
Abstract
Objective: The presence of the hexanucleotide repeat expansion (HRE) in C9orf72 gene is associated to the ALS/FTD spectrum, but also to parkinsonisms. We here describe an Italian family with the father diagnosed with Parkinson disease (PD) at the age of 67 and the two daughters developing FTD and ALS at 45 years of age. We searched for C9orf72 HRE with possible genetic and epigenetic modifiers to account for the intrafamilial phenotypic variability. Methods: C9orf72 mutational analysis was performed by fragment length analysis, Repeat-primed PCR and Southern blot. Targeted next generation sequencing was used to analyze 48 genes associated to neurodegenerative diseases. Promoter methylation was analyzed by bisulfite sequencing. Results: Genetic analysis identified C9orf72 HRE in all the affected members with a similar repeat expansion size. Both the father and the FTD daughter also carried the heterozygous p.Ile946Phe variant in ATP13A2 gene, associated to PD. In addition, the father also showed a heterozygous EIF4G1 variant (p.Ala13Pro), that might increase his susceptibility to develop PD. The DNA methylation analysis showed that all the 26 CpG sites within C9orf72 promoter were unmethylated in all family members. Conclusions: Neither C9orf72 HRE size nor promoter methylation act as disease modifiers within this family, at least in blood, not excluding HRE mosaicism and a different methylation pattern in the brain. However, the presence of rare genetic variants in PD genes suggests that they may influence the clinical manifestation in the father. Other genetic and/or epigenetic modifiers must be responsible for disease variability in this C9orf72 family case.
Keywords:
Introduction
A hexanucleotide repeat expansion (HRE) in C9orf72 gene is the most frequent cause of familial and sporadic amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Citation1,Citation2), ranging from 2–23 units in the normal population to >30–>4000 units in pathological conditions (Citation3). In contrast to other repeat expansion disorders, no clear association between HRE size and phenotype severity or disease state (ALS/FTD) has been demonstrated so far. Genetic anticipation is not an evident phenomenon and, within the same pedigree, individuals with a similar HRE may manifest indifferently ALS, FTD, or mixed phenotypes (Citation4–11). In addition, C9orf72 HRE has been reported in a heterogeneous array of neurological disorders, other than ALS and FTD, including parkinsonism and psychosis (Citation12,Citation13). However, also within the ALS/FTD disease spectrum, the wide heterogeneity of clinical features and symptoms even intra-familiarly suggests that modifiers, both genetic and epigenetic, may contribute to influence disease presentation in HRE carriers (Citation14). The presence of additional rare genetic variants in ALS-associated genes (Citation15–18) and of microdeletions in the low complexity region flanking the repeat sequence (Citation9,Citation19) have been described as consistent features in C9orf72 mutation carriers, possibly influencing disease presentation as well.
As regards epigenetic disease modifiers, C9orf72 gene promoter was described to be methylated in 30% of both ALS and FTD HRE carriers, in association to decreased V1 and V3 transcripts spanning the HRE and diminished pathological RNA foci number and dipeptide repeat protein (DPR) in autoptic brains (Citation20–24). C9orf72 promoter methylation was also associated to a reduced cerebral atrophy in neuroimaging data from C9orf72 mutation carriers (Citation25) and increased survival in C9orf72-FTD patients (Citation22), suggesting a neuroprotective role of this epigenetic modification.
Here we investigated possible genetic and epigenetic modifiers in a C9orf72-positive Italian family showing very different clinical presentations, including Parkinson disease (PD), ALS and FTD.
Materials and methods
C9orf72 genotyping
The family was clinically investigated at the Department of Neurology-Stroke Unit, Hospital of Crema, Italy. Informed consent was obtained and the study approved by the ethics committee at IRCCS Istituto Auxologico Italiano. Genomic DNA was isolated from whole blood using Wizard® kit (Promega). C9orf72 repeat expansion was evaluated by a combination of fluorescent amplicon-length analysis and repeat-primed PCR (RP-PCR) as previously described (Citation26). In the first amplification reaction we used primers flanking the repeat sequence (FAM-labeled forward 5′-TGTAAAACGACGGCCAGTCAAGGAGGGAAACAACCGCAGCC-3′ and reverse 5′-GCAGGCACCGCAACCGCAG-3′). For RP-PCR, three primers were used (FAM-labeled forward 5′-TGTAAAACGACGGCCAGTCAAGGAGGGAAACAACCGCAGCC-3′; reverse 5′-CAGGAAACAGCTATGACCGGGCCCGCCCCGACCACGCCCCGGCCCCGGCCCCGG-3′; reverse anchor M13 primer 5′-CAGGAAACAGCTATGACC-3′) with AccuPrime™ GC-Rich DNA Polymerase (Invitrogen) and 180µM 7-deaza-2-deoxy GTP (Roche). Amplicons were run on ABI Prism 3500 (Applied Biosystems) and visualized using Gene Mapper v.4 software (Applied Biosystems).
Southern blotting
Genomic DNA (10 µg) was XbaI-digested and run on 0.7% agarose gel. After gel washes in 0.25N HCl for 20 min, in 0.4N NaOH and 0.6M NaCl for 30 min and in 0.5M Tris pH 7.5 and 1.5M NaCl for 30 min, DNA was transferred to a Hybond N + membrane (Amersham) in 20X Saline Sodium Citrate (SSC) buffer. A unique-sequence probe (466bp) within C9orf72 first intron was PCR amplified (forward 5′-CTTTCTCCAGATCCAGCAGCCTCC-3′ and reverse 5′-CTGAGTTCCAGAGCTTGCTACAG-3′) and α-32P-dCTP radiolabelled by random primer labeling kit (Thermo Fisher). Probe hybridization was carried out in 50% formamide, 5X SSC, 5X Denhardt, 0.5% SDS and 25 µg/µl salmon sperm at 42 °C overnight. The membrane was washed in 2X SSC/0.1% SDS buffer at room temperature and in 0.5X SSC/0.1% SDS buffer at 60 °C for 20 min each and visualized by autoradiography.
For the non-radioactive Southern Blotting, the same probe was labeled using Digossigenin-dUTP and PCR DIG Probe Synthesis Kit (Roche). Probe hybridization was performed with Dig Easy Hyb buffer (Roche). The DIG Nucleic Acid Detection Kit (Roche) was used and hybridization signals were visualized by the image system Azure c200 (Azure Biosystems).
Bisulfite-sequencing of C9orf72 promoter
Blood genomic DNA (200 ng) was treated with sodium bisulfite using the EZ DNA Methylation™ Kit (Zymo Research). A semi-nested PCR was performed with methylation-specific primers (BSP_1F 5′-TTTATTAGGGTTTGTAGTGGAGTTTT-3′ or BSP_2F 5′-TATTAGGGTTTGTAGTGGAGTTTT-3′, and BSP_1R 5′-AAATCTTTTCTTATTCACCCTCAAC-3′). The methylation status of the 450bp-sequence encompassing 26 CpG sites was assessed by Sanger sequencing on ABI Prism 3500.
NGS target resequencing
The custom-designed NGS panel includes 48 genes associated to ALS (ALS2, ANG, DCTN1, FUS, HNRNPA1, HNRNPA2B1, MATR3, NEK1, OPTN, PFN1, SETX, SOD1, SPAST, SPG11, SQSTM1, TARDBP, TBK1, TUBA4A, UBQLN2, VAPB), FTD (CHMP2B, GRN, MAPT, PRNP, VCP), Alzheimer's disease (APOE, APP, PSEN1, PSEN2, TREM2) and PD (ATP13A2, DJ1, DNAJC6, EIF4G1, FBXO7, GBA, GCH1, LRRK2, PARK2, PINK1, PLA2G6, PRKRA, SNCA, TAF1, TH, UCHL1, VPS13C, VPS35). DNA libraries (Illumina) were sequenced on NextSeq 550 (Illumina).
For data analysis we considered only variants with a Non-Finnish European (NFE) MAF ≤0.001 in gnomAD database and predicted to be nonsense, missense, insertions/deletions and splicing.
Results
Clinical report
We report an Italian family with 4 members (parents and two offsprings; ) who came to our attention because the proband (ND68), at 45 years of age, started to manifest subacute delirium with fluctuating psycho-motor agitation, spatial/temporal disorientation, confusion, confabulation, paranoid/persecution type delusions and multimodal hallucinations. At onset the disorder was fluctuating with periods of remission, but then it became persistent with night worsening. No fever, headache, brain injury, previous infectious event, vaccinations or psychiatric disorders were reported, neither history of alcohol or psychoactive substances. Routine blood sampling and brain CT scan were normal. She was admitted to the psychiatric ward and treated with neuroleptic drug and benzodiazepine with gradual improvement of psychotic symptoms. She was diagnosed with a suspected major depressive episode but, due to late onset and atypical course, the possibility of a neurological disease was considered. Neurological examination evidenced a generalized and symmetric hypokinesia and extrapyramidal rigidity probably due to drug-induced parkinsonism. No tremor or MND signs were present. Antipsychotic drugs were reduced with reappearing of a severe confusional state. Neuroleptic drug was resumed with symptoms control. Routine blood tests, thyroid function, anti-thyroglobulin and thyroid peroxidase antibodies, ACTH, serum and urine cortisol, Vitamin B12, folate, ceruloplasmin, serum and urine copper were normal. Antibody tests for HBV, HCV, HIV 1-2 and Borrelia burgdorferi were negative. Serologic tests for systemic autoimmune diseases, neoplastic markers, onconeural antigens, and serum anti VGKC-complex, NMDAR and GAD antibodies were negative. Cerebrospinal fluid (CSF) analysis and cytology were normal with no oligoclonal bands. There was a minimal reduction of CSF beta-amyloid protein with Tau, P-Tau and 14.3.3 proteins in normal range. EEG was normal and brain MRI with gadolinium showed a mild widening of subarachnoid spaces without parenchymal signal alterations or contrast-enhancing lesions. Neuropsychological testing showed a mild cognitive impairment with executive dysfunction, attention and judgment deficit, visuospatial alterations, loss of empathy, motivation, planning and difficulty in performing multiple tasks. Short-term memory deficit was also detected. She did not develop MND signs. She was finally diagnosed with a form of FTD and we decided to further inspect her family.
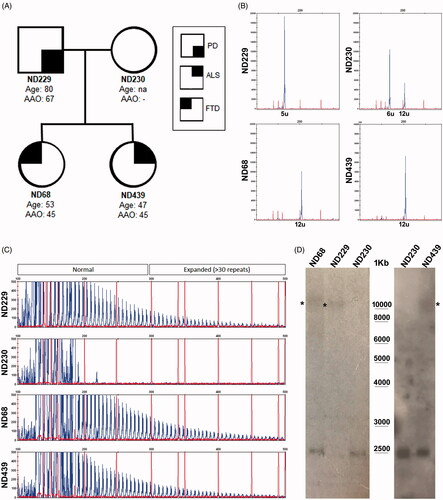
Figure 1 Mutational analysis of C9orf72 gene in the Italian family case. (A) The pedigree of the investigated family is shown together with the diseases state of each member (PD, FTD, ALS), the age at onset (AAO) and the age at last follow-up; (B) C9orf72 repeat sizing is shown by GeneScan run of fluorescent amplicons obtained using primers pairs flanking the hexanucleotide repeat; the number of repeat units is shown for each allele; (C) Electrophoregram of the fluorescent amplicons obtained by RP-PCR using a degenerate primer on the repeat sequence; the 6bp-interval peaks and the pathogenic threshold of 30 repeat units are clearly visible in the three affected members. (D) Southern blot analysis was first conducted by using a radioactive P32-labeled probe (left panel) and then with a DIG-labeled probe (right panel). Genomic DNA was XbaI-digested and the 2.3Kb wild-type allele is visible. * expanded alleles; the wild-type mother DNA (ND230) was used as a negative control in the assay.
Her father (ND229) was diagnosed with PD associated with delusions of paranoid type. When he was 67, he began presenting resting tremor to the right hand and minimal motor discomfort. He also complained of a memory deficit due to recent events, insomnia, and behavioral alterations. Neurological examination showed a fine tremor at rest and a shaded plastic hypertonus in the upper limbs prevalent on the right. Brain MRI showed suffering of the periventricular white substance as from chronic microcirculation disorders. He started therapy with pramipexole with improvement in tremor and insomnia and good control of motor and cognitive-behavioral symptoms for at least two years. At the age of 69 he developed psychiatric symptoms with delusional ideas. A history of nonspecific psychiatric disorder was reported also in his mother. Neuropsychological testing showed a mild cognitive impairment with executive dysfunction with difficulty in performing multiple tasks (MMSE: 21). In the next years disease worsened and DOPA was introduced with few beneficial effects. The patient began to show a progressive cognitive impairment with hallucinations and delirium. No MND signs were present until the last follow-up ().
The proband’s mother (ND230) did not report any neurological sign or family history of neurodegenerative diseases. Recently, also the proband’s sister (ND439) came to our attention at the age of 47 because in the last two years she started to show a disorder of deambulation due to muscle weakness in the legs with bilateral steppage. She presented a spinal form of ALS, more severe in lower limb. Spasticity was present in upper and lower limbs. Bilateral hand muscle wasting was evident, bulbar muscles were still spared. EMG showed active denervation in all limb muscles examined. Cerebral and cervical MRI were normal. She started oral therapy with riluzole. Neuropsychological testing showed a constructive apraxia with no global cognitive impairment. She did not develop extrapyramidal signs, at least at the last follow-up two years after disease onset.
Mutational analysis of C9orf72 gene
Given the clinical features of the ND68 proband and of her ND229 father () with a family history of psychiatric symptoms, we performed a mutational analysis of C9orf72 gene. We found that both ND229 and ND68 carried a single wild-type allele of 5 and 12 repeat units, respectively, and the pathological HRE, while the mother (ND230) carried two wild-type alleles (6 and 12 units) (). Since the proband showed an earlier disease onset compared to the father, we first evaluated HRE length. We performed a radioactive Southern Blot and found that both the proband and her father carried a similar HRE length (∼1500 repeats)(, ). When also the ALS-affected sister ND439 came later to our attention (), C9orf72 mutational analysis revealed a single 12-repeat wild-type allele and the HRE (). HRE sizing by DIG-labeled Southern Blot showed that she also carried ∼1500 repeat units-expansion ().
Table 1 Summary of phenotypic, genotypic and epigenetic features of the C9orf72 family members.
Analysis of C9orf72 genetic and epigenetic modifiers
We investigated possible genetic and epigenetic disease modifiers, already associated in literature to C9orf72, other than HRE size.
We first searched for additional rare variants by using a custom targeted resequencing gene panel which included 48 causative genes associated to ALS, FTD, AD and PD (gene list in Materials and Methods). The proband ND68 and her father ND229 carried a rare heterozygous ATP13A2 (PARK9) variant (p.Ile946Phe; MAF = 0.001)(). The father also carried a heterozygous variant in the PD-associated gene EIF4G1 (p.Ala13Pro), never reported before () and predicted to be “deleterious” and “possibly damaging” by SIFT and Polyphen2 prediction tools, respectively. No additional pathogenic variants were identified in the ALS-affected sibling ND439 ().
We then analyzed the epigenetic state of C9orf72 promoter by bisulfite sequencing. We found no DNA methylation either in the father or in the two siblings and in the wild-type mother at any of the 26 CpG sites analyzed in the promoter region ().
Discussion
We here describe an Italian family in which the presence of C9orf72 HRE segregates with different disease manifestations, including PD, FTD and ALS, and different ages of onset. By investigating whether genetic and/or epigenetic factors might act as disease modifiers, we first analyzed HRE size and found a similar pathological length in all mutated members. A clear relationship between HRE size and disease severity or clinical manifestations in C9orf72 mutation carriers is not consistently supported in literature (Citation5,Citation27–30), in contrast to the other repeat expansion disorders. Some studies have provided supportive evidence for disease anticipation in families with a C9orf72 mutation (Citation5,Citation31) although in contrast to other studies (Citation28) and to reports of HRE contraction across generations (Citation7), leaving this issue still inconclusive.
In our family the two siblings showed the same age of onset and HRE size, but different clinical manifestations, thereby excluding a role for HRE size in influencing disease presentation. However, given the high degree of HRE somatic mosaicism (Citation29), we can’t exclude a different HRE length in the affected brains compared to blood.
The size of the wild-type alleles in C9orf72 carriers has also been considered a possible risk factor because the >8-units alleles, rare in the normal population, are functionally associated to a decreased transcription of C9orf72 gene in a length-dependent manner (Citation5,Citation9). The 12-units wild-type alleles in the two daughters might represent a still undefined risk factor, since no evidence of a differential C9orf72 gene expression in association to the wild-type allele repeat size was demonstrated in ALS/FTD patients so far.
As a multifactorial disease, ALS is largely characterized by an oligogenic etiology also in familial cases where double mutations in ALS/FTD causative genes have been reported (Citation15–18). In our NGS-based mutational analysis of a wide array of genes implicated not only in ALS and FTD, but also in PD and AD, we identified two rare variants in heterozygous state in ATP13A2 and EIF4G1 genes. Of interest, the father carried both these variants, while the FTD daughter shared only the ATP13A2 one. Homozygous and compound heterozygous mutations in the ATP13A2 gene were shown to cause a rare, atypical form of recessive juvenile parkinsonism with dementia, known as Kufor–Rakeb syndrome (Citation32). ATP13A2 is a lysosomal protein found in Lewy bodies, whose loss-of-function leads to a-synuclein accumulation (Citation33). Single rare ATP13A2 variants were also associated to sporadic PD patients (Citation34,Citation35) who showed a decreased expression of ATP13A2 gene in dopaminergic neurons of the substantia nigra (Citation36). In contrast to the ATP13A2 p.Ile946Phe variant we identified, predicted to be tolerated and previously described also in a healthy control (Citation35), the novel EIF4G1 p.Ala13Pro variant in the father was predicted to be deleterious by in silico analyses. Although variants in EIF4G1 gene were initially identified in familial and sporadic PD (Citation37,Citation38), their association to PD is now disputed (Citation39). Therefore we can only speculate that the presence the two rare variants in ATP13A2 and EIF4G1 genes may confer a higher risk to the father to develop PD. Given that the ALS-affected sister did not carry any rare variant in the selected genes, we can exclude their contribution in influencing disease presentation within the family. However, our targeted NGS approach, although quite extended to comprehend 48 genes, may miss rare variants in still unknown genes for ALS/FTD and other neurodegenerative disorders. Only whole-exome or whole-genome sequencing will allow to definitely clarify whether additional genetic variants may act as disease modifiers in this C9orf72 family.
Finally, as a possible epigenetic disease modifier, we studied the methylation state of C9orf72 gene promoter whose hypermethylation occurs in about 30% of C9orf72 ALS and FTD carriers and acts as a potential neuropotective factor, being associated to decreased C9orf72 gene expression, DPR synthesis and cerebral atrophy (Citation23–25). Although C9orf72 promoter was unmethylated in all three family members, ruling out an association with this epigenetic modification, methylation is tissue-specific so that we can not completely exclude a different methylation pattern in the affected brains compared to the blood genomic DNA analyzed.
In conclusion, by investigating the possible role of different disease modifiers within a C9orf72 family, our study failed to find a clear association between disease presentation and HRE length, oligogenic inheritance or promoter methylation, suggesting that the high phenotypic variability of the mutation carriers likely relies on a more complex interplay of still unknown genetic factors, possibly including the wild-type allele repeat size, undefined environmental risk factors and other epigenetic modifications.
Declaration of interest
V.S. is in the Editorial Advisory Board of Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. All the other Authors report no conflicts of interest.
Additional information
Funding
References
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–68.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–56.
- Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet. 2013;92:345–53.
- Fournier C, Barbier M, Camuzat A, Anquetil V, Lattante S, Clot F, et al. Relations between C9orf72 expansion size in blood, age at onset, age at collection and transmission across generations in patients and presymptomatic carriers. Neurobiol Aging. 2019;74:234.e1–234.e8.
- Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Engelborghs S, De Bleecker J, et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry. 2016;21:1112–24.
- Esselin F, Mouzat K, Polge A, Juntas-Morales R, Pageot N, De la Cruz E, et al. Clinical phenotype and inheritance in patients with C9ORF72 hexanucleotide repeat expansion: results from a large French cohort. Front Neurosci 2020;14:316.
- Jackson JL, Finch NCA, Baker MC, Kachergus JM, Dejesus-Hernandez M, Pereira K, et al. Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol Neurodegener 2020;15:7.
- Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C. Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev. 2017;44:117–24.
- van der Zee J, Gijselinck I, Dillen L, Van Langenhove T, Theuns J, Engelborghs S, et al. A Pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat. 2013;34:363–73.
- O'Brien M, Burke T, Heverin M, Vajda A, McLaughlin R, Gibbons J, et al. Clustering of neuropsychiatric disease in first-degree and second-degree relatives of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2017;74:1425–30.
- Calvo A, Moglia C, Canosa A, Cistaro A, Valentini C, Carrara G, et al. Amyotrophic lateral sclerosis/frontotemporal dementia with predominant manifestations of obsessive-compulsive disorder associated to GGGGCC expansion of the c9orf72 gene. J Neurol. 2012;259:2723–5.
- Floris G, Borghero G, Cannas A, Di Stefano F, Costantino E, Murru MR, et al. Frontotemporal dementia with psychosis, parkinsonism, visuo-spatial dysfunction, upper motor neuron involvement associated to expansion of C9ORF72: a peculiar phenotype? J Neurol. 2012;259:1749–51.
- Cooper-Knock J, Kirby J, Highley R, Shaw PJ. The spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:326–39.
- Saracino D, Le Ber I. Clinical update on C9orf72: frontotemporal dementia, amyotrophic lateral sclerosis, and beyond. Adv Exp Med Biol. 2021;1281:67–76.
- Van Blitterswijk M, Van Es MA, Hennekam EAM, Dooijes D, Van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:3776–84.
- Morgan S, Shatunov A, Sproviero W, Jones AR, Shoai M, Hughes D, et al. A comprehensive analysis of rare genetic variation in amyotrophic lateral sclerosis in the UK. Brain. 2017;140:1611–8.
- Ross JP, Leblond CS, Laurent SB, Spiegelman D, Dionne-Laporte A, Camu W, et al. Oligogenicity, C9orf72 expansion, and variant severity in ALS. Neurogenetics 2020;21:227–42.
- McCann EP, Henden L, Fifita JA, Zhang KY, Grima N, Bauer DC, et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J Med Genet 2021;58:87–95.
- Corrado L, Tiloca C, Locci C, Bagarotti A, Hamzeiy H, Colombrita C, et al. Characterization of the c9orf72 GC-rich low complexity sequence in two cohorts of Italian and Turkish ALS cases. Amyotroph Lateral Scler Front Degener 2018;19:426–431.
- Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am J Hum Genet. 2013;92:981–9.
- Russ J, Liu EY, Wu K, Neal D, Suh ER, Irwin DJ, et al. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 2015;129:39–52.
- Xi Z, Rainero I, Rubino E, Pinessi L, Bruni AC, Maletta RG, et al. Hypermethylation of the CpG-island near the C9orf72 G4C2-repeat expansion in FTLD patients. Hum Mol Genet. 2014;23:5630–7.
- Belzil VV, Bauer PO, Gendron TF, Murray ME, Dickson D, Petrucelli L. Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Res. 2014;1584:15–21.
- Liu EY, Russ J, Wu K, Neal D, Suh E, McNally AG, et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014;128:525–41.
- McMillan CT, Russ J, Wood EM, Irwin DJ, Grossman M, McCluskey L, et al. C9orf72 promoter hypermethylation is neuroprotective: neuroimaging and neuropathologic evidence. Neurology 2015;84:1622–30.
- Akimoto C, Volk AE, van Blitterswijk M, Van den Broeck M, Leblond CS, Lumbroso S, et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet. 2014;51:419–24.
- Dols-Icardo O, García-Redondo A, Rojas-García R, Sánchez-Valle R, Noguera A, Gó mez-Tortosa E, et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum Mol Genet. 2014;23:749–54.
- van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 2013;12:978–88.
- Nordin A, Akimoto C, Wuolikainen A, Alstermark H, Jonsson P, Birve A, et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum Mol Genet. 2015;24:3133–42.
- Suh ER, Lee EB, Neal D, Wood EM, Toledo JB, Rennert L, et al. Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol. 2015;130:363–72.
- Van Mossevelde S, Van Der Zee J, Gijselinck I, Sleegers K, De Bleecker J, Sieben A, et al. Clinical evidence of disease anticipation in families segregating a C9orf72 repeat expansion. JAMA Neurol. 2017;74:445–52.
- Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91.
- Park JS, Blair NF, Sue CM. The role of ATP13A2 in Parkinson's disease: clinical phenotypes and molecular mechanisms. Mov Disord. 2015;30:770–9.
- Hopfner F, Mueller SH, Szymczak S, Junge O, Tittmann L, May S, et al. Rare variants in specific lysosomal genes are associated with Parkinson's disease. Mov Disord. 2020;35:1245–8.
- Djarmati A, Hagenah J, Reetz K, Winkler S, Behrens MI, Pawlack H, et al. ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord. 2009;24:2104–11.
- Dehay B, Martinez-Vicente M, Ramirez A, Perier C, Klein C, Vila M, et al. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy 2012;8:1389–91.
- Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89:398–406.
- Deng H, Wu Y, Jankovic J. The EIF4G1 gene and Parkinson's disease. Acta Neurol Scand. 2015;132:73–8.
- Saini P, Rudakou U, Yu E, Ruskey JA, Asayesh F, Laurent SB, et al. Association study of DNAJC13, UCHL1, HTRA2, GIGYF2, and EIF4G1 with Parkinson's disease. Neurobiol Aging. 2021;100:119.e7–e13.