
Abstract
Objective
Amyotrophic Lateral Sclerosis is one major disease in the group of neurodegenerative conditions. As with most other neurodegenerative diseases, clinical signs of the disease usually show among the elderly population, and most commonly around 60–65 years of age. Therefore the disease is not expected to impact the fertility of ALS patients. When examined from an evolutionary medicine and evolutionary biology perspective, there should be no selection pressure on the patient population due to the late onset of ALS. Methods: In this study, we tested the hypothesis that ALS does not affect fertility on a group of patients with ALS that we collected in a multi-center study. We recruited 511 patients diagnosed with ALS according to the revised El Escorial criteria, and 236 control cases without a neurodegenerative disease. We compared the ALS group’s number of offspring with the control group in three consecutive generations. Results: No statistically significant difference was found between the number of siblings of ALS and control groups (p = 0.44). A statistically significant difference was found between the number of children of ALS and control groups (p < 0.001), indicating ALS patients had more children than controls. When the number of children is assessed by gender, for women, there was no statistically significant difference between the number of children of ALS and control groups (p = 0.067). Conclusions: This finding supports the view that ALS does not have a negative selection pressure on the patient population’s fertility.
Introduction
Amyotrophic Lateral Sclerosis (ALS) is predominantly an adult onset neurodegenerative disease, which mostly affects individuals of older age. The incidence is 2 per 100,000 in a year, and the prevalence is around 3–4 per 100,000 population globally (Citation1). The disease is most common between 60 and 65 years of age and is observed 1.2–2 times more in males than in females (Citation2,Citation3). Considering the age range and peak age of the disease, the disease usually develops at an age after fertility. Because of this, it is considered that the disease has no effect on the fertility of the affected individual (Citation4).
When recent studies are reviewed, the average age of diagnosis varies between 54 and 69 (Citation5–13). Familial ALS (FALS) cases start approximately ten years earlier than sporadic ALS (SALS) cases (Citation14,Citation15). C9Orf72 is the best-known gene associated with ALS, which explains 40% of familial cases and 8% of sporadic cases in European and North American societies (Citation16). C9Orf72 gene prevalence in ALS cases is below 4% in the East (Japan) (Citation17), 5.9% in familial cases in South Asia (Iran), and 1.6% in sporadic cases (Citation18). In Asian countries, this gene is not considered one of the main causes of ALS. In contrast, the prevalence of C9Orf72 is very high in Finland, which is also one of the countries with the highest incidence of ALS in the world (Citation19).
Natural selection determines the tendency of features to increase or decrease in frequency in a population, depending on the reproductive success of those exhibiting that feature. Positive selection pressure increases the frequency of preferred alleles, negative selection pressure eliminates harmful alleles, and compensation selection supports diversity (Citation20). The theory of evolution predicts that negative reproductive selection pressure will quickly remove a dysfunctional and highly pathogenic gene from the genome (Citation21).
ALS gives the impression of a neurodegenerative disease with either simple monogenetic transition or a complex genetic origin. When this genetic character and the age of occurrence are evaluated together, the disease is expected to have no effect on fertility (Citation4). The disease affects both female and male individuals after the reproductive age, and therefore, the number of children is not expected to be affected. In the most neurodegenerative diseases that manifest after the age of fertility such as Huntington’s, Alzheimer’s, and Parkinson’s diseases, the disease is not expected to affect the average number of children a patient has (Citation22). There were previous studies on the fecundity of ALS (Citation23,Citation24), but the neurodegenerative diseases’ effect on fertility has not been explored sufficiently. In the OnWebDUALS project, we have the chance to test the effect of ALS on fertility. We recorded the number of siblings and children in our patient cases and compared them in three consecutive generations with the control group with no neurodegenerative disease. Thus, we were able to review the changes, and compare the number of children between the ALS and the control group in three consecutive generations, and discuss our findings on the effect of ALS on fertility.
Methods
ALS cases and controls were recruited by the clinician members of the consortium who participated in the OnWebDUALS project (Akdeniz University, Antalya, Turkey; Hannover Medical School Neurology Clinic, Germany; Jena University Medical School Neurology Clinic, Germany; Faculty of Medicine, University Hospital of Lisbon, Portugal; Medical University of Warsaw Neurology Clinic, Poland). Members of OnWebDuals Project prepared the patients’ data form before recruiting the patients, and the procedure was harmonized for all the partners involved. Two questionnaires were carried out by face-to-face interview with the patients and controls. Ethical permission for the study was granted by the respective Clinical Ethics Committees of the participating centers, and each patient gave written informed consent for the collection and analysis of their medical data.
The cases were analyzed in terms of their number of children in three consecutive generations. The number of siblings of the cases and the number of children of the patients represent the two successive generations, and the number of siblings of the patients’ parents represents the 3rd generation.
To avoid a selection bias, a standardized clinical assessment protocol was used at each center, which was developed for structured and systematic data recording (Citation25). Control subjects were individuals who did not have any neurodegenerative disease, and had no familial affinity with the ALS patients. Anonymised data were entered in Excel files and kept in a cloud file at Jena University.
Statistical analysis
Quantitative data were presented with median value and range, and qualitative data were presented as absolute numbers and percentages. To assess the difference between categorical variables, Chi-square test was used. In the crosstabs, if the expected value was less than 5 in at least one cell, Fisher’s Exact Test was used. The normal distribution of the data was examined using Shapiro–Wilk normality test. Mann–Whitney U test was applied to determine whether there was a statistically significant difference between two independent groups. A value of p < 0.05 was used to assess the significance for all statistical analyses. The statistical analysis was carried out with IBM SPSS Version 21.0 (SPSS Inc., Chicago, IL, USA) software and the R statistical programming environment (Citation26).
Results
Between March 2015 and December 2018, a total of 1376 patients and 822 control cases were included. Of the 1376 recruited ALS patients, 233 were later excluded from analysis; 77 because they were considered not to fulfill the entry criteria for revised El Escorial diagnosis, and 156 because there was missing data related to the disease, leaving 1143 patients. Secondly, 632 were excluded from the analysis because of missing data on the number of siblings for mothers or fathers, or the number of children of the patients, leaving 511 patients.
Of the 822 recruited control subjects, 300 were excluded from the analysis because of data inconsistencies, or missing data on the number of siblings for mothers or fathers, or the number of children of the controls, or having another neurodegenerative disease (Parkinson, Alzheimer); and 286 were excluded from the analysis because of being a relative or spouse of the patient: leaving altogether 236 control subjects. Familial ALS patients were 7.9% of all cases. The comparison of the ALS and the control groups is shown in .
Table 1. General characteristics of the dataset and Comparison of ALS and Control groups.
The median age was 63 (25–87) in the ALS group and 60 (30–91) in the control group (p = 0.095).
The median number of mother’s siblings was 4 (1–13) and 3 (1–13) in the ALS group and the control group, respectively. A statistically significant difference was found between the ALS and the control groups in the numbers of mother’s siblings (p = 0.003).
The median number of father’s siblings was 3 (1–14) and 4 (1–15) in the ALS and control group, respectively. There was no statistically significant difference between the ALS and the control groups in the number of father’s siblings (p = 0.186).
The median number of siblings was 3 (1–14) and 3 (1–14) in the ALS group versus the control group. There was no statistically significant difference between the number of siblings of ALS and control groups (p = 0.440).
The median number of children was 2 (0–14) in the ALS group and 2 (0–9) in the control group. A statistically significant difference was found between the number of children of ALS and control groups (p < 0.001). The number of children of the ALS and the control groups is shown in .
Figure 1. Number of children of the ALS and the control groups.
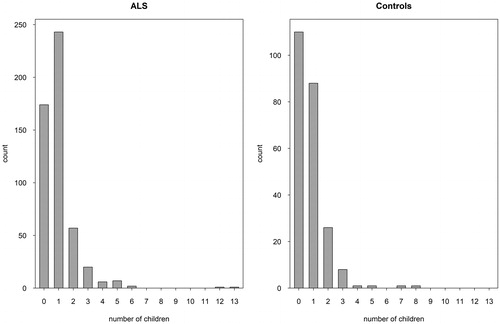
When the number of children is assessed by gender, for women (which is defined as the classic fertility rate); the median number of children was 2 (0–13) in the ALS group and 2 (0–8) in the control group. There was no statistically significant difference between the number of children of ALS and control groups (p = 0.067).
When we analyzed the data for the five cities in the study separately, we found that the number of children in the ALS group was statistically different from the control group of Lisbon and Antalya but was not statistically different from the control groups of Warsaw and Jena-Hannover. A comparison of ALS and control groups in different cities is shown in .
Table 2. Comparison of ALS and control groups in different cities.
When we analyzed ALS patients as familial ALS and sporadic ALS groups, the median number of mother’s siblings was 4 (1–13) in the FALS group, and 4 (1–13) in the SALS group. There was no statistically significant difference between the FALS and the SALS groups in the numbers of mother’s siblings (p = 0.703). The median number of siblings was 3 (1–8) and 3 (1–14) in the FALS group versus the SALS group. There was no statistically significant difference between the number of siblings of FALS and SALS groups (p = 0.409). The median number of children was 2 (0–5) in the FALS group and 2 (0–14) in the SALS group. There was no statistically significant difference between the number of children of FALS and SALS groups (p = 0.179).
Discussion
Human beings live in an era where mortality is decreasing and life expectancy is increasing to 70 years or more (Citation27). This is due to many advances, including the application of microbe theory, the development of medical practices such as advanced nutrition and public health measures. Complex chronic degenerative diseases are distinguished by an indeterminate etiology, long time delay, long-term illness, functional impairment, and in many cases the lack of a cure. One argument is that evolutionary forces are neutral on these diseases, since neurodegenerative diseases occur during a post-reproductive period of the human life cycle (Citation4). The presence of neurodegenerative diseases in the population over 50 can mean that there is no strong evolutionary selection pressure against these diseases (Citation21).
The fact that we did not observe a change in the number of offspring in two consecutive generations in this study is consistent with the above-mentioned approach of evolutionary biology. The lack of selection pressure related to the genetic infrastructure that causes ALS can perhaps explain the complexity of the disease. We can consider that the studies claiming that the disease is related to the development process of Homo sapiens can be established in this respect (Citation28,Citation29). If there was a selection pressure, we would expect the neurodegenerative diseases observed in older ages, such as ALS, to eventually disappear in the population. The diversity of the genetic mechanisms that cause neurodegenerative diseases supports the idea that there is no selection pressure on them.
Johnson et al. (Citation24) reported that “there were fewer children per SOD1 gene-carrier male (2.42) than per gene-carrier female (3.25) for ALS”. Their suggestion was that fecundity in ALS gene-carriers was reduced in males (Citation24).
In another study carried out in Ireland back in 2014, Byrne et al. (Citation23) aimed to determine whether ALS is associated with reduced fertility. They included 172 patients with definite, probable, or possible ALS, and 192 age and gender-matched controls in the study. They reported that the ALS group had a median of two children, and the control group had a median of three children but this result was not statistically significant (p = 0.67). Nevertheless, the 14 cases carrying the C9orf72 repeat expansion of females had one or fewer children compared to female controls (Citation23). In our study, however, we did not observe a significant change in fertility rates when we compare both familial and non-familial cases. We also did not observe a difference in the fecundity characteristics of male cases.
The “Granny hypothesis” (Citation30) states that survival beyond fertility and a long life expectancy are distinctive human adaptations, selected because grandmothers contribute to their offspring’s chance of survival. Neurodegenerative diseases that appear in post-reproductive ages reduce this added chance of survival, hence resulting in the gradual elimination of the disease from the population (Citation31). Our findings in this study do not confirm or deny this hypothesis.
In theory, the minimum time interval required for the micro-evolutionary change of the gene pool is two generations (Citation4), but there is no restriction on the minimum time interval for an adaptive phenotypic change. Generations overlap largely in living populations due to a lengthy human fertility life of about 30 years. The classical definitions of evolutionary processes point to long-time periods, therefore we may expect that there may be a change in gene frequencies for a century. Currently, the average age for having children is between 20 and 40 years of age, and three generations can spread over a century, providing each generation with an opportunity to change gene frequencies. This change can be rapid in the presence of a certain evolutionary force. For example, the gene pool of a particular geographic area can change via migration in a matter of a few decades (Citation30).
Cases of ALS increase with age, reaching a peak between 65 and 75 years, and tend to decrease for men and later for women from the age of 75 (3). When the life expectancies are analyzed, the life expectancy in many areas with high population density (South and East Asia, Africa, South America) has not reached these ages as of now, but there is a rapid aging process in all societies. In the near future, the age distribution of the population in these regions will develop in line with the countries of Northern Europe and North America. This will mean a significant population increase in the age group with a high risk of developing ALS, leading to an increase in the incidence and prevalence of ALS. A new study involving ten regions (China, Europe, Iran, Japan, Libya, New Zealand, Serbia, Taiwan, the USA, and Uruguay) and data from 34% of the world’s population foresees a major increase in the prevalence of ALS (Citation31).
We acknowledge that this study has some limitations. Firstly, the fertility rates of each country change over the years and there are differences between the countries. Therefore, there could be a drawback in evaluating data from different countries together. However, when we analyzed the data for each city separately, we found that the number of children in the ALS group was statistically different from the control group in Lisbon and Antalya but was not different in Jena-Hannover and Warsaw (). Furthermore, the difference was that ALS patients in Lisbon and Antalya had more children than their control groups. In Lisbon and Antalya, there was no indication regarding the decrease in the number of children of the ALS group. We think that the population dynamics in these cities may be somehow different from the other two countries. Secondly, the number of patients and controls in our study may not be sufficient to explain the population dynamics of the disease. Nevertheless, we know the difficulty of forming groups with large numbers for a neurodegenerative disease with high mortality, such as ALS. Thirdly, there are individuals in both the patient and control groups who still have the chance to have children, at the time of the study. The menstrual status of female cases was not asked. In addition, we did not question the socio-economic status of the cases in the study. These factors may have an effect on the number of children that cases have. As the study includes a long questionnaire, not all the questions were answered in full and so the amount of missing information was higher than expected. Therefore, we believe, there is need for further studies with larger patient and control groups that may eliminate some of these shortcomings, and establish the effect of ALS on fertility.
Acknowledgements
The authors thank Philippa Price for making the English revisions of the article in her native language.
Declaration of interest
The authors do not report any conflict of interest regarding this manuscript.
Additional information
Funding
References
- Longinetti E, Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol. 2019;32:771–6.
- Collaborators GBDMND. Global, regional, and national burden of motor neuron diseases 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17:1083–97.
- Marin B, Boumediene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL, et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol. 2017;46:57–74.
- Medawar PB. The Uniqueness of the Individual. London: Methuen; 1957.
- Longinetti E, Regodon Wallin A, Samuelsson K, Press R, Zachau A, Ronnevi LO, et al. The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:528–37.
- Palese F, Sartori A, Verriello L, Ros S, Passadore P, Manganotti P, et al. Epidemiology of amyotrophic lateral sclerosis in Friuli-Venezia Giulia, North-Eastern Italy, 2002-2014: a retrospective population-based study. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:90–9.
- Benjaminsen E, Alstadhaug KB, Gulsvik M, Baloch FK, Odeh F. Amyotrophic lateral sclerosis in Nordland county, Norway, 2000-2015: prevalence, incidence, and clinical features. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:522–7.
- Jun KY, Park J, Oh KW, Kim EM, Bae JS, Kim I, et al. Epidemiology of ALS in Korea using nationwide big data. J Neurol Neurosurg Psychiatry. 2019;90:395–403.
- Turgut N, Varol SaraCoglu G, Kat S, Balci K, B GU, Birgili O, et al. An epidemiologic investigation of amyotrophic lateral sclerosis in Thrace, Turkey, 2006-2010. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:100–6.
- Leighton DJ, Newton J, Stephenson LJ, Colville S, Davenport R, Gorrie G, et al.; CARE-MND Consortium. Changing epidemiology of motor neurone disease in Scotland. J Neurol. 2019;266:817–25.
- Dorst J, Chen L, Rosenbohm A, Dreyhaupt J, Hubers A, Schuster J, et al. Prognostic factors in ALS: a comparison between Germany and China. J Neurol. 2019;266:1516–25.
- Goutman SA, Boss J, Patterson A, Mukherjee B, Batterman S, Feldman EL. High plasma concentrations of organic pollutants negatively impact survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90:907–12.
- Ryan M, Zaldivar Vaillant T, McLaughlin RL, Doherty MA, Rooney J, Heverin M, et al. Comparison of the clinical and genetic features of amyotrophic lateral sclerosis across Cuban, Uruguayan and Irish clinic-based populations. J Neurol Neurosurg Psychiatry. 2019;90:659–65.
- Li TM, Alberman E, Swash M. Comparison of sporadic and familial disease amongst 580 cases of motor neuron disease. J Neurol Neurosurg Psychiatry. 1988;51:778–84.
- Camu W, Khoris J, Moulard B, Salachas F, Briolotti V, Rouleau GA, et al. Genetics of familial ALS and consequences for diagnosis. French ALS Research Group. J Neurol Sci. 1999;165: S21–S6.
- Pliner HA, Mann DM, Traynor BJ. Searching for Grendel: origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. 2014;127:391–6.
- Konno T, Shiga A, Tsujino A, Sugai A, Kato T, Kanai K, et al. Japanese amyotrophic lateral sclerosis patients with GGGGCC hexanucleotide repeat expansion in C9ORF72. J Neurol Neurosurg Psychiatry. 2013;84:398–401.
- Alavi A, Nafissi S, Rohani M, Shahidi G, Zamani B, Shamshiri H, et al. Repeat expansion in C9ORF72 is not a major cause of amyotrophic lateral sclerosis among Iranian patients. Neurobiol Aging. 2014;35:267.e1.
- Murros K, Fogelholm R. Amyotrophic lateral sclerosis in Middle-Finland: an epidemiological study. Acta Neurol Scand. 1983;67:41–7.
- Karlsson EK, Kwiatkowski DP, Sabeti PC. Natural selection and infectious disease in human populations. Nat Rev Genet. 2014;15:379–93.
- Crow JFK. Motoo. An introduction to population genetics theory. Caldwell, NJ: The Blackburn Press; 2009.
- Carleton GD. The Transmissible Dementias and Other Brain Disorders Caused by Unconventional Viruses. In: Edouard Kurstak ZJL, P. V. Morozov, editor. Viruses, Immunity, and Mental Disorders Springer US; 1987.
- Byrne S, Heverin M, Bede P, Elamin M, Hardiman O. Fecundity in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:204–6.
- Johnson WG, Lucek PR, Chatkupt S, Furman Y, Lustenberger A, Lazzarini A. Reduced fecundity in male ALS gene-carriers. Am J Med Genet. 1995;59:149–53.
- De Carvalho M, Ryczkowski A, Andersen P, Gromicho M, Grosskreutz J, Kuzma-Kozakiewicz M, et al. International survey of ALS experts about critical questions for assessing patients with ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:505–10.
- R: A language and environment for statistical computing [Internet]. Vienna, Austria: R Foundation for Statistical Computing; 2013. Available at: http://www.R-project.org/.
- Human Development Report 2019 [Internet]. December 12, 2019. Available at: http://www.hdr.undp.org/en.
- Henderson RD, Garton FC, Kiernan MC, Turner MR, Eisen A. Human cerebral evolution and the clinical syndrome of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90:570–5.
- Ying W. A new hypothesis of neurodegenerative diseases: the deleterious network hypothesis. Med Hypotheses. 1996;47:307–13.
- Ruhli FJ, Henneberg M. New perspectives on evolutionary medicine: the relevance of microevolution for human health and disease. BMC Med. 2013;11:115.
- Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun. 2016;7:12408.