
Abstract
Objective: To assess the efficacy of tirasemtiv, a fast skeletal muscle troponin activator, vs. placebo in patients with amyotrophic lateral sclerosis. Methods: VITALITY-ALS (NCT02496767) was a multinational, double-blind, randomized, placebo-controlled clinical trial. Participants tolerating 2 weeks of open-label tirasemtiv (125 mg twice daily) were randomized 3:2:2:2 to placebo or one of three target tirasemtiv dose levels, using an escalating dosage protocol lasting 28 days. The primary outcome measure was changed in slow vital capacity (SVC) at 24 weeks. Secondary endpoints included a change in muscle strength and time to respiratory milestones of disease progression. Results: Of 744 participants, 565 tolerated open-label tirasemtiv and received randomized treatment. By 24 weeks, 23 (12.2%) placebo-treated participants discontinued study treatment vs. 129 (34.2%) randomized to tirasemtiv. SVC declined by 14.4% (95% CI: −16.8, −11.9) in the placebo group and 13.4% (95% CI: −15.3, −11.6) in the tirasemtiv group (p = 0.56). Secondary endpoints did not show significant differences. However, participants who tolerated tirasemtiv at their randomized dose showed a numeric trend toward a dose-related slowing of decline in SVC (p = 0.11). Dizziness, fatigue, nausea, weight loss, and insomnia occurred more frequently on tirasemtiv. Serious adverse events were similar across groups. Conclusions: Tirasemtiv did not alter the decline of SVC or significantly impact secondary outcome measures. Poor tolerability of tirasemtiv may have contributed to this result. However, participants tolerating their intended dose exhibited a trend toward treatment benefit on SVC, suggesting the underlying mechanism of action may still hold promise, as is being tested with a different fast skeletal muscle troponin activator (NCT03160898).
Trial registration: ClinicalTrials.gov identifier: NCT02496767.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder causing progressive weakness and death, on average 3–5 years after diagnosis and most often as a result of respiratory failure (Citation1–4).
Most therapeutic approaches to treating ALS have targeted neurodegeneration, although skeletal muscle is also a plausible target (Citation5). Tirasemtiv, a fast skeletal muscle troponin activator (FSTA), increases skeletal muscle contractility by sensitizing the sarcomere to calcium (Citation6,Citation7).
A large, multinational, randomized, double-blind, placebo-controlled, phase IIb clinical trial (BENEFIT-ALS) (Citation8) randomized 605 participants with ALS who tolerated 1 week of open-label tirasemtiv 125 mg twice a day (BID). Participants randomized to tirasemtiv underwent weekly dose escalation to a maximum tolerated dose of 250 mg BID in the 12-week, double-blind phase of the study. No treatment effect was noted in the primary endpoint (change from baseline in ALS Functional Rating Scale-Revised [ALSFRS-R]). However, slow vital capacity (SVC) and muscle strength declined significantly more slowly in tirasemtiv-treated participants (Citation8). Tirasemtiv reduced the slope of decline in SVC by ∼50% during the 12-week treatment phase (Citation9). Dizziness was the most commonly reported adverse event (AE) (Citation8).
A phase III trial (VITALITY-ALS) was designed to extend and confirm the findings of BENEFIT-ALS. Double-blind treatment was extended to 48 weeks, with the primary outcome, change in SVC, evaluated at 24 weeks. To improve tolerability, the open-label period was extended to 2 weeks, and dose escalation occurred every 2 weeks rather than weekly. Three target dose levels were studied.
Methods
Participants
Adults with possible, probable, or definite ALS in accordance with the revised El Escorial criteria (Citation10) were enrolled from 79 sites in 11 countries in North America and Europe. Inclusion criteria required an upright SVC ≥70% predicted for age, height, and sex. No participants were taking edaravone during this study, and prior exposure to tirasemtiv was exclusionary.
Standard protocol approvals, registrations, and patient consents
All participants in VITALITY-ALS provided written informed consent, and institutional review board approvals were received at all sites before enrollment. The study was conducted in accordance with the Declaration of Helsinki. An independent data and safety monitoring board monitored safety throughout the study. This study was registered with ClinicalTrials.gov (NCT02496767).
Study design
Following a screening period of up to 14 days, eligible participants were enrolled in the trial. The study included an open-label phase; a 48-week, double-blind, placebo-controlled phase; and then a 4-week, double-blind, placebo-controlled tirasemtiv withdrawal phase intended to assess for a potential rebound effect (none was observed, and not further discussed here). Participants who successfully completed 2 weeks of treatment with open-label tirasemtiv 125 mg BID were randomized (using a centralized IWRS system) 3:2:2:2 to matching placebo or one of three different target daily dose levels of tirasemtiv (250, 375, or 500 mg) stratified by riluzole use vs. nonuse. It was felt that patients who could not tolerate 125 mg BID as an initial dose would be unlikely to complete the study, so we did not titrate to this dose. All participants on riluzole took 50 mg daily from their personal supply in the morning and a blinded dose of riluzole in the evening in the same manner as in BENEFIT-ALS. All participants randomized to double-blind tirasemtiv continued at 250 mg/day (125 mg BID) for the first 2 weeks of double-blind treatment. Participants randomized to a target daily dosage of 375 mg (125 mg in the morning and 250 mg in the evening) or 500 mg (250 mg BID) of tirasemtiv had their dose adjusted in 125-mg increments every 2 weeks until the target dose was reached or signs of intolerance emerged, in which case the dose was not escalated or was down-titrated to 250 mg daily of tirasemtiv or matching placebo as necessary due to AEs. This titration period was not included in the open-label phase, as differential drop out would have rendered the 3 groups unequal.
The safety analysis set consisted of all participants who received any dose of study drug. The full analysis set (FAS) included participants who received at least one dose of study medication during the randomized, double-blind, placebo-controlled phase and had at least one post-randomization efficacy assessment. There were a total of six post randomization assessments through 24 weeks. The per-protocol set (PPS) consisted of all participants who completed ≥20 weeks of double-blind, placebo-controlled treatment, had at least one post-randomization efficacy assessment on double-blind study treatment, and had no major protocol violations.
Assessments
The last assessment obtained before the first dose of the open-label phase was considered baseline. The primary endpoint was change in percent predicted SVC from baseline to week 24 of the double-blind, placebo-controlled phase. Secondary endpoints included muscle strength assessed by hand-held dynamometry, the respiratory subscales of the ALSFRS-R, and time to certain respiratory milestones of disease progression, such as the initiation of assisted ventilation during the 48 weeks of randomized, double-blinded treatment. Safety assessments included physical examinations, clinical laboratory evaluations, vital signs, and monitoring of AEs and serious AEs (SAEs).
Statistical methods
The primary global null hypothesis was that no treatment difference existed in the change from baseline in percent predicted SVC at week 24 between participants in the FAS randomized to placebo and those randomized to tirasemtiv (pooled three-target dosage levels) during placebo-controlled, double-blind treatment. The original protocol sample size estimation indicated that ∼360 participants were needed to complete the 24 weeks of double-blind treatment to provide 90% power to detect a treatment difference from placebo in percent predicted SVC change from baseline to the end of the first 24-week phase of 6% (an ∼30% change from placebo) for all tirasemtiv target dose groups pooled using a common standard deviation of 17% with a two-tailed alpha of 0.05. Dropout rates of 16% at 24 weeks for placebo and 25% for all tirasemtiv target dose groups combined were assumed, necessitating ∼600 participants enrolled in the study and ∼477 participants randomized to placebo and the three different target dose levels of tirasemtiv in an allocation ratio of 3:2:2:2 in the double-blind, placebo-controlled treatment phase, stratifying based on riluzole use. The final sample size was slightly higher after adjusting for the aggregate blinded standard deviation and dropout rate during the study. Subjects withdrawing from active treatment were encouraged to attend all scheduled study visits and complete assessments and, if unable to attend, were contacted by phone on a monthly basis to obtain vital status and respiratory status through 48 weeks.
The primary analysis was performed using a repeated-measures mixed model with restricted maximum likelihood method (PROC MIXED default; SAS®, Cary, NC). The model included terms for treatment, baseline, pooled site, visit, and riluzole use/nonuse as well as interaction terms for treatment-by-visit and baseline-by-visit with an unstructured covariance matrix. Multiple imputation by randomized treatment dosage group was performed for missing data prior to modeling. The repeated-measures mixed model was applied to other change from baseline secondary endpoints. Cox regression models were used to estimate the hazard ratios of time to event endpoints between tirasemtiv and placebo, stratified by randomization strata.
Data availability statement
In collaboration with the ALS association, this study also included a biofluids collection at every visit. All demographic and efficacy data will be migrated to NeuroBANK, a database maintained by the NEALS Biorepository. Biofluids and phenotypic data will be available through application to the NEALS Biorepository Committee.
Results
Baseline characteristics
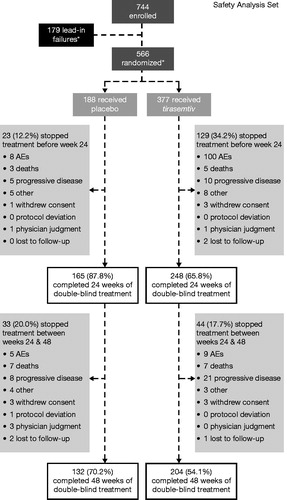
Of 866 participants screened, 744 were enrolled and 565 received randomized treatment (one patient was randomized but did not receive the treatment). After successfully completing the open-label phase, 188 patients received at least one dose of placebo and 377 received at least one dose of tirasemtiv. Demographics at study entry () were similar to another recent phase III trials (Citation8,Citation11–15). The majority of participants (74.5% [554/744]) were taking riluzole at the start of the study. Baseline characteristics between riluzole and non-riluzole strata were similar.
Table 1 Demographics and baseline disease characteristics.
Open-label phase
In the open-label phase of the study, 86.0% (640/744) of participants experienced AEs, most commonly dizziness (48.5% [361/744]), fatigue (27.4% [204/744]), and nausea (14.5% [108/744]). Only 1.5% (11/744) of participants (4 not taking and 7 taking riluzole) experienced SAEs; 23.7% (176/744) of participants discontinued from the study during the open-label phase, with a similar discontinuation rate among participants taking riluzole (26.7% [148/554]) and those not taking riluzole (31.1% [59/190]) (). AEs were the most common reason for discontinuation, with 11.6% of participants (86/744) discontinuing because of dizziness, 8.1% (60/744) because of fatigue, and 4.2% (31/744) because of nausea. Riluzole did not significantly affect the frequency or type of AE resulting in discontinuation. The baseline demographics and disease characteristics of participants completing the open-label phase and randomized into the double-blind, placebo-controlled phase were similar to those at the time of entry into the open-label phase ().
Figure 1 Patient disposition. *One patient was randomized but did not receive study drug. AE: adverse event.
Double-blind, placebo-controlled phase
Participant disposition is shown in . Overall, 183 participants were randomized to placebo; 165 (87.8%) completed 24 weeks of treatment, and 177 (94.1%) completed the week 24 visit. In contrast, 248 (65.8%) participants randomized to tirasemtiv completed 24 weeks of treatment and 311 (82.5%) completed the week 24 visit. There were 97 (77.0%), 60 (47.6%), and 39 (31.2%) patients who tolerated and achieved their randomized target doses of 250 mg/day, 375 mg/day, and 500 mg/day at week 24, respectively. AEs were the most common reason for discontinuing therapy and were more common in participants randomized to tirasemtiv. Termination from treatment primarily occurred during the first 12 weeks and in relation to dose titration (); the frequency of study drug discontinuation was strongly related to the target dosage level. Down-titrations also occurred early but were not effective in deterring study drug discontinuation. Following 12 weeks of treatment with the study drug, participants on placebo and on tirasemtiv stopped treatment at similar rates (). AEs and SAEs are shown in and , respectively; the AE profile was similar to the previously reported phase II trial. Rates of SAEs and deaths were not different between tirasemtiv- and placebo-treated groups. Seventeen patients died during active treatment on placebo [9.0%] as compared with 26 deaths during active treatment on tirasemtiv [6.9%] after >1 year of exposure to study medication.
Figure 2 Probability of (A) staying on the assigned treatment dose and (B) staying on treatment.
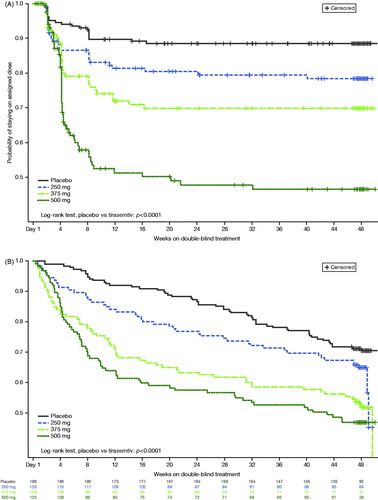
Table 2 Most common TEAEs during the 48-week, double-blind, placebo-controlled phase.
Table 3 Serious AEs occurring in >1% of participants during the 48-week double-blind phase.
At 24 weeks, there was no significant difference in the decline in percent predicted SVC in the placebo-treated group compared with all tirasemtiv-treated participants (14.4% for placebo, 13.4% for all tirasemtiv; p = 0.56). Decline in percent predicted SVC was 12.6% for the 250-mg target dose group, 13.7% for the 375-mg target dose group, and 13.9% for the 500-mg target dose group (p = 0.38, 0.76, and 0.84, respectively). There was no significant difference between placebo and all tirasemtiv-treated participants on the following secondary endpoints: ALSFRS-R respiratory domain score change from baseline to week 48, slope of Muscle Strength Mega-Score from baseline to week 48, time to the first occurrence of a decline from baseline in percent predicted SVC ≥20 percentage points or the onset of respiratory insufficiency or death to week 48, time to the first occurrence of a decline in SVC ≤50% predicted or the onset of respiratory insufficiency or death over 48 weeks, change from baseline in the ALSFRS-R total score to the end of week 48, and time to first use of mechanical ventilator assistance or death to week 48 (p = 0.92, 0.75, 0.46, 0.76, 0.61, 0.65, respectively).
To assess whether a potential benefit of tirasemtiv was masked by poor tolerability, two separate analyses were performed on more restrictive data sets. The PPS included participants who completed ≥20 weeks of study medication and contributed outcome measures at the 24-week time point, when the primary outcome measure was assessed. It eliminated participants who had stopped study medication for >4 weeks and includes no imputations for missing data. However, participants who reduced their dose were still included within their target dose group. In this data set, the point estimate of effect was greater than for the FAS; for the 250-mg target dosage group in the PPS, percent predicted SVC declined by 10.0 percentage points, compared with 13.1 percentage points for the placebo group (). This difference was not statistically significant (p = 0.13), and there was an inverse dose-response similar to the primary analysis, with participants randomized to higher dosage groups performing less well than the 250-mg target dosage group.
Table 4 Percent predicted SVC change from baseline to week 24 in PPS, mixed model for repeated measures.
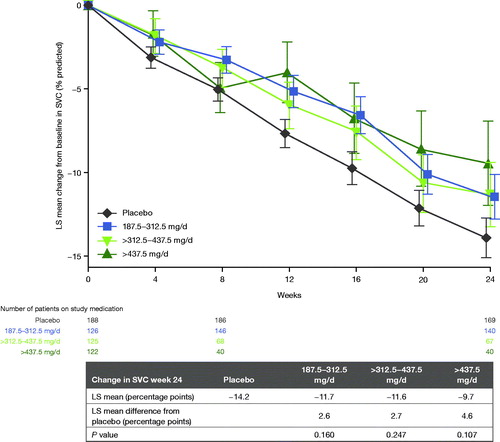
A second analysis evaluated whether participants who tolerated their target dose level well enough to stay at the target dose showed a greater positive impact of tirasemtiv. Change in SVC was evaluated as a function of average daily maintenance dosage from the end of week 8 for dose titration to the end of week 24. In this analysis, all patients who remained on study drug through 24 weeks were included. The greatest estimate of effect was noted in participants randomized to the highest daily dosage (500 mg) who were able to tolerate study drug at that dose (). Participants in this group showed a decline in percent predicted SVC of 9.7 percentage points compared with a drop of 14.2 percentage points in placebo-treated participants (p = 0.11). This difference represents a 32% slower decline in SVC for those treated with tirasemtiv at a daily dosage of 500 mg compared with placebo treatment; if this point estimate was a true effect of tirasemtiv in participants who tolerated this dose, such a difference likely would be clinically important. Every 100 mg of daily tirasemtiv was associated with a 0.19-percentage point (p = 0.019) improvement in change from baseline of percent predicted SVC per month. This analysis suggested that there was an expected dose response for those patients who were able to tolerate tirasemtiv at their assigned target dosage level, such that participants receiving lower dosages experienced less of an effect than those receiving higher dosages of tirasemtiv. No treatment effect was observed on ALSFRS-R and hand-held dynamometry endpoints. When the baseline characteristics of patients who remained on double-blind treatment (whether at their target doses or after down-titration) were compared to those who did not, those who discontinued double-blind treatment were, on average, slightly older, had lower vital capacities and ALSFRS-R total scores (and were less likely to be scored as 4 on the dyspnea and orthopnea domains), were less likely to be on riluzole, and were more likely to use caffeine.
Figure 3 Change from baseline in percent predicted SVC in patients completing 24 weeks of treatment. Error bars represent standard error of the mean. LS: least squares; SVC: slow vital capacity.
Discussion
In the primary analysis of the FAS, no significant differences were seen in the primary outcome measure or any secondary measures comparing all tirasemtiv-treated participants with placebo-treated participants. There appeared to be an inverse dose-response relationship with respect to the primary outcome measure. However, this is likely spurious, as many higher-dosage participants had been reduced to the lowest dose, and more had also stopped study drug at higher dosages but remained in the study providing outcome measure data without receiving therapy.
Results demonstrated that tolerability was not improved by the dosing strategies used in this study, indicating that overall tolerability was dose-dependent and not strongly improved by a slower dose escalation. A slightly higher percentage of participants withdrew during the open-label phase as compared with BENEFIT-ALS. However, of those who were randomized and continued into the double-blind phase, approximately the same percentage of participants stopped study drug by 12 weeks as compared with BENEFIT-ALS by 12 weeks. In both studies, study drug discontinuation occurred early, usually within 1 week of dose escalation. Dose reductions also occurred early in VITALITY-ALS but did not improve retention. Participants who were randomized to higher dosages but were down-titrated to 125 mg BID were much more likely to ultimately stop study drug than those originally randomized to the same dosage. Participants on tirasemtiv also experienced more AEs, most notably dizziness, fatigue, and nausea. Importantly, however, there was no increase in SAE rates in patients on active treatment and no indication of increased mortality.
Despite the impact of poor tolerability on the results of this study, 2 analyses prespecified in the protocol suggested a benefit in patients able to tolerate tirasemtiv. In the PPS, participants who were randomized to the 250-mg target daily dose group of tirasemtiv declined in SVC 24% more slowly than those on placebo (). This change, if true, would be clinically important; the fact that the difference between active treatment and placebo was smaller than in BENEFIT-ALS may be partially explained by the fact that the cohort as a whole declined more slowly than expected (discussed below). The observation that the lower dosages were associated with larger treatment differences was consistent with the inverse dose-response relationship in tolerability among randomized dosage groups. However, when participants were evaluated according to the maintenance dose from the end of week 8 to the end of week 24, those who tolerated the highest average daily dosage (∼500 mg of tirasemtiv daily), showed an SVC decline that was 32% slower than those on placebo in the FAS, with a positive dose-response relationship. Overall, the poor tolerability of tirasemtiv appears to have masked the potential treatment benefit with regard to SVC. These results suggest that the underlying mechanism of action of tirasemtiv still has promise in patients with ALS.
This study did not meet its primary endpoint. Although there was a trend toward benefit in tirasemtiv-treated participants, it was not statistically significant and not considered clinically meaningful. There are several reasons why a potential positive effect was obscured in this study. First, as discussed above, tolerability was a significant issue, with >50% more patients stopping study drug on active treatment as compared with placebo. When patients who tolerated tirasemtiv were evaluated separately, much larger and potentially important estimates of effect were seen. These effects did not reach statistical significance at least in part because of the small sample sizes in these groups. Secondly, participants recruited to this study performed differently than other recent ALS cohorts, at least with regard to SVC rate of change. The decline in SVC was lower than expected. Over 24 weeks, the decline in SVC was 14.2% or 0.080% per day in the placebo group for VITALITY-ALS. For comparison, the rate of decline for placebo-treated ALS patients in the phase III trial of dexpramipexole was 0.088% per day or 16.9% over 24 weeks and 0.090% per day for the phase II trial of tirasemtiv. The slower progression rate in the current study may have contributed to the finding of no significant treatment effect. However, as was noted in the phase 2 study (BENEFIT ALS), SVC was the measure that seemed to show the largest signal among all outcome measures, confirming that the choice in primary outcome measure was justified.
Thus, although this phase III study failed to show a meaningful effect of tirasemtiv on SVC, subanalyses suggest that tirasemtiv had a biological effect on SVC, such that fast skeletal muscle troponin activation remains worthy of further study. A next-generation FSTA (reldesemtiv), lacking central nervous system effects and having the potential to produce a larger pharmacodynamic effect (Citation16), has been reported to show promising effects in spinal muscular atrophy (Citation17) and is being tested in a large phase II trial in ALS patients (NCT03160898).
Declaration of interest
JS serves as a consultant/advisor to Biogen, Biohaven, Cytokinetics, MT Pharma America, Orphazyme, AveXis, and has received grant funding from Biogen, Cytokinetics, Amylyx, the ALS Association, the ALS Finding a Cure Foundation, Muscular Dystrophy Association. MC serves as a consultant/advisor to Biogen, Biohaven, Cytokinetics, Lilly, Takeda Aclipse, and MT Pharma. OH is an investigator for Cytokinetics and has served as a consultant for Biogen, Cytokinetics, Novartis, Roche, Merck, and Mitsubishi. She is the editor-in-chief of the journal Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. JA serves as a consultant/advisor for Cytokinetics, Biohaven, and Avexis, and is an investigator for Neuraltus, Roche, Orion, and Biogen. BC, JL, FM, LM, SR, and AW are shareholders and/or full-time employees of Cytokinetics, Inc.
Additional information
Funding
Related Research Data
References
- Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5:140–7.
- Goyal NA, Mozaffar T. Respiratory and nutritional support in amyotrophic lateral sclerosis. Curr Treat Options Neurol. 2014;16:270.
- Rooney J, Byrne S, Heverin M, Corr B, Elamin M, Staines A, et al. Survival analysis of Irish amyotrophic lateral sclerosis patients diagnosed from 1995-2010. PLoS One. 2013;8:e74733.
- Shefner JM, Watson ML, Meng L, Wolff AA, The Neals/Cytokinetics STUDY Team. A study to evaluate safety and tolerability of repeated doses of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:574–81.
- Shefner JM. Muscle as a therapeutic target in amyotrophic lateral sclerosis. Exp Neurol. 2009;219:373–5.
- Hwee DT, Kennedy A, Ryans J, Russell AJ, Jia Z, Hinken AC, et al. Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL-SOD1G93A ALS mouse model. PLoS One. 2014;9:e96921.
- Russell AJ, Hartman JJ, Hinken AC, Muci AR, Kawas R, Driscoll L, et al. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat Med. 2012;18:452–5.
- Shefner JM, Wolff AA, Meng L, Bian A, Lee J, Barragan D, on behalf of the BENEFIT-ALS study group, et al. A randomized, placebo-controlled, double-blind phase IIb trial evaluating the safety and efficacy of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17:426–35.
- Shefner J, Andrews J, Meng L, Bian A, Wolff AA. Relationships between riluzole and tirasemtiv levels on outcomes in the BENEFITS-ALS trial. Presented at the Presented at the 25th International Symposium on ALS/MND; December 5–7, 2014; Brussels, Belgium.
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Aggarwal SP, Zinman L, Simpson E, McKinley J, Jackson KE, Pinto H, et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:481–8.
- Cudkowicz ME, Shefner JM, Schoenfeld DA, Zhang H, Andreasson KI, Rothstein JD, et al. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol. 2006;60:22–31.
- Cudkowicz ME, Titus S, Kearney M, Yu H, Sherman A, Schoenfeld D, et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:1083–91.
- Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013;12:1059–67.
- Macchi Z, Wang Y, Moore D, Katz J, Saperstein D, Walk D, et al. A multi-center screening trial of rasagiline in patients with amyotrophic lateral sclerosis: possible mitochondrial biomarker target engagement. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:345–52.
- Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, et al. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018;57:729–34.
- Rudnicki SA, Andrews JA, Malik FI, Wolff AA, Day JW. Update of CY 5021: a phase 2 clinical trial of reldesemtiv, a fast skeletal muscle troponin activator (FSTA), for the potential treatment of spinal muscular atrophy. Presented at the Presented at CURE SMA; June 14–17, 2018; Dallas, TX.
The VITALITY-ALS Study Group
Belgium: Philip Van Damme, MD, PhD, Universitair Ziekenhuis Leuven, Leuven, [email protected].
Canada: Lawrence Korngut, MD, FRCPC, Heritage Medical Research Clinic, Calgary, Alberta, [email protected]; Wendy Johnston, MD, Kaye Edmonton Clinic, Edmonton, Alberta, [email protected]; Colleen O’Connell, MD, River Valley Health - Stan Cassidy Foundation, Stan Cassidy Fredericton, New Brunswick, [email protected]; Ian Grant, MD, FRCPC, QEII Health Sciences Centre, Halifax, Nova Scotia, [email protected]; John Turnbull, MD, McMaster University, Hamilton, Ontario, [email protected]; Christen Shoesmith, MD, London Health Sciences Centre, London, Ontario, [email protected]; Lorne Zinman, MD, Sunnybrook Health Sciences Centre, Toronto, Ontario, [email protected]; Stephan Botez, MD, CHUM Hospital Notre-Dame, Montreal, Quebec, [email protected]; Angela Genge, MD, FRCP(c), Montreal Neurological Institute and Hospital, Montreal, Quebec, [email protected]; Annie Dionne, MD, CHU de Québec, Hôpital de l'Enfant-Jésus, Québec City, Quebec, [email protected].
France: Philippe Couratier, MD, PhD, CHU - Hôpital Dupuytren, Limoges, [email protected]; Shahram Attarian, MD, CHU - Hôpital de la Timone, Marseilles, [email protected]; Jean Pouget, MD, CHU - Hôpital de la Timone, Marseilles, [email protected]; William Camu, MD, PhD, Hôpital de Chauliac, Montpellier, [email protected]; Claude Desnuelle, MD, PhD, CHU de Nice – Hopital de l’Archet 1, Nice, [email protected]; François Salachas, MD, Hôpital La Pitié Salpétrière, Paris, [email protected]; Philippe Corcia, MD, Centre SLA de Tours, Tours, [email protected].
Germany: Thomas Meyer, MD, Charite - Campus Virchow-Klinikum (CVK), Berlin, [email protected]; Susanne Petri, MD, Medizinische Hochschule Hannover, Hannover, [email protected]; Albert Ludolph, MD, Universitätsklinikum Ulm, Ulm, [email protected].
Ireland: Orla Hardiman, MD, Trinity Biomedical Sciences Institute, Trinity College, Dublin, [email protected].
Italy: Andrea Calvo, MD, Ospedale Molinette, AOU Città della Salute e della Scienza di Torino, Torino, [email protected]; Christian Lunetta, MD, Ospedale Niguarda Ca' Granda, AO, Milan, [email protected]; Vincenzo Silani, MD, Ospedale S. Luca, Istituto Auxologico Italiano-IRCCS Istituto, Milan, [email protected].
Netherlands: Leonard van den Berg, MD, PhD, Universitair Medisch Centrum Utrecht, locatie Academisch Zie, Utrecht, [email protected].
Portugal: Mamede de Carvalho, MD, H. Santa Maria, Centro Hospitalar de Lisboa Norte, Lisbon, [email protected].
Spain: Jesús Mora Pardina, MD, H. San Rafael, Madrid, [email protected].
United Kingdom: Carolyn Young, MD, Walton Centre for Neurology and Neurosurgery, Liverpool, [email protected]; Ammar Al-Chalabi, MD, King’s College Hospital, London, [email protected]; Aleksander Radunovic, PhD, FRCP, Barts and the London MND Centre, London, [email protected]; Clemens Hanemann, MD, FRCP, Plymouth Hospitals NHS Trust, Plymouth, [email protected].
United States: Jeremy M. Shefner, MD, PhD, Barrow Neurological Institute, Phoenix, AZ, [email protected]; Shafeeq Ladha, MD, St. Joseph's Hospital and Medical Center, Phoenix, AZ, [email protected]; Namita Goyal, MD, University of California Irvine, Irvine, CA, [email protected]; John Ravits, MD, University of California San Diego Altman Clinical and Translational Research Institute, La Jolla, CA, [email protected]; Richard Lewis, MD, Cedars-Sinai Medical Center, Los Angeles, CA, [email protected]; Nanette Joyce, MD, University of California Davis Medical Center, Sacramento, CA, [email protected]; Bjorn Oskarsson, MD, University of California Davis Medical Center, Sacramento, CA, [email protected]; Jonathan S. Katz, MD, California Pacific Medical Center, San Francisco, CA, [email protected]; Yuen So, MD, PhD, Stanford Neuroscience Health Center, Stanford, CA, [email protected]; Bettina M. Cockroft, MD, MBA, Cytokinetics, Inc., South San Francisco, CA, [email protected]; Jacqueline H. Lee, Cytokinetics, Inc., South San Francisco, CA, [email protected]; Fady I. Malik, MD, PhD, Cytokinetics, Inc., South San Francisco, CA, [email protected]; Lisa Meng, PhD, Cytokinetics, Inc., South San Francisco, CA, [email protected]; Stacy A. Rudnicki, MD, Cytokinetics, Inc., South San Francisco, CA, [email protected]; Andrew A. Wolff, MD, Cytokinetics, Inc., South San Francisco, CA, [email protected]; Dianna Quan, MD, University of Colorado Hospital - Anschutz Outpatient Pavilion, Aurora, CO, [email protected]; Kevin Felice, MD, Hospital for Special Care, New Britain, CT, [email protected]; Elham Bayat, MD, George Washington University, Washington, DC, [email protected]; Kevin Boylan, MD, Mayo Clinic Jacksonville, Jacksonville, FL, [email protected]; Michael G. Benatar, MD, University of Miami UHealth Professional Arts Center, Miami, FL, [email protected]; Tuan Vu, MD, University of South Florida, Tampa, FL, [email protected]; Jonathan Glass, MD, Emory University School of Medicine, Atlanta, GA, [email protected]; Robert Sufit, MD, Northwestern University - Feinberg School of Medicine, Chicago, IL, [email protected]; Cynthia Bodkin, MD, Indiana University, Indianapolis, IN, [email protected]; Andrea Swenson, MD, University of Iowa Hospitals & Clinics, Iowa City, IA, [email protected]; Jeffrey Statland, MD, University of Kansas Medical Center, Kansas City, KS, [email protected]; Nicholas Maragakis, MD, Johns Hopkins Medicine - Transverse Myelitis Center, Baltimore, MD, [email protected]; Merit E. Cudkowicz, MD, Massachusetts General Hospital, Boston, MA, [email protected]; James Berry, MD, Massachusetts General Hospital, Boston, MA, [email protected]; Robert Brown, MD, UMass Memorial Medical Center - University Campus, Worcester, MA, [email protected]; Johnny Salameh, MD, UMass Memorial Medical Center - University Campus, Worcester, MA, [email protected]; Stephen Goutman, MD, University of Michigan, Ann Arbor, MI, [email protected]; Daniel S. Newman, MD, Henry Ford Hospital, Detroit, MI, [email protected]; Gaurav Guliani, MD, Hennepin County Medical Center, Twin Cities ALS Research Consortium, Minneapolis, MN, [email protected]; Samuel Maiser, MD, Hennepin County Medical Center, Twin Cities ALS Research Consortium, Minneapolis, MN, [email protected]; Alan Pestronk, MD, Washington University, St. Louis, MO, [email protected]; Ghazala Hayat, MD, Saint Louis University, St. Louis, MO, [email protected]; Gary Pattee, MD, Neurology Associates, P.C., Lincoln, NE, [email protected]; Jeffrey Cohen, MD, Dartmouth Hitchcock Medical Center, Lebanon, NH, [email protected]; Benjamin Brooks, MD, Carolinas Neuromuscular/ALS MDA Center, Charlotte, NC, [email protected]; Richard Bedlack, MD, Duke University Medical Center, Durham, NC, [email protected]; Jinsy A. Andrews, MD, The Neurological Institute Columbia University, New York, NY, [email protected]; James Caress, MD, Wake Forest University Baptist Medical Center, Winston-Salem, NC, [email protected]; Hiroshi Mitsumoto, MD, Columbia University Medical Center, New York, NY, [email protected]; Dale Lange, MD, Weill Medical College of Cornell University, New York, NY, [email protected]; Deborah Bradshaw, MD, State University of New York (SUNY), Syracuse, NY, [email protected]; Stephen J. Kolb, MD, PhD, Ohio State University Medical Center, Columbus, OH, [email protected]; Chafic Karam, MD, Oregon Health & Science University, Portland, OR, [email protected]; Julie Khoury, MD, Oregon Health & Science University, Portland, OR, [email protected]; Kimberly Goslin, MD, Providence Brain and Spine Institute ALS Center, Portland, OR, [email protected]; Zachary Simmons, MD, Penn State Milton S. Hershey Medical Center, Hershey, PA, [email protected]; Leo McCluskey, MD, Pennsylvania Hospital, Philadelphia, PA, [email protected]; Terry Heiman-Patterson, MD, Temple University School of Medicine, Philadelphia, PA, [email protected]; Peter Donofrio, MD, Vanderbilt University Medical Center, Nashville, TN, [email protected]; Daragh Heitzman, MD, Texas Neurology, Dallas, TX, [email protected]; Yadollah Harati, MD, Baylor College of Medicine, Houston, TX, [email protected]; Carlayne Jackson, MD, FAAN, University of Texas Health Science Center San Antonio, San Antonio, TX, [email protected]; Lawrence Phillips, MD, University of Virginia, Charlottesville, VA, [email protected]; Michael Weiss, MD, University of Washington, Seattle, WA, [email protected]; Christopher Nance, MD, West Virginia University, Morgantown, WV, [email protected]; Shumaila Sultan, MD, West Virginia University, Morgantown, WV, [email protected]; Paul Barkhaus, MD, Medical College of Wisconsin Milwaukee, WI, [email protected].