
Abstract
Objective: To estimate amyotrophic lateral sclerosis (ALS) prevalence, 5-year survival, and explore factors associated with survival in a Medicare population. Methods: A validated administrative claims algorithm was used to classify individual’s ages 18–89 years at index date (first claim with a diagnosis of motor neuron disease or ALS between 1 January 2007 and 31 December 2011) with Medicare Advantage prescription drug coverage into mutually exclusive categories: ALS, no ALS, and possible ALS. Crude prevalence and cumulative survival from index date to the date of death, disenrollment or end of the study were calculated. Cox-proportional hazards were used to estimate and explore factors associated with survival. Results: Of 2631 eligible individuals, the algorithm identified 1271 (48 %), 1157 (44 %), 203 (8 %) as ALS, no ALS and possible ALS, respectively. The 5-year period prevalence and the 2011 point prevalence of ALS were 20.5 and 11.8 per 100,000, respectively. Evidence of death was documented in 81%, 35%, and 1.6% of the ALS, no ALS or possible ALS groups, respectively. Unadjusted median survival time was 388, 542 and 1473 days for the ALS, no ALS and possible ALS groups, respectively. Seeing a psychiatrist or neurologist at the index visit, having respiratory or genitourinary comorbidities, and the number of pre-index acute inpatient admissions were associated with shorter survival. Conclusions: Surveillance data from a Medicare population demonstrated a higher prevalence of ALS. Results highlight the need for effective ALS treatment options and resources for patients with ALS who will likely face limited therapeutic choices and care options at the end of life.
Introduction
Amyotrophic lateral sclerosis (ALS) is a heterogeneous, degenerative, neuromuscular disorder characterized by muscle weakness that progresses to death, frequently from respiratory failure, within a few years of symptom onset (Citation1). The disease predominantly affects white males, and has an average age of onset of 60 (Citation2,Citation3). Establishing a differential diagnosis of ALS is difficult as the signs and symptoms of this disease mimic other motor neuron disorders (Citation4). Without a cure, patients are limited to symptomatic treatment and palliative care options. Although efforts to identify effective new treatments and care modalities are ongoing, riluzole, and most recently edaravone, are the only United States’ (U.S.) Federal Drug Administration approved pharmacological therapies. When used appropriately they may delay disease progression and extend survival on average ∼2–3 months (Citation5,Citation6).
Incidence, prevalence, and survival of ALS have been reported in several studies worldwide, but vary widely by geography and study design (Citation2,Citation7,Citation8). In the US, the primary source for evaluating ALS incidence, prevalence, mortality, and associated risk factors is the National ALS Registry, an organization initiated by the Agency for Toxic Substances and Disease Registry (ATSDR) by congressional mandate (Citation9). In 2014, a report from the National ALS registry indicated a US prevalence of 5.0 cases of ALS per 100,000 people, which aligned with the global median prevalence of 5.4 cases per 100,000 people reported in a meta-analysis; however, the National ALS rate was higher than the crude rates (2.9 and 3.9/100,000) described in the two studies from the US included in the global meta-analysis (Citation2,Citation3,Citation10,Citation11).
To aid in validating National ALS registry findings, local surveillance projects were conducted in three states comprising eight metropolitan areas and the results were reported in 2015, as well as in subsequent studies using the same data (Citation12–15). These studies described ALS incidence and prevalence across the United States, but were limited to specific populations within certain geographies, and used only three years of data collection (2009–2011). Some of the potential risk factors for ALS that came out of these studies included age, gender, lifestyle, and genetic factors. Other researchers have reported a number of additional potential risk factors, including results from a recent survey of registrants of the US National ALS Registry. The survey evaluated demographic characteristics, risk behaviors, and military and occupational history as potential risk factors for ALS (Citation14). Furthermore, a review of 53 ALS studies found older age and the bulbar onset of disease consistently indicated a poorer prognosis, but data on gender were inconsistent (Citation16). The review noted that respiratory and nutritional support improved survival, while psychological distress had a negative impact.
While some controversy remains around prognostic factors associated with ALS, the administrative data algorithm used to populate the National ALS Registry has advanced knowledge and research of ALS in the US (Citation14,Citation16,Citation17). Using the validated algorithm employed by the National ALS Registry, the present study sought to add to the body of knowledge regarding ALS by estimating ALS prevalence, describing population characteristics, survival time and factors associated with mortality in a large national sample of individuals with Medicare Advantage prescription drug (MAPD) coverage from a single health and well-being organization, which is a unique subset of the overall US population.
In 1965, a law was signed into effect that established Medicare and Medicaid in the US as part of Title XVIII and Title XIX of the Social Security Administration as a means to ensure all residents and citizens have access to health care coverage. Medicare was originally offered to persons aged 65 and above; however, in 1972 Medicare was expanded to cover people with disabilities under age 65 while requiring a 24-month waiting period before Medicare coverage begins. There are two exceptions to this rule allowing patients with ESRD or ALS to have immediate Medicare coverage (Citation18).
Methods
Data source and subject selection
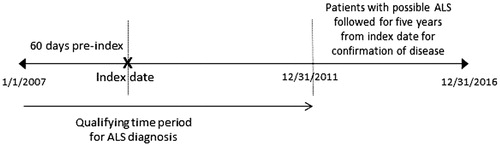
This retrospective study utilized health plan enrollment information, and medical, pharmacy, and laboratory claims data between 1 January 2007 and 31 December 2011, to identify a population of individuals ages 18–90 years with evidence of motor neuron disease (MND), and MAPD coverage. International Classification of Diseases, 9th Revision (ICD-9) 335.20-335.24, 335.29 or 10th Revision (ICD-10) diagnosis codes G12.21, G12.22, G12.8, G12.29 were used to identify MND (Supplementary Appendix A). The administrative claims-based algorithm developed and validated by Kaye et al., which has high sensitivity (0.87) and specificity (0.85), was adopted and applied to mitigate misclassification of ALS with other motor neuron diseases, as well as classify individuals as “ALS”, “no ALS” or “possible ALS” (Supplementary Appendix B). Patients fell into the possible ALS category if they had one but not both requirements for the ALS or no ALS category where applicable. Since the cause of death was not available, the algorithm applied in this study was modified to require only death notification data from CMS. The date of the first medical claim for ALS or other motor neuron disease (for the no ALS group) was considered the index date. Anyone classified as possible ALS, either the date of first ALS diagnosis (when available) or MND diagnosis served as the index date. The possible ALS group was followed for an additional 5-years post-index (through 31 December 2016) to observe whether an ALS diagnosis was confirmed (). Assessment of survival and all independent variables required at least 60 days continuous health plan enrollment prior to the index date.
Figure 1 Study design. ALS: amyotrophic lateral sclerosis.
Outcome measures
Period prevalence
A cross-sectional analysis was completed to evaluate the prevalence of ALS between 1 January 2007 and 31 December 2011. Crude period prevalence was calculated as the number of individuals with ALS divided by the total MAPD plan population per 100,000.
Point prevalence
The 2011 prevalence of ALS was also reported per 100,000 and was calculated as the number of patients that were alive as of 1 January 2011 and were identified as ALS anytime between 2007 and 2011 divided by a total number of members alive as of 1 January 2011.
Age-adjusted point and period prevalence
The direct method referencing the 2014 US National population was used.
Survival
Mortality could have been observed anytime between the index date and 31 December 2016, the last day of study follow-up. Cumulative days of survival were calculated from the index date to the date of death, disenrollment, or end of the study, whichever occurred first.
Independent variables
Demographics
The following demographic characteristics were obtained and measured from the enrollment file on the index date: age; sex; race; health plan type (health maintenance organization, preferred provider organization, point-of-service, fee-for-service, and other); indicator of low income subsidy (LIS) and/or dual Medicare/Medicaid eligibility (categorized as LIS, dual eligible, both or neither); geographic region based on state (categorized as Northeast, Midwest, South and West); and population density grouped by zip code (rural, suburban, urban, and unknown) (Citation19–21).
Pre-index healthcare utilization
Healthcare resource utilization was assessed based on place of service from the administrative claims (i.e. inpatient hospitalizations, emergency department [ED] visits, physician office visits, and outpatient visits). Utilization was reported as the number of visits per 1000 individuals.
Clinical characteristics
The Clinical Classification Software (CCS) published by the Agency of Healthcare Research and Quality was used to group pre-index clinical characteristics from administrative claims. CCS is a diagnosis and procedure categorization scheme that collapses ICD-9/10 diagnosis codes and procedure codes into two related classification systems: (1) single-level CCS categories and (2) multi-level CCS which further describes the single level into more specific categories (Citation22).
Provider characteristics
Provider specialty was identified using National Provider Identifier codes associated with the index pharmacy or medical claims. Provider specialty and practice type were assessed using the Healthcare Provider Taxonomy Code Set, which is a hierarchical set of codes, descriptions, and definitions that categorizes the type, classification, and/or specialization of health care providers (Citation23). Provider practice type on the index date was categorized as an individual, group, solo practice, or unknown.
Statistical analysis
Continuous outcome measures were described using the mean with standard deviation (SD) or median with interquartile range (IQR) as appropriate. Categorical variables were described using frequency counts and percentages. Parametric or non-parametric tests of statistical differences between subgroups were performed based on the variables underlying distribution.
A Kaplan–Meier curve was used to compare survival patterns among those with ALS, no ALS, and possible ALS, and a log-rank test was used to assess the potential differences between groups. Cox-proportional hazards models were used to evaluate the adjusted risk estimates for mortality, while controlling for demographic, baseline healthcare utilization and costs, and clinical characteristics. The proportional hazards model also identified factors associated with the probability of dying at a given time, assuming patients had survived to that point.
A parsimonious Cox-proportional hazards model was built using a backward stepwise elimination process, which through multiple steps eliminated the least significant variables (i.e. those with highest p values), and only variables significant at p < 0.15 were retained in the final model (Citation24,Citation25).
Results
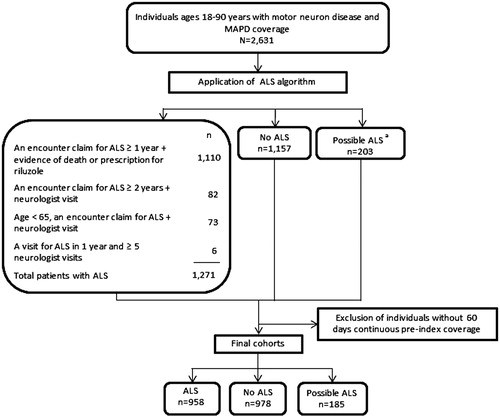
A study population of 2631 individuals aged 18–90 years with an MND-related diagnosis and MAPD coverage were identified. The algorithm classified 1271 (48%) individuals as ALS, 1157 (44%) as no ALS, and 203 (8%) as possible ALS ().
Figure 2 Attrition diagram. MAPD: Medicare Advantage Prescription Drug Program; ALS: amyotrophic lateral sclerosis. aThe possible ALS category represents patients that were identified by claims analysis but did not qualify for the ALS or No ALS categories. For example, a patient with an encounter claim for ALS within 2 years, but without evidence of a visit to a neurologist, would fall into the possible ALS category.
ALS prevalence
The 5-year period prevalence of ALS was 20.5 (95% CI 19.4–21.7) and the 2011 point prevalence of ALS was 11.8/100,000 (95% confidence interval [CI] 11.0–12.6) in the study population (). ALS prevalence in this Medicare population decreased over the 5th and 6th decades of life and then increased again in the oldest age bands, (70–79, and ≥80 years). The period and point prevalence of ALS was higher for males (26.2 and 16.2 per 100,000, respectively) compared with females (15.8 and 10.6 per/100,000, respectively).
Table 1 Prevalence of ALS compared with national prevalence in 2011 and 2014.
After excluding 510 individuals without 60 days of continuous pre-index health plan coverage, 2121 individuals were followed through 31 December 2016. Median follow up for this group was 701 days. Nearly half of the population (n = 958, 45%) was classified as ALS, with 46% (n = 978) classified as no ALS, and only 9% (n = 185) as possible ALS. Individuals classified as possible ALS were older compared to those classified as no ALS, and had a higher proportion of males and dual-eligibility for Medicare/Medicaid (). In the pre-index period, those with no ALS had a significantly higher proportion of patients with diseases of the circulatory, genitourinary, musculoskeletal, endocrine systems; neoplasms; injury and poisoning; and mental illness. Emergency department visits and hospitalizations were higher for individuals classified as no ALS, while physician office and outpatient visits were higher for those classified as ALS, whereas those classified with ALS demonstrated higher annual testing services and overall procedures ().
Table 2 Characteristics of individuals with Medicare advantage prescription drug coverage and 60 days pre-index continuous enrollment.
Survival
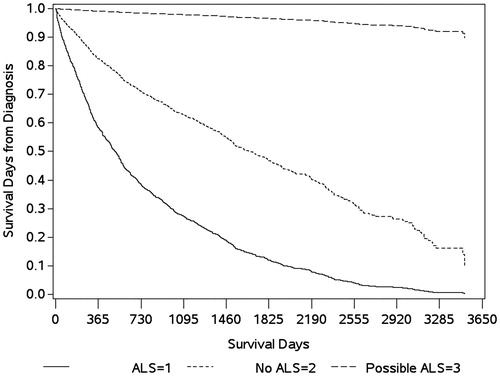
For the 2121 individuals assessed for survival, 47% survived from the index date to 31 December 2016. Cumulative survival was lowest (19%) for individuals classified as ALS compared to those with no ALS (65%) and possible ALS (98%, ). The unadjusted median survival time was 388, 542, and 1473 days for the ALS, no ALS, and possible ALS groups, respectively ().
Figure 3 Kaplan–Meier curve for cumulative survival. ALS: amyotrophic lateral sclerosis.
After controlling for baseline characteristics identified through backward elimination, having a classification of ALS was associated with a 2.85 times higher risk of mortality (95% CI: 2.48–3.27; p < 0.0001) at any given time compared to the no ALS group. A number of other variables were found to be significantly associated with an increased risk of mortality in this MAPD population (). Of the seven pre-index comorbid conditions significantly associated with survival time, the respiratory and genitourinary disease was most notable. Number of pre-index acute inpatient admissions was also significantly associated with mortality and survival time.
Table 3 Factors associated with time to death in an MAPD population.
With an additional five years of study follow-up, none of the 185 individuals classified as possible ALS could be confirmed as having ALS. Three individuals died during the additional five years of follow-up and the remaining 182 remained classified as possible ALS. Those classified as no ALS, were not followed as they were determined not to be in the pool of patients that might develop disease from the outset of the study.
Discussion
The present study used an algorithm for application to healthcare claims data published by Kaye and others in 2014 which identifies patients with ALS in administrative claims data to characterize ALS prevalence and patient characteristics in a specific MAPD population. The crude and adjusted 2011 point prevalence of ALS was 11.8 and 9.7 cases/100,000 persons, respectively, which is over or nearly twice that of the overall US prevalence of 5/100,000 in 2014, but closer to the prevalence reported in the 60–69 and 70–79 age bands (14.3 and 20, respectively) (Citation3). This likely reflects the concentration of older adults in our study population. Another consideration in variation between these results and those reported by the National ALS registry is systematic selection bias as demonstrated in the higher prevalence rates of those patients that would qualify for Medicare through disability. However, as this specific study focuses on Medicare Advantage, government-funded health coverage managed by a private insurer, those patients aging into Medicare and electing Medicare Advantage opposed to standard fee-for-service Medicare have less comorbidity (Citation27). This increase in the denominator within the 60–69 age band may have underestimated the prevalence. While the National ALS Registry demonstrated increasing ALS prevalence with older age until age ≥80 years, prevalence in our MAPD population dropped during the 5th and 6th age decades, then increased for both 70–79 and ≥80 age bands.
One possible explanation for the contrast in our results with the National ALS Registry is related to the lack of data on specific cause of death. A death certificate specifying motor neuron disease was a core component of the algorithm developed by Kaye et al., but that level of specificity was not available from the death notifications from CMS, which was the source of mortality information in this study (Citation28). Given this difference, our ALS prevalence may have been overestimated as ALS classification was based on a combination of an ALS diagnosis and notification of death.
We found that musculoskeletal, circulatory, and endocrine comorbidities were prevalent in the MAPD study population, and more frequently noted in the no ALS group compared with the ALS group. This supports the view that the pathogenesis of ALS may be different in older individuals, presenting significant challenges in differential diagnosis from comorbid conditions affecting muscle strength and mobility (Citation29). To further complicate diagnosis, muscle pain, and fatigue are commonly reported complaints in older adults (Citation30). Other common comorbidities in this study population included diseases of the circulatory and endocrine systems. This is not surprising given that overall muscle weakness could decrease mobility resulting in edema, and impairments of the autonomic nervous system could compromise the circulatory system.
Median survival for ALS patients in this study was just over a year, which is shorter than other published survival estimates that typically range from 20–48 months (Citation2); however, direct comparisons of these results must be tempered by differences in study design and populations. Published estimates largely describe survival from symptom onset or date of diagnosis (incidence), whereas this study reports survival from date of first ALS-related claim (prevalence), which may not be a true representation of symptom onset or initial diagnosis. Additionally, the association between older age and shorter survival supports the shorter survival demonstrated in this older study population (Citation29). Other than age, certain comorbidities were also associated with a shorter survival time. The significant effect of musculoskeletal, respiratory, and mental illness is supported by other finding parallels a review of prognostic factors for ALS that found poor respiratory function, cognitive function, and psychosocial factors to have a negative effect on ALS outcomes (Citation16). Although genitourinary and endocrine diseases were also significantly associated with survival time, this may be related to a higher prevalence of these conditions in an older population. Frequent hospitalization was also associated with shorter survival, which could simply reflect a more severe disease or an older more clinically complex population.
Applying this algorithm to administrative claims may identify patients with “possible ALS,” and represents an opportunity to reach out to this population. As healthcare providers develop programs to support patients with this disease, this method can help find patients that may benefit from specific interventions. Providing earlier treatment through a multidisciplinary clinical care team may improve the overall quality of life and survival.
This study should be considered in the context of key limitations. As with all claims-based studies, coding errors, incomplete or missing data, and the inability to account for factors not captured in claims are inherent. Differences in post-index follow up time could have introduced a mortality bias into survival estimates and the classification of confirmed ALS diagnosis. The inability to link with national death records made it impossible to confirm a diagnosis of ALS as the cause of death for individuals in the possible ALS group who died during follow up. Additionally, the analysis included individuals enrolled in healthcare plans from Humana, Inc, which has a higher number of members residing in the Southern states of US as compared to other parts of the country, and thus may not be generalizable across the entire US or to those not enrolled in other Medicare Advantage health plans. Finally, this study was limited to a single payer’s administrative data, in contrast to the Kaye and others study, which drew from multiple databases; however, the drive to use the single database was tested an algorithm that could be used by any health insurer to find patients with ALS or possible ALS ensuring appropriate resource to include end-of-life planning and support are available to these individuals and their caregivers.
Conclusions
While national data estimates ALS prevalence at 5/100,000, surveillance data from this MAPD population demonstrates a higher prevalence. Additional studies are needed to demonstrate the feasibility of using single payer data for identifying patients with ALS.
Declaration of interest
R. Bhattacharya, R. Harvey, K. Abranham, and J. Rosen are employed by Humana Inc, which is a health insurance provider for ALS patients. No other conflicts are reported. P. Mehta is employed by the Centers for Disease Control and Prevention (CDC)/Agency for Toxic Substances and Disease Registry (ATSDR) and is a federal employee. No conflicts are reported.
Supplementary_Appendicies.docx
Download MS Word (51.1 KB)Acknowledgments
The authors thank Charron Long and Wendy Kaye for their scientific guidance and appraisal of this manuscript.
Data availability statement
Due to contracting requirements, the data and associated materials cannot be made publically available.
Related Research Data
References
- Gordon PH. Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. A&D. 2013;04:295–310.
- Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118–30.
- Mehta P, Kaye W, Raymond J, Wu R, Larson T, Punjani R, et al. Prevalence of amyotrophic lateral sclerosis - United States, 2014. Mmwr Morb Mortal Wkly Rep. 2018;67:216–8.
- Ghasemi M. Amyotrophic lateral sclerosis mimic syndromes. Iran J Neurol. 2016;15:85–91.
- Dorst J, Ludolph AC, Huebers A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther Adv Neurol Disord. 2017;11:1–16.
- Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;3:CD001447.
- Kaye WE, Wagner L, Wu R, Mehta P. Evaluating the completeness of the national ALS registry, United States. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:112–7.
- Marin B, Boumédiene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol. 2017;46:57–74.
- Antao VC, Horton DK. The national amyotrophic lateral sclerosis (ALS) registry. J Environ health. 2012;75:28–30.
- McGuire V, Longstreth WT, Jr., Koepsell TD, van Belle G. Incidence of amyotrophic lateral sclerosis in three counties in western Washington state. Neurology. 1996;47:571–3.
- Sorenson EJ, Stalker AP, Kurland LT, Windebank AJ. Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925 to 1998. Neurology. 2002;59:280–2.
- Wagner L, Rechtman L, Jordan H, Ritsick M, Sanchez M, Sorenson E, et al. State and metropolitan area-based amyotrophic lateral sclerosis (ALS) surveillance. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17:128–34.
- Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. 2015;7:181–93.
- Bryan L, Kaye W, Antao V, Mehta P, Muravov O, Horton DK. Preliminary results of national amyotrophic lateral sclerosis (ALS) registry risk factor survey data. PLoS One. 2016;11:e0153683.
- Yu B, Pamphlett R. Environmental insults: critical triggers for amyotrophic lateral sclerosis. Transl Neurodegener. 2017;6:15.
- Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10:310–23.
- Malek AM, Stickler DE, Antao VC, Horton DK. The National ALS Registry: a recruitment tool for research. Muscle Nerve. 2014;50:830–4.
- Center for Medicare and Medicaid Services. Medicare Provider-Supplier Enrollment- Taxonomy [online]. Available from: https://data.cms.gov/Medicare-Enrollment/CROSSWALK-MEDICARE-PROVIDER-SUPPLIER-to-HEALTHCARE/j75i-rw8y. Accessed August 8, 2018
- Hart LG, Larson EH, Lishner DM. Rural definitions for health policy and research. Am J Public Health. 2005;95:1149–55.
- West AN, Weeks WB, Wallace AE. Rural veterans and access to high-quality care for high-risk surgeries. Health Serv Res. 2008;43:1737–51.
- Barsevick A. Defining the symptom cluster: how far have we come? Semin Oncol Nurs. 2016;32:334–50.
- Fisch MJ, Lee J-W, Weiss M, Wagner LI, Chang VT, Cella D, et al. Prospective, observational study of pain and analgesic prescribing in medical oncology outpatients with breast, colorectal, lung, or prostate cancer. J Clin Oncol. 2012;30:1980–8.
- Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45:613–9.
- Hosmer DW, Lemeshow S. Applied survival analysis: regression modeling of time to event data. New York, N.Y.: Wiley; 1999.
- Hosmer DW, Lemeshow S. Applied logistic regression. 2nd ed. New York, N.Y.: Wiley; 2000.
- Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J, et al.Prevalence of Amyotrophic Lateral Sclerosis – United States, 2015. MMWR MorbMortal Wkly Rep. 2018;67:1285–1289.
- Miller EA, Decker SL, Parker JD. Characteristics of medicare advantage and fee-for-service beneficiaries upon enrollment in medicare at Age 65. J Ambul Care Manage. 2016;39:231–41.
- Kaye WE, Sanchez M, Wu J. Feasibility of creating a National ALS Registry using administrative data in the United States. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:433–9.
- Beghi E, Logroscino G, Chiò A, Hardiman O, Mitchell D, Swingler R, et al. The epidemiology of ALS and the role of population-based registries. Biochim Biophys Acta. 2006;1762:1150–7.
- Sha MC, Callahan CM, Counsell SR, Westmoreland GR, Stump TE, Kroenke K. Physical symptoms as a predictor of health care use and mortality among older adults. Am J Med. 2005;118:301–6.