct clinical groups based on rates of FVC decline (>97 mL/30 days vs. <97 mL/30 days). Patients with slower rates of pulmonary function loss had a 2-fold increase in survival duration compared with patients with a more rapid loss when referenced to date of dyspnea onset or time from bi-level positive airway pressure (BiPAP) (Citation34). Delineating the relationship between dyspnea onset and VC values, therefore, would help clinicians help their patients better understand and prepare for how their disease will likely progress.
A limitation of this study is that the data were analyzed retrospectively and these analyses were performed for only one dataset. Moreover, respiratory questions in the respiratory subdomain of the ALSFRS-R are very influenced by patient motivation, mobility, and time of NIV initiation, which is quite variable among ALS centers. This could explain some discrepancies in results, such as the lack of statistical significance for testing correlation between rate of SVC decline and questions 10 and 12 of the score. Finally, VC may not be the optimal maker for respiratory dysfunction in ALS; the sigmoidal shape of the lung pressure/volume curve may result in the preservation of lung volumes even with muscle weakness, causing mild-to-moderate respiratory weakness to only reduce VC slightly.
In conclusion, this study uses two of the most commonly employed assessments of ALS disease progression to relate values of objective respiratory tests, used by clinicians to make treatment decisions, with specific symptoms such as dyspnea and orthopnoea that are meaningful to patients. Moreover, the higher correlation between percent predicted SVC value and specific symptom score on the ALSFRS-R in patients whose disease is progressing more rapidly versus those whose disease is progressing more slowly underscores the importance of continually monitoring the respiratory function of patients with ALS to better associate the values to outcomes that are more meaningful to patients and their caregivers.
Declaration of inerest
Jackson: Consultant/advisor for Cytokinetics, MT Pharma, and OneWorld Meds; scientific studies/trials for Cytokinetics, Brainstorm, Mallinckrodt, FLEX Pharma and Neuraltis; Board Member/officer/trustee on the American Academy of Neurology Board of Directors.
de Carvalho: Consultant/advisor to Biogen, Cytokinetics, and Kedrion; investigator for the Institute of Molecular Medicine; scientific studies/trials for AB Science and Cytokinetics; board member/officer/trustee of Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, Clinical Neurophysiology-Neurophysiologie Clinique, and Neurology Research International.
Genge: Investigator for AB Sciences, Alexion, AL-S Pharma, Baxter, Bioblast, Biogen, CSL Behring, Cytokinetics, Genzyme, Grifols, Ionis, Novartis, Roche, Sanofi, and UCB.
Heiman-Patterson: Consultant/advisor to Cytokinetics and MT Pharma America; investigator for Cytokinetics; scientific studies/trials for Cytokinetics, FLEX Pharma, mallinckrodt, and SanBio; board member/officer/trustee of the ALS Hope Foundation.
Shefner: Consultant/advisor to Biogen, Biohaven, Cytokinetics, Denali, MT Pharma America, and Neuraltus Pharmaceuticals; grant funding from Biogen, Cytokinetics, the ALS Association, the ALS Finding a Cure Foundation, Muscular Dystrophy Association, and Neuraltus.
Wei: Shareholder/full-time employee of Cytokinetics.
Wolff: Shareholder/full-time employee of Cytokinetics.
Additional information
Funding
Related Research Data
References
- EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis, Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J Neurol. 2012;19:360–75.
- Gordon PH. Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 2013;04:295–310.
- Rocha JA, Reis C, Simoes F, Fonseca J, Mendes Ribeiro J. Diagnostic investigation and multidisciplinary management in motor neuron disease. J Neurol. 2005;252:1435–47.
- Gil J, Funalot B, Verschueren A, Danel-Brunaud V, Camu W, Vandenberghe N, et al. Causes of death amongst French patients with amyotrophic lateral sclerosis: a prospective study. Eur J Neurol. 2008;15:1245–51.
- Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218–26.
- Schiffman PL, Belsh JM. Pulmonary function at diagnosis of amyotrophic lateral sclerosis. Rate of deterioration. Chest. 1993;103:508–13.
- Pinto S, de Carvalho M. The R of ALSFRS-R: does it really mirror functional respiratory involvement in amyotrophic lateral sclerosis? Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:120–3.
- Bourke SC. Respiratory involvement in neuromuscular disease. Clin Med (Lond). 2014;14:72–5.
- Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. J Neurol Neurosurg Psychiatry. 2006;77:390–2.
- Paganoni S, Cudkowicz M, Berry JD. Outcome measures in amyotrophic lateral sclerosis clinical trials. Clin Investig (Lond). 2014;4:605–18.
- Pinto S, de Carvalho M. Correlation between forced vital capacity and slow vital capacity for the assessment of respiratory involvement in amyotrophic lateral sclerosis: a prospective study. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:86–91.
- Chhabra SK. Forced vital capacity, slow vital capacity, or inspiratory vital capacity: which is the best measure of vital capacity? J Asthma. 1998;35:361–5.
- Caress JB, Paladenech CC. Respiratory therapy. In: Bedlack RS, Mitsumoto H, ed. Amyotrophic lateral sclerosis: a patient care guide for clinicians. New York: Demos Medical Publishing, 2013:137–50.
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169:13–21.
- Kaufmann P, Levy G, Thompson JL, DelBene ML, Battista V, Gordon PH, et al. The ALSFRSr predicts survival time in an ALS clinic population. Neurology. 2005;64:38–43.
- Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013;12:1059–67.
- Fitting JW, Paillex R, Hirt L, Aebischer P, Schluep M. Sniff nasal pressure: a sensitive respiratory test to assess progression of amyotrophic lateral sclerosis. Ann Neurol. 1999;46:887–93.
- Pinto S, de Carvalho M. Comparison of slow and forced vital capacities on ability to predict survival in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:528–33.
- Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5:140–7.
- Vandoorne E, Vrijsen B, Belge C, Testelmans D, Buyse B. Noninvasive ventilation in amyotrophic lateral sclerosis: effects on sleep quality and quality of life. Acta Clin Belg. 2016;71:389–94.
- Vrijsen B, Buyse B, Belge C, Robberecht W, Van Damme P, Decramer M, et al. Noninvasive ventilation improves sleep in amyotrophic lateral sclerosis: a prospective polysomnographic study. J Clin Sleep Med. 2015;11:559–66.
- Burkhardt C, Neuwirth C, Sommacal A, Andersen PM, Weber M. Is survival improved by the use of NIV and PEG in amyotrophic lateral sclerosis (ALS)? A post-mortem study of 80 ALS patients. PLoS One. 2017;12:e0177555.
- Gordon PH, Salachas F, Bruneteau G, Pradat PF, Lacomblez L, Gonzalez-Bermejo J, et al. Improving survival in a large French ALS center cohort. J Neurol. 2012;259:1788–92.
- Leonardis L, Dolenc Grošelj L, Vidmar G. Factors related to respiration influencing survival and respiratory function in patients with amyotrophic lateral sclerosis: a retrospective study. Eur J Neurol. 2012;19:1518–24.
- Oh J, Kim JA. Information-seeking behavior and information needs in patients with amyotrophic lateral sclerosis: analyzing an online patient community. Comput Inform Nurs. 2017;35:345–51.
- Kaufmann P, Levy G, Montes J, Buchsbaum R, Barsdorf AI, Battista V, et al. Excellent inter-rater, intra-rater, and telephone-administered reliability of the ALSFRS-R in a multicenter clinical trial. Amyotroph Lateral Scler. 2007;8:42–6.
- Leigh PN, Swash M, Iwasaki Y, Ludolph A, Meininger V, Miller RG, et al. Amyotrophic lateral sclerosis: a consensus viewpoint on designing and implementing a clinical trial. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5:84–98.
- Marin B, Bianchi E, Pupillo E, Lunetta C, Tremolizzo L, Logroscino G, et al. Non-self-sufficiency as a primary outcome measure in ALS trials. Amyotroph Lateral Scler Frontotemporal Degener. 2015;17:77–84.
- Cipolletta S, Gammino GR, Palmieri A. Illness trajectories in patients with amyotrophic lateral sclerosis: how illness progression is related to life narratives and interpersonal relationships. J Clin Nurs. 2017;26:5033–43.
- Traynor BJ, Zhang H, Shefner JM, Schoenfeld D, Cudkowicz ME; on behalf of the NEALS Consortium. Functional outcome measures as clinical trial endpoints in ALS. Neurology. 2004;63:1933–5.
- Voustianiouk A, Seidel G, Panchal J, Sivak M, Czaplinski A, Yen A, et al. ALSFRS and appel ALS scores: discordance with disease progression. Muscle Nerve. 2008;37:668–72.
- Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler. 2010;11:178–80.
- Rooney J, Burke T, Vajda A, Heverin M, Hardiman O. What does the ALSFRS-R really measure? A longitudinal and survival analysis of functional dimension subscores in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2017;88:381–5.
- Vender RL, Mauger D, Walsh S, Alam S, Simmons Z. Respiratory systems abnormalities and clinical milestones for patients with amyotrophic lateral sclerosis with emphasis upon survival. Amyotroph Lateral Scler. 2007;8:36–41.