
Abstract
Introduction: More insight is needed into participation in daily activities and autonomy among patients with amyotrophic lateral sclerosis (ALS). Aims of this study were (1) to describe the course of participation restrictions and autonomy in participation during the first 10 months after diagnosis; (2) to study the influence of the rate of ALS progression on the course of participation. Methods: Secondary analysis of data from the longitudinal multicenter FACTS-2-ALS study. Self-report questionnaires were administered at inclusion (T0; n = 71), at 4 months (T1), 7 months (T2), 10 months (T3) after inclusion. Median duration of follow-up was 10.0 months. Participation restrictions were assessed using the sum of the Mobility Range and Social Behavior subscales of the Sickness Impact profile-68 (SIPSOC). Autonomy in participation was assessed using the Impact on Participation and Autonomy (IPA) Questionnaire. Fast disease progression was defined as an increase of 1.1 points per month or more on the ALS Functional Rating Scale. Results: Patients reported participation restrictions in all subscales while having mild physical limitations. There was a decrease of participation over time (restrictions and autonomy). This decrease was greatest in patients with fast disease progression. Disease progression negatively influenced movement-related participation more than social interaction domains. Rate of disease progression was more strongly related to SIPSOC scores compared to IPA scores. Discussion: Preserving participation may be an important determinant of quality of care for patients with ALS. Rate of progression of the disease should be taken into account as it was found to be significantly associated with the level of participation.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal, progressive, neurodegenerative disorder. Despite extensive research, no curative treatment is currently available. Daily care focuses on symptom management and preserving participation and health-related quality of life (HRQOL) (Citation1). In the International Classification of Functioning, Disability, and Participation (ICF) (Citation2), participation is defined as involvement in a life situation and covers an individual's experience in life activities and social roles, for example, work, leisure activities, and involvement in the community. It is a broad concept that can be evaluated from different perspectives, such as experienced restrictions in daily and social activities, or in terms of (loss of) experienced autonomy and control (Citation3,Citation4).
The variety and severity of impairments and disabilities that accompany ALS in relation to HRQOL have been described extensively (Citation5–7). There has, however, been less focus on preserving participation among ALS patients or on how multidisciplinary care might help optimize their participation during disease progression. Previous studies revealed that ALS patients experienced a withdrawal from many social activities during all disease stages and that physical decline, psychological factors, and communication disorders were associated with participation restrictions in ALS (Citation8,Citation9). Previous studies also demonstrated that restrictions in participation are associated with decreased HRQOL among patients with progressive neurological diseases, including ALS (Citation10–12). However, no longitudinal data about the impact of ALS on participation are available.
It is, therefore, important to know more about the course of participation restrictions, the way autonomy in participation is upheld in relation to disease progression, and whether participation restrictions increase in parallel with physical decline, or follow a different pattern. This knowledge may help optimize supportive care for patients with ALS. The aims of this study were (1) to determine the course of participation restrictions and autonomy in participation in the first 10 months after diagnosis of ALS and (2) to investigate the influence of the rate of ALS progression on the course of participation.
Materials and methods
Patients
This study concerns a secondary analysis of data from the longitudinal multicenter FACTS-2-ALS study (Citation13). Recruitment took place between 2009 and 2015. Patients could be included for two interventions, cognitive behavioral therapy (CBT) and aerobic exercise therapy (AET), or usual care (control group). As neither intervention proved effective, we studied these patients as one group (Citation5, Citation14). Inclusion criteria were: aged between 18 and 80 years; diagnosis of probable or definite ALS; life-expectancy of more than 1 year (estimate based on the clinical view of the rehabilitation physician), predicted forced vital capacity (FVC) of at least 80%; at least one month post-diagnosis; and able to walk and cycle. Exclusion criteria were: cognitive impairment (whether or not related to ALS, sufficiently serious to prevent the study from being completed) and psychiatric disorder, both assessed using the Cumulative Illness Rating Scale (CIRS) (Citation15). Eligibility criteria were confirmed by the rehabilitation physician. All participants who filled in questionnaires at T0 (N = 71) were included in the analyses.
Methods
Data for the current study were collected at inclusion (T0), and 4 months (T1), 7 months (T2), and 10 months (T3) thereafter. The initial assessment took place within approximately two weeks of enrollment and included self-administered questionnaires to be completed at home. Within the same week, FVC was measured by trained research assistants. The same procedure was followed at T1, T2, and T3. The Medical Ethics Committees from all participating centers approved the study protocol and informed consent was obtained from all patients.
Instruments
Experienced participation restrictions were assessed using the 68-item Sickness Impact profile (SIP68) (Citation16). The questionnaire comprises six subscales, two of which measure participation restrictions: Mobility Range (10 questions; range of actions to which a person has (limited) capabilities given his or her health status, such as shopping, house-cleaning, and taking care of personal business affairs) and Social Behavior (12 items; possible consequences of a health disorder in a person's functioning in relation to other people involving sexual activity, visiting friends, and activities in groups of people). We used the sum of these two subscales, the SIPSOC, to measure participation restrictions (Citation17). The SIPSOC asks patients to confirm or deny 22 statements about possible restrictions in participation. A higher score indicates more participation restrictions. The SIPSOC has been proven to be valid and reliable in individuals with disabilities and SCI (Citation18,Citation19).
The Impact on Participation and Autonomy (IPA) Questionnaire assesses autonomy in participation (Citation20). This measure consists of 32 items in six subscales: Autonomy Indoors (seven items, mobility indoors, and self-care), Family Role (seven items, responsibilities, and performing tasks at home), Autonomy Outdoors (five items, visiting friends/neighbors, engaging in social activities outdoors), Social Life (seven items, personal interaction with loved ones and friends), and Work/Education (six items). All items are graded on a five-point rating scale with discrete responses, ranging from 0 (very good) to 4 (very poor). For each domain, the participation score is calculated by summing the item scores. Higher scores denote more limitations in participation and autonomy. The validity, consistency and reliability of the instrument are good. This has been tested in patients with a wide range of conditions, in particular, neuromuscular disease, spinal cord injuries, traumatic head injuries, multiple sclerosis, stroke, and rheumatoid arthritis (Citation21–23).
Demographic variables (age, gender), time since symptom onset and site of onset were collected at inclusion. Disease severity was assessed using the revised ALS Functional Rating Scale-Revised (ALSFRS-R) (Citation24). The ALSFRS-R is a valid, reliable and sensitive instrument for assessing physical functioning. It consists of 12 items to evaluate bulbar function, gross and fine motor function and respiratory function; each item is scored on a scale of 0–4. Higher scores indicate better physical functioning.
FVC, as a determinant of lung-capacity, was measured with a spirometer (MicroRPM; PT Medical, Leek, The Netherlands) and the score was expressed as a percentage of the predicted score based on the patient’s gender, weight, race, and height. In case of insufficient lip closure, a face mask was used. Each participant made two attempts and the highest score was recorded.
Analyses
Descriptive statistics were used to describe characteristics of the study population. Rate of disease progression per month was calculated as the difference between two ALSFRS-R scores at T0 and a second measurement (the last available measurement in time) divided by the time between these measurements in months. The median of the difference score was calculated and used as cutoff to define two subgroups: “slow” progression and “fast” progression. Mann–Whitney tests were performed at T0 to describe differences in participant characteristics between these two subgroups.
To provide greater insight into changes in participation, scores were calculated for individual items, subscales of the SIPSOC and the subscales of the IPA. To analyze the course of participation between T0 and T3, random coefficient analysis (multi-level analysis) was applied. With this technique, all available data could be used. SIPSOC scores, SIPSOC Social Behavior and Mobility, and all IPA subscales were separately used as the dependent variable, resulting in eight different models. First, unconditional means models were fitted with a random intercept to account for nested data within individuals (due to the repeated measures). Next, the models were expanded by adding a random intercept to account for nested data within the intervention conditions (CBT, AET, or usual care). Likelihood ratio tests were used to assess model fit. For all SIPSOC and IPA subscales, adding a random intercept for intervention condition did not result in a significantly better model. Therefore, in all cases, we used the models with only a random intercept to account for repeated measures within individuals.
Subsequently, time was entered into the models as a set of three dummy variables with T0 as reference. Finally, the dichotomous disease progression variable was added to the model to study the effect of disease progression and the interaction effect between disease progression rate and participation time. In case the effect was significant, Cohen’s effect size was calculated, to determine the impact of progression and (d=difference between scores at T3/SD of baseline score). Using Cohen’s rule of thumb, an effect size of 0.12 was considered “small”, of 0.30 “medium”, and of 0.50 “large” (Citation25). SPSS version 25 for Windows was used for all statistical analyses (SPSS Inc., Chicago, IL).
Results
Seventy-one patients were included. Of these 71 patients, 10 patients were allocated to CBT, 26 patients were allocated to AET, and 35 patients were allocated to the control group. Seven patients died during the course of the study and 14 dropped out because they experienced the study as too burdensome (total 21 patients). Of these 21 patients, we included data of 13 patients, of which there were data of at least two measurements in time.
Patient characteristics are displayed in . Progression of disease was calculated for 63 patients who filled in questionnaires, at least at T0 and T1. The median progression rate was 1.1 points per month. We defined slow progression as <1.1 points per month and fast progression as ≥1.1 points per month. Time since onset of ALS was the only variable showing a significant difference between the patients with slow progression versus those with fast progression.
Table 1 Patient characteristics.
SIPSOC item scores over time from the complete case analysis are presented in and in the Supplement. The proportion of patients who experience participation restrictions over time (affirmative answer to the questions) increases for almost all items. Over time, most patients (40–72%) reported restrictions in daily work and chores around the house, in participating in social activities, and from onset about 40% of the patients reported restrictions in sexual activity and community activities. Four groups of SIPSOC items can be distinguished: (1) “Minor restrictions” is a category with items to which hardly any patients report restrictions throughout the study (indoor mobility, taking care of personal business). (2) “Restrictions from the start of the study” is a category of items with a substantial amount of patients reporting restrictions from onset and which increases over time (sexual activity, community activities, regular daily work around the house, doing heavy work around the house). (3) Items showing a strong increase of patients reporting restrictions during the study (activities around the house, household activities, going out at night, visiting friends). (4) Items that were difficult to categorize. also shows mean IPA scores over time from the complete case analyses. Autonomy Indoors scores increase most over time, followed by Autonomy Outdoors. Social life and relationships scores are lowest from onset and throughout the course of the study.
Table 2 SIPSOC and IPA scores over time.
Rate of change in participation over time (longitudinal models)
shows the estimated SIPSOC (participation restrictions) and IPA (autonomy in activities) scores, based on estimates of fixed effects. Overall, the SIPSOC and the IPA scores increased significantly over time ( and ).
Figure 1 (a) Estimated SIPSOC scores, based on estimates of fixed effect over time. Higher score means more restrictions in participation. (b) Estimated IPA scores, based on estimates of fixed effect over time. Higher score implies less autonomy.
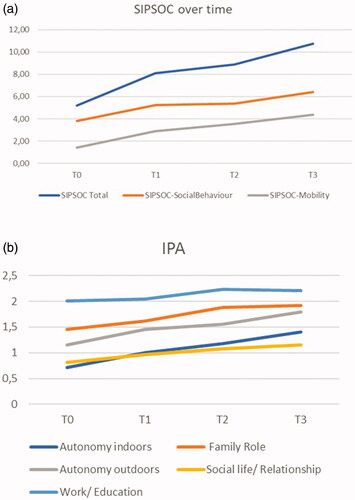
Table 3 Rate of change in participation (measured by SIPSOC scores, Social Behavior and Mobility) over 10 months after diagnosis of ALS (N = 71) and associations with disease progression (n = 63).
Table 4 IPA.
Associations with disease progression
Disease progression and time by progression interaction effects were calculated in a subgroup of 63 patients. Significant time by progression interaction effects were observed with regard to the total SIPSOC and the subscales Mobility Range and Social Behavior (the latter only between T0–T1, T0–T3), showing that the number of participation restrictions increased more in patients with faster progression of disease (). We additionally calculated the effect size, regarding the impact of progression. Cohen’s effect size was respectively 0.97 for the SIPSOC scale (large effect), 0.93 for subscale Mobility Range (large effect), and 0.77 subscale Social Behavior (medium effect).
Regarding the IPA, significant time by progression interaction effects were observed for Autonomy Indoors and Autonomy Outdoors (both between T0–T2, T0–T3) (). Cohen’s effect size was large, respectively 0.89 and 1.0. No interaction effect of progression of disease was observed for Family Role and Social Life (IPA), meaning that in our population, patients with fast progressive disease do not experience less autonomy in participation with regard to Family Role or Social Life compared to patients with slow progression. We cannot draw conclusions about Work and Education, because of the large number of missing values for this subdomain of the IPA.
Discussion
ALS patients reported participation restrictions in all subscales shortly after diagnosis, while experiencing relatively mild physical limitations. Participation decreased over time (restrictions and autonomy), to the greatest extent in patients with a more rapidly progressive disease course. Over time, rate of disease progression negatively influenced the participation domains related to movement indoors/outdoors more than those related to social interaction domains. Rate of disease progression also negatively influenced experienced restrictions in activities (SIPSOC scores) more than the sense of autonomy (IPA).
As stated before, after onset 34% of the patients were already experiencing restrictions in sexual activity, 36% in community activities, and 72% in heavy work around the house. These percentages did not change significantly over time, which could suggest that physical decline is not associated with these factors. On the other hand, it could also suggest that ALS care in The Netherlands is very effective in arranging adequate auxiliary tools during those 10 months. Our results on sexuality are consistent with previous studies, showing that sexuality plays a crucial role in personal well-being (Citation26). These results imply that dealing with restrictions in sexuality, experienced by ALS patients and their partners, may be an important early topic for multidisciplinary care.
A substantial number of the patients experience restrictions in community activities (church, voluntary work, clubs), indicating that this is an important group to identify to prevent social isolation (Citation27,Citation28).
In over two-thirds of patients, restrictions in being able to do regular and/or heavy housework were reported, meaning that support from caregivers, family, and/or neighbors was needed. This is also reflected in the IPA subscale “Family Role”. Throughout the study, participants reported the least autonomy in these activities. Compared to patients with spinal cord injury, mean SIPSOC scores showed that patients with ALS reported fewer participation restrictions shortly after the diagnosis of ALS, but considerably more restrictions 4 and 10 months later (Citation29). At onset, patients in our study were able to walk and exercise on a home-trainer, but experienced physical decline due to progression of the disease. In contrast, following spinal cord injury (paraplegic and tetraplegic), patients immediately experience severe physical restrictions which do, however, remain stable.
Taking IPA scores over time, mean scores increased most in the subdomain of Autonomy Indoors. When we compare our mean IPA scores with results from studies in patients with MS, stroke, spinal cord injury, and a mixed group (neuromuscular disease and brain injury), ALS patients experienced less autonomy in participation in all domains, 10 months after diagnosis (Citation30–35). Shortly after diagnosis, scores of ALS patients were comparable to those of patients with MS, and patients following stroke, spinal cord injury, and brain damage.
All this underlines the large impact of ALS on participation and related thereto quality of life of patients, starting shortly after diagnosis.
Rate of progression of the disease seems to influence the motor domains more than the social interaction domains and activities at home. Despite a rapid decline in physical functions, patients can apparently still maintain autonomy in activities and responsibilities at home and in personal interactions with loved ones, friends. They depend heavily on their caregivers who facilitate all aspects of their everyday lives. This means that caregivers must also adapt in order to support the autonomy of a patient, a loved one. There is, therefore, an increasing relevance of seeking support from family members (Citation36); we must be aware of the burden on caregivers, already in the first 10 months after the diagnosis. When comparing the interaction coefficients, the rate of progression seems to negatively influence experienced restrictions in activities (SIPSOC scores) more than the sense of autonomy (IPA scores). Patients can become overwhelmed by the ongoing decline in function, especially those with rapidly progressive disease, but apparently this has more impact on experienced restrictions. Apparently, many patients are successful in maintaining a sense of autonomy, a sense of control, even when it becomes more difficult to perform certain activities. Our study suggests that for those patients with a more progressive disease course, it is harder to find a new equilibrium, but not impossible. We know that QOL in patients with ALS, when considered in its broadest sense, does not correspond well to physical function, and is maintained by psychological, existential, and support factors. Perhaps the relative maintenance of social interaction domains and autonomy is what preserves QOL in these individuals, as described by McCaffrey et al. (Citation37–39).
This study has a number of strengths and limitations. Follow-up started directly after diagnosis and has given us insight into the interaction of rate of progression from 1 month after diagnosis. Having insight in rate of progression can help determine which patients are more “at risk” of restrictions in participation at the beginning of this palliative care process. This would improve the personalized care we aim to give our patients.
As patients included in the FACTS-2-ALS trial needed to be able to participate in physical exercise, less impaired patients were selected at baseline. One could argue, therefore, that participation restrictions were possibly underestimated at baseline. However, slope of disease progression in our patients is comparable to that of patients in other studies (Citation40). Hence, we do not believe that patients with a fast progressive disease course are under-represented in this study. This is a secondary analysis of data of the FACTS-2-ALS study. Follow-up of this study is 10 months which is relevant but relatively short. Future studies should have a follow up throughout the disease course giving us important knowledge adjuvant to natural course studies on physical complaints.
We used multilevel analysis to deal with the dropouts. All 13 patients who dropped out during the study, but who were included in analysis, were in the fast progression group. Had we not used multilevel analysis, then data of these patients would not have been included and our results would have been set to high and we would have underestimated the levels of participation restrictions.
We did not focus on possible other determinants of participation, such as communicational problems. This should be the focus of future studies (Citation41).
We made the assumption of a linear course of disease progression measured by the ALS-FRSR. However, the rate of progression of ALS, based on change in ALSFRS-R, is not necessarily linear. Yet, the rate of progression calculated for our patients is not always calculated over the same interval of time. We choose to follow standard practice for clinical trials where both trial design and the analysis virtually always assume a linear trend in ALSFRS-R rate of decline. Additionally, we conducted a multilevel analysis with progression as the outcome and time as predictor to investigate the course of progression. The results supported our choice to assume a linear trend.
In conclusion, ALS patients in the first 10 months after diagnosis experienced an increase of participation restrictions and loss of autonomy over time, which was highest in patients with rapidly progressive disease. Over time, rate of progression of the disease negatively influenced the participation domains related to movement indoors/outdoors, more than those related to social interaction domains, and negatively influenced experienced restrictions in activities (SIPSOC scores) more than the sense of autonomy (IPA).
Our results indicate that, from day one, focus on participation is an important determinant for optimal multidisciplinary care of ALS patients and their caregivers. Professionals must be aware that even patients with relatively mild physical limitations experience restrictions (in sexuality, community activities) and loss of autonomy in important activities. Prioritizing a patient’s participation in social and meaningful activities is one of the characteristics of person-centered care (Citation42) and improves quality of life.
Supplemental Material
Download MS Word (16.7 KB)Declaration of interest
E. Th. Kruitwagen-van Reenen, E.W.M. Scholten, and A. C. van Groenestijn report no disclosures. L.H. van den Berg serves on scientific advisory boards for Cytokinetics, Orion, Orphazyme, Denali; serves on the editorial board of Amyotrophic Lateral Sclerosis, The Journal of Neurology, Neurosurgery and Psychiatry. M.W.M. Post and J.M.A. Visser-Meily report no disclosures.
Additional information
Funding
References
- Hobson EV, McDermott CJ. Supportive and symptomatic management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2016;12:526–38.
- WHO. International classification of functioning, disability and health. Geneva: World Health Organization; 2001.
- Cardol M, de Haan RJ, de Jong BA, van den Bos GAM, de Groot IJM. Psychometric properties of the Impact on Participation and Autonomy Questionnaire. Arch Phys Med Rehabil. 2001;82:210–6.
- Perenboom RJ, Chorus AM. Measuring participation according to the International Classification of Functioning, Disability and Health (ICF). Disabil Rehabil. 2003;25:577–87.
- van Groenestijn AC, Schröder CD, van Eijk RPA, Veldink JH, Kruitwagen-van Reenen ET, Groothuis JT, et al. Aerobic exercise therapy in ambulatory patients with ALS: a randomized controlled trial. Neurorehabil Neural Repair. 2019;33:153–64.
- Simmons Z. Patient-perceived outcomes and quality of life in ALS. Neurotherapeutics. 2015;12:394–402.
- Ilse B, Prell T, Walther M, Hartung V, Penzlin S, Tietz F, et al. Relationships between disease severity, social support and health-related quality of life in patients with amyotrophic lateral sclerosis. Soc Indic Res. 2015;120:871–82.
- Morley D, Dummett S, Kelly L, Fitzpatrick R, Jenkinson C. Predictors of activity and participation across neurodegenerative conditions: a comparison of people with motor neurone disease, multiple sclerosis and Parkinson's disease. BMC Neurol. 2018;18:19.
- Van Groenestijn AC, Schröder CD, Kruitwagen-Van Reenen ET, Van Den Berg LH, Visser-Meily JMA. Participation restrictions in ambulatory amyotrophic lateral sclerosis patients: physical and psychological factors. Muscle Nerve. 2017;56:912–8.
- Duncan RP, Earhart GM. Measuring participation in individuals with Parkinson disease: relationships with disease severity, quality of life, and mobility. Disabil Rehabil. 2011;33:1440–6.
- Thordardottir B, Nilsson MH, Iwarsson S, Haak M. "You plan, but you never know"—participation among people with different levels of severity of Parkinson's disease . Disabil Rehabil. 2014;36:2216–24.
- Sandstedt P, Johansson S, Ytterberg C, Ingre C, Holmqvist LW, Kierkegaard M. Predictors of health-related quality of life in people with amyotrophic lateral sclerosis. J Neurol Sci. 2016;370:269–73.
- van Groenestijn AC, van de Port IGL, Schröder CD, Post MWM, Grupstra HF, Kruitwagen ET, et al. Effects of aerobic exercise therapy and cognitive behavioural therapy on functioning and quality of life in amyotrophic lateral sclerosis: protocol of the FACTS-2-ALS trial. BMC Neurol. 2011;11:70.
- van Groenestijn AC, Schröder CD, Visser-Meily JM, Reenen ET, Veldink JH, van den Berg LH. Cognitive behavioural therapy and quality of life in psychologically distressed patients with amyotrophic lateral sclerosis and their caregivers: results of a prematurely stopped randomized controlled trial. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:309–15.
- Linn BS, Linn MW, Gurel L. Cumulative illness rating scale. J Am Geriatr Soc. 1968;16:622–6.
- Post MW, de Bruin A, de Witte L, Schrijvers A. The SIP68: a measure of health-related functional status in rehabilitation medicine. Arch Phys Med Rehabil. 1996;77:440–5.
- de Bruin AF, Buys M, de Witte LP, Diederiks JP. The sickness impact profile: SIP68, a short generic version. First evaluation of the reliability and reproducibility. J Clin Epidemiol.1994;47:863–71.
- de Bruin AF, Diederiks JP, de Witte LP, Stevens FC, Philipsen H, de Bruin AF, et al. Assessing the responsiveness of a functional status measure: the Sickness Impact Profile versus the SIP68. J Clin Epidemiol. 1997;50:529–40.
- Post MW, de Witte LP, van Asbeck FW, van Dijk AJ, Schrijvers AJ. Predictors of health status and life satisfaction in spinal cord injury. Arch Phys Med Rehabil. 1998;79:395–401.
- Cardol M, de Haan RJ, van den Bos GA, de Jong BA, de Groot IJ. The development of a handicap assessment questionnaire: the Impact on Participation and Autonomy (IPA). Clin Rehabil. 1999;13:411–9.
- Kersten P, Cardol M, George S, Ward C, Sibley A, White B, Kersten P, et al. Validity of the impact on participation and autonomy questionnaire: a comparison between two countries. Disabil Rehabil. 2007;29:1502–9.
- Cardol M, Beelen A, van den Bos GAM, de Jong BA, de Groot IJM, de Haan RJ. Responsiveness of the impact on participation and autonomy questionnaire. Arch Phys Med Rehabil. 2002;83:1524–9.
- Magasi S, Post MC. A comparative review of contemporary participation measures’ psychometric properties and content coverage. Phys Med Rehabil. 2010;91:17–28.
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functioning rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169:13–21.
- Cohen J. Statistical power analysis for the behavioural sciences. 2nd ed. New York: Academic Press; 1988.
- Poletti B, Carelli L, Solca F, Pezzati R, Faini A, Ticozzi N, et al. Sexuality and intimacy in ALS: systematic literature review and future perspectives. J Neurol Neurosurg Psychiatry. 2019;90:712–9.
- Gibbons CJ, Thornton EW, Ealing J, Shaw PJ, Talbot K, Tennant A, et al. Assessing social isolation in motor neurone disease: a Rasch analysis of the MND Social Withdrawal Scale. J Neurol Sci. 2013;334:112–8.
- Rigby SA, Thornton EW, Tedman S, Burchardt F, Young CA, Dougan C. Quality of life assessment in MND: development of a social withdrawal scale. J Neurol Sci. 1999;169:26–34.
- Kilkens OJ, Post MW, Dallmeijer AJ, van Asbeck FW, van der Woude LH. Relationship between manual wheelchair skill performance and participation of persons with spinal cord injuries 1 year after discharge from inpatient rehabilitation. J Rehabil Res Dev. 2005;42:65–73.
- Blikman LJ, van Meeteren J, Twisk JW, de Laat FA, de Groot V, Beckerman H, et al. Effectiveness of energy conservation management on fatigue and participation in multiple sclerosis: a randomized controlled trial. Mult Scler. 2017;23:1527–41.
- Palstam A, Sjödin A, Sunnerhagen KS. Participation and autonomy five years after stroke: a longitudinal observational study. PLoS One. 2019;14:e0219513.
- van der Zee CH, Baars-Elsinga A, Visser-Meily JM, Post MW. Responsiveness of two participation measures in an outpatient rehabilitation setting. Scand J Occup Ther. 2013;20:201–8.
- Törnbom K, Hadartz K, Sunnerhagen KS. Self-perceived participation and autonomy at 1-year post stroke: a part of the Stroke Arm Longitudinal Study at the University of Gothenburg (SALGOT Study). J Stroke Cerebrovasc Dis. 2018;27:1115–22.
- Kwiatkowski A, Marissal JP, Pouyfaucon M, Vermersch P, Hautecoeur P, Dervaux B. Social participation in patients with multiple sclerosis: correlations between disability and economic burden. BMC Neurol. 2014;14:115.
- Piatt JA, Van Puymbroeck M, Zahl M, Rosenbluth JP, Wells MS. Examining how the perception of health can impact participation and autonomy among adults with spinal cord injury. Top Spinal Cord Inj Rehabil. 2016;22:165–72.
- Tramonti F, Bongioanni P, Fanciullacci C, Rossi B. Balancing between autonomy and support: coping strategies by patients with amyotrophic lateral sclerosis. J Neurol Sci. 2012;320:106–9.
- Simmons Z, Bremer BA, Robbins RA, Walsh SM, Fischer S. Quality of life in ALS depends on factors other than strength and physical function. Neurology. 2000;55:388–92.
- Robbins RA, Simmons Z, Bremer BA, Walsh SM, Fischer S. Quality of life in ALS is maintained as physical function declines. Neurology. 2001;56:442–4.
- McCaffrey N, Bradley S, Ratcliffe J, Currow DC. What aspects of quality of life are important from palliative care patients' perspectives? A systematic review of qualitative research. J Pain Symptom Manage. 2016;52:318–28.e5.
- Takei K, Tsuda K, Takahashi F, Hirai M, Palumbo J. An assessment of treatment guidelines, clinical practices, demographics, and progression of disease among patients with amyotrophic lateral sclerosis in Japan, the United States, and Europe. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:88–97.
- Felgoise SH, Zaccheo V, Duff J, Simmons Z. Verbal communication impacts quality of life in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17:179–83.
- Wade DT. What is rehabilitation? An empirical investigation leading to an evidence-based description. Clin Rehabil. 2020;34:571–83.