
Abstract
Objective
A recent case-series described patients with ALS to improve and/or stabilize after treatment with intravenous high-dose Penicillin G/Hydrocortisone (PenGH). In this study, we determine the safety and efficacy of intravenous PenGH versus placebo in combination with riluzole in patients with ALS.
Methods
Patients diagnosed with ALS according to the El Escorial criteria were randomized double-blind to four quarterly cycles of 21 d of intravenous PenGH or placebo in a 5:3 ratio. The primary outcome was change from baseline to week 48 in Amyotrophic Lateral Sclerosis Functional Rating Scale—Revised (ALSFRS-R). Secondary outcomes were lung function, muscle strength, plasma creatinine, clinical stage, gastrostomy placement, quality of life and occurrence of adverse of events.
Results
In total, 16 patients were randomized (10 PenGH and 6 placebo), of which 6 (40%) completed the study. Patients treated with PenGH progressed with 2.2 (95% CI 1.1–3.3) ALSFRS-R points per month and PenGH treatment did not halt disease progression (p = 0.002). No significant differences were found between PenGH or placebo (mean difference 0.5, 95% CI −1.01 to ∞, p = 0.28). Although PenGH was well-tolerated, 6 patients (38%, 3 in each arm) had thrombotic complications due to the intravenous administration method.
Conclusions
Treatment with PenGH does not halt disease or reverse progression in patients with ALS and showed no statistical difference with those who received placebo. Prolonged intravenous administration therapies may inflate thrombosis risk.
Introduction
A recent case-series described three ALS patients that were treated with 21-d rounds of intravenous penicillin G and hydrocortisone (PenGH) at 10-week intervals, in response to which symptoms appeared to improve and/or stabilize (Citation1). Patient 1 was wheelchair bound, but within days he apparently regained the ability to walk and within weeks could walk 650 m unaided. He reportedly also experienced improvements in speech, swallowing and limb strength. In the weeks thereafter he became wheelchair-bound again, but improvement of finger movement, speech, and swallowing appeared to be sustained. The other patients also reported improvement of speech and swallowing shortly after starting PenGH.
The rationale for PenGH comes from two opinion articles by a pharmacologist (Citation2,Citation3), in which an absence of epilepsy in ALS is reported, despite high glutamate levels. He reasons elevated glutamate should cause seizures, but that in ALS these are suppressed by compensatory GABA-overactivity. Considering GABA-ergic drugs (e.g. benzodiazepines) induce “ALS-like”-symptoms, such as weakness and dysarthria, he suggests that this GABA-overactivity causes motor symptoms. Subsequently, he offers an alternative interpretation of a case-series on five Moroccan syphilis patients with “ALS-like” features that improved after PenGH treatment (Citation2). He suggests that they did not have neurosyphilis, but both ALS and syphilis. Further stating that syphilis was a co-morbid condition, endemic to Morocco, but without relevance to the neurological symptoms. As PenG is a potent GABA-antagonist, as well as an antibiotic, he proposes that these patients were cured from ALS by suppressing GABA.
ALSUntangled reviewed the evidence for PenGH and raised several concerns (Citation4); (a) multiple publications report on seizures in ALS. Moreover, in frontotemporal dementia (FTD), which forms a spectrum with ALS (Citation5), there is clearly an increased risk of seizures. (b) Animal models of ALS show decreased (not increased) GABA in the motor cortex. (c) MR-spectroscopy also demonstrates lower GABA-levels in the motor cortex in ALS compared to controls. (d) TMS studies in ALS show cortical hyper-excitability, rather than hypo-excitability and (e) Dysarthria and weakness have many underlying causes, including benzodiazepine overuse, but in ALS result from motor neuron death. Overall, they conclude there is no evidence for GABA-overload in ALS.
However, PenG might have relevance in ALS through a different mechanism. In SOD1 mouse models β-lactam antibiotics, such as ceftriaxone and PenG, increase EAAT2 levels in the spinal cord and slows disease progression (Citation6). This formed the basis for the phase III clinical trial with ceftriaxone in ALS, which unfortunately was negative (Citation7). Despite, this result upregulating EAAT2 remains an interesting treatment strategy.
Although one can debate the GABA-hypothesis and if this justifies treating patients with PenGH, the reported results are nothing short of spectacular. Patients appeared to stabilize or even improve in response to treatment with readily available and safe drugs (Citation1). The PenGH case-series attracted considerable media attention and an increasing number of patients and physicians started to enquire about this treatment. At least 20 ALS patients in The Netherlands and Belgium were subsequently treated with PenGH by various physicians (mostly non-neurologists) and off-label treatment with PenGH was approved in The Netherlands by the national health authority (IGJ, see statement on PENGH).
Despite, the reportedly positive clinical results and a plausible mechanism (inducing EAAT2 expression) for PenGH, in the past astonishing improvements have all too often been reported in uncontrolled studies which could subsequently not be replicated. We therefore performed a randomized, placebo-controlled trial to investigate whether PenGH treatment is indeed capable of halting or even reversing ALS symptoms.
Methods
Trial design, patients and oversight
This was a mono-center, randomized, double-blind, placebo-controlled, parallel group, phase 2 trial. Given the prior case-series with anecdotal evidence in especially bulbar affected patients, the eligibility criteria were targeted to replicate the treatment response in this selected subgroup of patients. Eligible patients were ≥18 years; diagnosed according to revised El Escorial criteria with laboratory supported, clinically probable or definite ALS (Citation8); disease duration <24 months; slow vital capacity (SVC) ≥80%; bulbar involvement (either dysarthria or ≥1 point drop on questions 1–3 of the ALS functional rating scale [ALS-FRS-R] (Citation9)); and on a stable dose of riluzole for ≥30 d. We excluded patients with concomitant FTD, on noninvasive ventilation (NIV) or with tracheotomy, or (a medical history of) syphilis, epilepsy, currently on corticosteroids or with contraindications for either penicillin or steroids. Having a feeding tube in place was not an exclusion criterion. Trial participants were recruited from the outpatient clinic and through a web-based platform (TRICALS). Patients were informed about the study, given time to consider and ask questions. All patients provided written informed consent for the study.
At baseline, we randomly assigned patients in a 5:3 ratio to PenGH or placebo using a predefined randomized list. The patients were stratified according to the ALSFRS-R total score (≤ 40). Patients and site staff members were unaware of the trial-group assignments, and trial medication was identical in appearance. The trial was conducted in accordance with the provisions of the Declaration of Helsinki, the International Conference on Harmonization guidelines for Good Clinical Practice, and approved by the institutional review board of the UMC Utrecht. The trial was registered under EudraCT number 2017-001983-39 and overseen by a data safety and monitoring board (DSMB). The full study protocol is available in the Supplementary Material.
Trial procedures and assessments
Patients assigned to the PenGH group received intravenous penicillin G (1–20 million units in escalating dose) during 3 weeks and 100 mg hydrocortisone during the first 2 weeks. Patients assigned to the placebo arm received intravenous 0.9% saline solution for 3 weeks. Treatment cycles were repeated at quarterly intervals over the course of 48 weeks (four cycles). On day 1 of each treatment cycle, a PICC line was inserted. Subsequently, study medication was administered over the course of 8 h during 21 consecutive days at the neurology ward of the UMC Utrecht. Study medication and matching placebo i.v. bags were prepared by an unblinded pharmacist and nurse that were not involved in patient care or outcome assessment. The study medication was a clear, transparent solution similar to saline and indistinguishable from one another. Additionally, all i.v. bags were masked and sealed in order to ensure blinding of patients and study staff. At the end of each treatment cycle the PICC line was removed. During each cycle, we obtained muscle strength measurements (hand-held dynamometry), SVC and weight at day 1 and day 21. The ALSFRS-R and quality of life (EQ-5D-5L) were collected once during each treatment cycle at day 13. Between cycles, we collected safety and ALSFRS-R data at 4-week intervals.
End points
The primary end point was change from baseline in the ALSFRS-R total score using all available data up to week 48. Secondary end points were overall survival defined as the time from randomization to the date of documented death, date of tracheotomy or use of NIV for 22 h/d, change from baseline in SVC, isometric muscle strength, plasma creatinine level, weight and quality of life using all available data up to week 48. Adverse events (AEs) and serious adverse events (SAEs) were assessed throughout the study. Additionally, we explored the effect of treatment on time to gastrostomy placement and time to progression on King’s clinical staging system. Data from one endpoint, Center for Neurological Study—Bulbar Functional Scale (CNS-BFS), was affected by a translational error and excluded from the analysis.
Sample size
Our study was powered based on the prior case-series indicating a complete stop or reversion of the progression rate in patients with ALS (Citation1). Given that patients decline, on average, 1 ALSFRS-R point per month (Citation10), we expected after 12 months a 12-point difference between placebo and active treated patients (SD 8.1). Enrolling 5 placebo and 10 active patients would provide 72% power to detect this difference with a one-sided alpha of 5% based on an independent two-sample t-test. Using an ANCOVA model with correction for baseline ALSFRS-R, power was estimated at 80% using a simulation-based approach. We predicted a lost-to-follow-up of 20% in the placebo arm and, therefore, enrolled one additional patient.
Statistical analysis
Continuous variables were expressed as mean and standard deviation for normally distributed variables and median and range for non-normally distributed variables. Categorical variables were presented as number of cases and percentages. Baseline characteristics were compared between the placebo and intervention arm without significance testing. For each patient we determined the prognostic risk profile using the ENCALS survival prediction model (Citation11). The primary outcome, change in ALSFRS-R at 48 weeks follow-up, was analyzed using an ANCOVA linear mixed effects model to incorporate all available ALSFRS-R data. The fixed part of the model contained an effect for time, the baseline ALSFRS-R, treatment and the interaction between treatment and time. A random intercept and slope for time per individual were incorporated to account for dependencies in the data. Significance testing was done using a one-sided likelihood ratio test evaluating the interaction coefficient for time by treatment. Linear mixed models are flexible when missing data is present. Nevertheless, as death of participants may lead to informative missing data, we performed one sensitivity analysis to account for survival time using the Joint Modeling Framework (Citation12). Secondary continuous outcomes were analyzed in a similar fashion as the primary outcome. Time-to-event end points were analyzed using Cox proportional hazard models.
For all analyses, the one-sided type I error rate (α) was set to 0.05; p-values <0.05 were considered to be significant. In order to control type one error due to multiple comparisons, we applied a hierarchical testing procedure for the primary and key secondary endpoints (i.e. we continued statistical testing until an end point exceeded p > 0.05). Outcome hierarchy was predefined and was evaluated as (Citation1) ALSFRS-R total score, (Citation2) ALSFRS-R bulbar domain score, (Citation3) SVC, (Citation4) muscle strength, (Citation5) plasma creatine and (Citation6) weight. If the testing procedure did not exceed the significance threshold, only effect sizes and unadjusted 95% confidence intervals are reported.
In order to rule out a large negative or positive effect of the method of drug administration, one additional analysis was performed to compare the progression rate of the enrolled cohort to historical data (PRO-ACT) (Citation13). PRO-ACT was used rather than a historical Dutch trial population due to the restrictive inclusion criteria and the resulting small number of eligible patients. Of the 2084 PRO-ACT patients with complete data, 302 (14%) fulfilled the eligibility criteria. We used propensity matching (1 PHALS patient to 2 PRO-ACT patients) to match on age, sex, symptom duration, vital capacity, diagnostic delay, site of onset and ΔFRS (Citation14), ALSFRS-R total score and ALSFRS-R bulbar score; all factors have been associated with survival previously (Citation11,Citation15).
Results
Twenty-two patients were screened between November 2017 and May 2018; six patients did not meet the inclusion criteria and 16 patients were randomized. Of the 16 patients who received at least one dose of study treatment, 6 (38%) patients died before the end of the trial and, ultimately, 6 (38%) patients completed all 4 treatment cycles (). The arms were well-balanced for prognostic characteristics as assessed by the ENCALS risk profile (baseline characteristics in ).
Figure 1. Study profile.
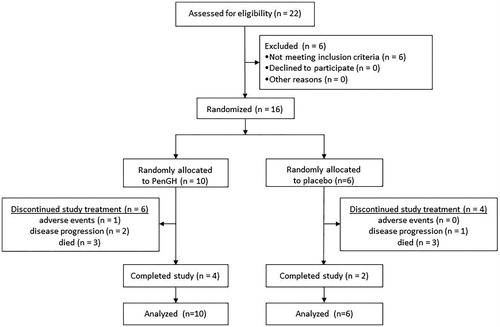
Table 1. Baseline characteristics.
Efficacy endpoints
The individual patterns of decline on the ALSFRS-R, are shown in . Patients allocated to either placebo or PenGH exhibited disease progression, with no signs of long-lasting functional improvements. On average, patients declined with 2.2 (95% CI 1.1–3.3) ALSFRS-R points/month in the PenGH arm and 2.7 (95% CI 1.2–4.3) points in the placebo arm, respectively. The primary hypothesis that PenGH would stop disease progression (i.e. rate of progression ≤ 0 during a 12-month follow-up) could be rejected (p = 0.002). There was no statistically significant difference between placebo and PenGH (mean difference 0.53, 95% CI −1.01 to ∞, p = 0.28) (). The mean difference between placebo and treatment was similar when adjusting for informative censoring due to death (mean difference 0.56, 95% CI −0.93 to ∞, p = 0.27). During the 12-month follow-up period, we noted no significant difference for survival (3 deaths in both arms, unadjusted HR 0.46, 95% CI −∞ to 1.79), time to gastrostomy (unadjusted HR 1.25, 95% CI −∞ to 3.43) or time to progression on King’s clinical staging system (unadjusted HR 0.45, 95% CI −∞ to 1.38). Results for the other secondary endpoints are presented in , all confirming continued disease progression under PenGH treatment. Due to the hierarchical testing procedure, and the non-significant difference in the primary end point, we only report 95% CI for the secondary end points.
Figure 2. Individual changes in ALSFRS-R total score since randomization. Individual patient ALSFRS-R total scores (y-axis) over time since randomization in months (x-axis). Prognosis is based on the ENCALS survival model (Citation11).
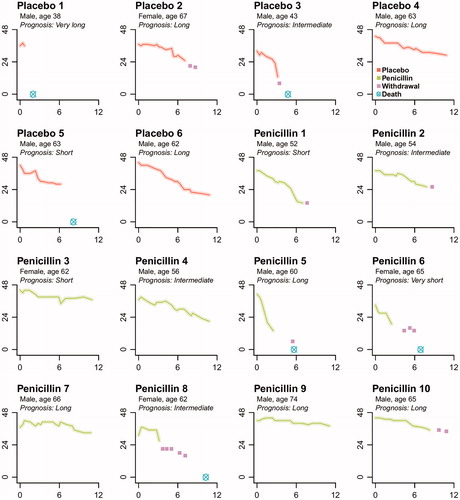
Table 2. Primary and secondary efficacy endpoints.
Safety
In a number of patients (placebo #3, penicillin #1, #5, #6 and #8, ) we observed relatively rapid disease progression after experimental treatment was initiated. This prompted us to request a number of additional safety analyses from the DSMB. After reviewing unblinded data of individual ALSFRS-R scores and line listing of all reported AEs and SAEs, the DSMB unanimously concluded each time that there was no evidence indicating clear benefit or harm for one of the treatment arms and therefore recommended to continue the trial as planned. In terms of side-effects, PenGH was well-tolerated, despite an increased rate of gastrointestinal disorders (80% vs 17% for PenGH and placebo respectively) (). All deaths that occurred during study follow-up were deemed unrelated to the study medication and due to ALS disease progression (with exception of one placebo patient (#1) who chose to undergo euthanasia). Six patients (38%, 3 in each arm) developed a deep line thrombosis, which is a complication of the mode of drug administration, and required additional medical interventions.
Table 3. Safety profile.
Comparison with PRO-ACT
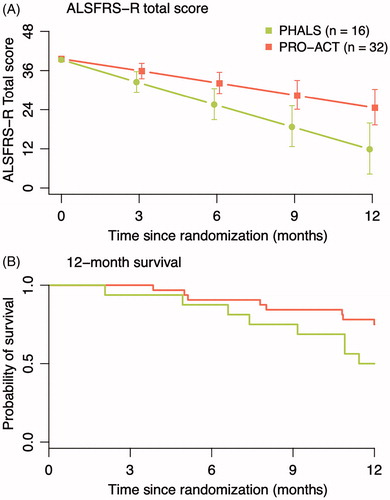
The PHALS population progressed, on average, with 2.4 (95% CI 1.5–3.3) ALSFRS-R points/month, which is nearly twice as fast as observed in previous clinical trials (Citation10). As exploratory analysis, we performed a propensity matched analysis to compare observed progression rates to historical rates observed in 32 matched PRO-ACT patients fulfilling the PHALS criteria () (Citation13). The mean difference in ALSFRS-R decline between populations was −1.03 points per month (95% CI −1.71 to −0.37, p = 0.003), indicating that PHALS patients exhibited an 83.2% increase in their progression rate. In terms of survival, PHALS patients had a 132% increase in mortality rate during the 12 months of follow-up, albeit not statistically significant (HR 2.32, 95% CI 0.87–6.20, p = 0.093).
Figure 3. Propensity matched analysis of PHALS vs. PRO-ACT. Patients were matched 1:2 with placebo patients in the PRO-ACT cohort. The matched sample from PRO-ACT consisted of eligible patients for the PHALS criteria and complete data on all prognostic variables (N = 302). In the matched analysis, the mean difference in ALSFRS-R decline between PHALS and PRO-ACT was −1.03 points per month (−1.72 to −0.37, p = 0.003).
Additional observations on neuropsychological changes and patients that received off-label treatment are available in Supplementary Appendix 2.
Discussion
Currently there is no effective treatment for ALS. Many patients are willing to try experimental therapies, but unfortunately most do not have the opportunity to enroll into clinical trials (Citation10). As a result, patients search the internet in desperate hope of finding treatment options and many self-experiment. There are indeed many reports of patients that appear to have benefited from off-label treatments, supplements and alternative medicine, although the level of evidence supporting efficacy is generally low (mostly anecdotal). Nonetheless, several of these therapies have been investigated in clinical studies (e.g. Lunasin (Citation16)) and unfortunately proved to be ineffective. Therefore, caution is warranted when interpreting results from uncontrolled studies and proper clinical trials are required before treatments are approved. The interest in PenGH was also sparked by a study with a low class of evidence (case-series; level V), but with spectacular results with drugs that could be made available to ALS patients rapidly (Citation1). Considering an increasing number of patients were being treated with PenGH and off-label treatment was even approved in The Netherlands, we strongly felt a clinical trial was warranted.
Unfortunately, this trial shows that ALS patients do not stabilize or improve in response to PenGH treatment. All patients declined on the ALSFRS-R and all other secondary outcome measures. Although this study was powered to a detect large effect, it seems highly unlikely that PenGH slows disease progression given the relatively high rate of decline on the ALSFRS-R in this trial. Strikingly, rate of decline was significantly higher for the PHALS study population compared to matched PRO-ACT controls, which begs the question whether the intervention (hospitalization with i.v. infusions) itself may even have been harmful. Patients felt immobilized and spent most of their time in or close to their hospital beds. Furthermore, we observed line thrombosis in 38% of our patients and 80% of the PenGH group experienced gastro-intestinal side-effects.
This is reminiscent of the discussion on Edaravone (Citation17–19). It has been argued that the statistically significant difference seen in a subgroup of ALS cases in favor of Edaravone compared to placebo, does not prove it is superior to no intervention at all. Indeed, in the Edaravone trial the rate of decline in the lead-in phase was less than after randomization to Edaravone (0.61 vs. 0.91 ALSFRS-R points/month) (Citation17). Similarly, the rate of decline for Edaravone trial cases is higher than for matched PRO-ACT cases (0.91 vs. 0.79 ALSFRS-R units/month) (Citation18).
A 1992 case report describes an ALS patient experiencing rapid improvements after receiving ceftriaxone (Citation20). He regained many functions and was able to sit up, drive a car, shave himself and speak and swallow better. A few months later, the patient developed pancreatitis and the treatment was stopped, after which he quickly deteriorated and restarting the drug did not have effect. The authors therefore reported that their conclusions had been premature (Citation21).
Nonetheless, other physicians treated patients with ceftriaxone or searched for evidence of efficacy in medical records of ALS patients that received ceftriaxone for other reasons. Subsequently, 6 reports were published in the early nineties, none of which found evidence for a beneficial effect of ceftriaxone and interest subsided (Citation22). Interest in beta-lactam antibiotics, ceftriaxone in particular, resurfaced in 2005 after the discovery that this class of antibiotics induces EAAT2 expression and thus potentially alleviates excitotoxicity (Citation23). This resulted in a large-scale phase 1–3 clinical trial on ceftriaxone in ALS (Citation7). During stages 1 and 2 ALSFRS-R decline was slower (±0.5 points/month) for ceftriaxone compared to placebo. These results were however not confirmed in stage 3, in which no significant differences were observed in survival or ALSFRS-R decline. Similar to our study, patients receiving ceftriaxone had significantly more gastrointestinal and hepatobiliary/pancreas-related side-effects as well as thrombotic complications (Citation7).
ALS patients understandably want access to potentially beneficial therapies and in particular when reports emerge that existing drugs might be effective. As these drugs have already been approved for other indications, the impression is often that they are safe and that there is nothing to lose. Unfortunately, this is an oversimplification. In ALS there have been multiple studies with existing treatments that were (possibly) harmful to patients (topiramate, minocycline, diaphragm pacing) or with considerable side-effects (e.g. lithium). Our study illustrates that the mode of study drug administration may also be associated with potentially serious complications (line thrombosis) or that prolonged immobilization may be harmful.
We agree with the view that novel treatments should not only be superior to placebo, but also to no intervention at all (Citation18). The use of historical controls from databases such as PRO-ACT or prognostic models may prove value to offer exploratory comparisons with usual care, in addition to adequate placebo control of the experimental arm. Studying the effect of the procedure itself should become an area of focus as many novel therapeutic approaches, although promising, are also more invasive.
Conclusion
Treatment with PenGH does not halt disease progression in patients with ALS and showed no statistical difference with those who received placebo. Prolonged intravenous administration therapies may inflate thrombosis risk.
iafd_a_1788093_sm9663.docx
Download MS Word (16.6 KB)iafd_a_1788093_sm9662.pdf
Download PDF (642.6 KB)Acknowledgments
The authors would like to thank the patients and their families for participating in this study. Also thanks goes out to ALS patients connected, Spierziekten Nederland, Stichting ALS Nederland, the members of the data safety and monitoring board and our trial staff.
Declaration of interest
MAvE received grants from the Netherlands Organization for Health Research and Development (Veni scheme), Joint Program Neurodegeneration (JPND), the Thierry Latran foundation, FIGHT-MND, MNDA and the Netherlands ALS foundation (Stichting ALS Nederland). He received travel grants from Shire (formerly Baxalta) and has consulted for Biogen. LHvdB serves on scientific advisory boards for Biogen, Cytokinetics, Orion, and Sarepta. LHvdB reports grants from ALS Foundation Netherlands, The Netherlands Organization for Health Research and Development (Vici scheme), The Netherlands Organization for Health Research and Development (SOPHIA, STRENGTH, ALS-CarE project) funded through the EU JPND, personal fees from Shire (previously Baxalta), Biogen, Cytokinetics, Prinses Beatrix SpierFonds and from the Thierry Latran foundation, outside the submitted work. RPAvE and TMB report no conflict of interest.
Additional information
Funding
References
- Tuk B, Jousma H, Gaillard PJ. Treatment with penicillin G and hydrocortisone reduces ALS-associated symptoms: a case series of three patients. F1000Res. 2017;6:410.
- Tuk B. Syphilis may be a confounding factor, not a causative agent, in syphilitic ALS. F1000Res. 2016;5:1904.
- Tuk B. Overstimulation of the inhibitory nervous system plays a role in the pathogenesis of neuromuscular and neurological diseases: a novel hypothesis. F1000Res. 2016;5:1435.
- Bedlack R, ALSUntangled Group. ALSUntangled 46: penicillin G/hydrocortisone. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:126–31.
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:2084–98.
- Hoye ML, Regan MR, Jensen LA, Lake AM, Reddy LV, Vidensky S, et al. Motor neuron-derived microRNAs cause astrocyte dysfunction in amyotrophic lateral sclerosis. Brain. 2018;141:2561–75.
- Cudkowicz ME, Titus S, Kearney M, Yu H, Sherman A, Schoenfeld D, et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:1083–91.
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169:13–21.
- van Eijk RPA, Westeneng HJ, Nikolakopoulos S, Verhagen IE, van Es MA, Eijkemans MJC, et al. Refining eligibility criteria for amyotrophic lateral sclerosis clinical trials. Neurology 2019;92:e451–60.
- Westeneng HJ, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17:423–33.
- van Eijk RP, Eijkemans MJ, Rizopoulos D, van den Berg LH, Nikolakopoulos S. Comparing methods to combine functional loss and mortality in clinical trials for amyotrophic lateral sclerosis. Clin Epidemiol. 2018;10:333–41.
- Atassi N, Berry J, Shui A, Zach N, Sherman A, Sinani E, et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology 2014;83:1719–25.
- Kimura F, Fujimura C, Ishida S, Nakajima H, Furutama D, Uehara H, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006;66:265–7.
- Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10:310–23.
- Bedlack RS, Wicks P, Vaughan T, Opie A, Blum R, Dios A, et al. Lunasin does not slow ALS progression: results of an open-label, single-center, hybrid-virtual 12-month trial. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:285–93.
- Writing G, Edaravone A. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2017;16:505–12.
- Turnbull J. Is edaravone harmful? (A placebo is not a control). Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:477–82.
- Turnbull J. Author response to a Letter to the Editor entitled: edaravone administration in pivotal clinical study 19 (Authors: Genge, Angela; Brooks, Benjamin). Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:300–2.
- Smith LG. Improvement of patient with amyotrophic lateral sclerosis given ceftriaxone. Lancet. 1992;339:1417.
- Smith LG. Failure of ceftriaxone for amyotrophic lateral sclerosis. Lancet. 1992;340:379.
- Norris FH. Ceftriaxone in amyotrophic lateral sclerosis. Arch Neurol. 1994;51:447.
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005;433:73–7.