
Abstract
Objective: To provide a detailed and differentiated description of the path to receiving the correct amyotrophic lateral sclerosis (ALS) diagnosis, including delay times, referrals, alternate diagnoses, and clinical progression.
Methods: Medical records until the date of ALS diagnosis were reviewed and linked to the Swedish Motor Neuron Disease Quality Registry.
Results: The study included 353 Stockholm ALS patients diagnosed in 2016–2021. Patients were divided into four groups: 117 (33.1%) with lower extremity (LE), 85 (24.1%) with upper extremity (UE), 136 (38.5%) with bulbar, and 15 (4.2%) with respiratory onset. The time from onset to diagnosis was 16.0 (9.4–27.5) months in LE, 12.9 (8.8–17.8) months in UE, 11.7 (7.4–16.0) months in bulbar, and 8.3 (4.7–15.6) months in respiratory onset. Patients with UE or LE onset were often referred to orthopedics or a spinal/hand surgery clinic (29.3% for LE and 41.8% for UE), while bulbar patients were more frequently referred to ENT (66.3%). For those with LE or UE onset, the most common alternate diagnosis was spinal/foraminal stenosis whereas myasthenia gravis and stroke were more common for bulbar onset patients. For the respiratory group, cardiopulmonary diagnoses predominated. The proportion of all patients in King’s stage 3 or 4 increased from 11.3% to 46.1% from the initial health care visit to diagnosis.
Conclusions: There was great variation in the path to ALS diagnosis according to the onset clinical phenotype. In all groups, the diagnostic delay and clinical progression was substantial. We identified subgroups where the delay was the longest and might be reduced.
Keywords:
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive motor neuron disease (MND) of unknown etiology (Citation1). The initial clinical presentation typically consists of focal weakness that generalizes to widespread motor dysfunction, eventually resulting in feeding difficulties, respiratory failure, and death (Citation2). About a third of ALS patients present with bulbar symptoms such as dysarthria or dysphagia, and a small proportion with respiratory difficulties. In addition, almost 50% of patients with ALS have some degree of cognitive impairment and up to 15% meet the criteria for frontotemporal dementia (Citation3). There is currently no effective treatment for ALS and the expected survival from onset is short, averaging about 2–4 years (Citation4). For the clinician, diagnosing the disease is difficult due to the heterogeneity and non-specific nature of the initial symptoms (Citation5). The lack of strong risk factors and established non-invasive biomarkers further increases the difficulty of correct and early diagnostics. Also, for most non-neurologists, clinical experience of the disease is limited due to its low incidence: estimated to 2.8/100,000 persons per year in the European population (Citation6), and research has shown that many patients are initially misdiagnosed (27–44%) (Citation7–9). Consequently, the delay from symptom onset to diagnosis is substantial, averaging about a year (Citation7,Citation9–12), resulting in frustration and anxiety among patients, caregivers, and physicians alike (Citation13). Moreover, diagnostic delay is associated with increased costs for the health care system (Citation14) and might preclude timely engagement with a multidisciplinary team as well as enrollment in clinical trials (Citation15).
There are previous studies of relatively small size showing a substantial diagnostic delay associated with a protracted investigational course (Citation16). We aimed at exploring this further, conducting an extensive study in a large patient cohort including diagnostic delay, referrals, alternate diagnoses, and importantly also symptom progression. Because of the heterogeneity of clinical characteristics and course, patients were divided into four groups based on the predominant presenting symptoms: lower extremity (LE), upper extremity (UE), bulbar, and respiratory onset.
Methods
Study population
We conducted a population-based, retrospective cohort study including a total of 353 ALS patients, diagnosed between January 2016 and May 2021 at the ALS Clinical Research Center (ALS CRC) at the Karolinska University Hospital in Stockholm, Sweden. The ALS CRC is the only tertiary center for ALS in the Stockholm region with a population of approximately 2 million inhabitants, and cares for all ALS patients in the region. All patients had a diagnosis of probable, possible, or definite ALS, according to the El Escorial criteria (Citation17).
The study was approved by the Ethical Review Board in Stockholm, Sweden (DNRs 2017/1895-31/1) and information was collected on an opt-out basis.
Data
We reviewed the medical records of each patient from most medical providers until the date of ALS diagnosis. The majority of providers in the Stockholm area (including primary care providers [PCPs], hospitals, and others) use a common interconnected medical record system. Among approximately 15% of the patients, all data were not available in the common system and had to be obtained through ancillary records or other medical documents. In a few cases (approximately 3%), date and neurological status at the initial health care visit was estimated from this information.
In addition, using the unique Swedish personal identification number, the participants were linked to the Swedish MND Quality Registry, which since 2015 has recruited MND patients in Sweden at the time of diagnosis. It includes the prospective collection of information on clinical measures, biological samples, and quality of life outcomes and as of 2017, this registry had a 99% coverage of all MND patients in Stockholm (Citation18).
Measures and definitions
Alternate and differential diagnoses
Any non-ALS diagnosis ending an investigation cycle was reported as an alternate diagnosis. Other diagnoses specifically mentioned in medical records as considerations were reported as differential diagnoses.
Clinical status
Spread of symptoms to a specific area was defined by the presence of weakness, functional impairment, or prominent atrophy. Fasciculations alone were not enough to classify as spread of symptoms, unless in the bulbar area (tongue). The revised ALS functional rating scale (ALSFRS-R) (Citation19) and King’s staging (Citation20) were used as quantitative measures of clinical status and disease progression.
Comorbidity
Several comorbid chronic conditions present at the time of the initial health care visit were reported based on clinical relevance. Some conditions were reported separately while others were grouped due to low prevalence or similar clinical characteristics. Patients were classified as suffering from multimorbidity if there were three or more documented conditions.
Specialist ALS clinic
This term refers to the ALS CRC at the Karolinska University Hospital, the only tertiary center for ALS in Stockholm.
Investigation cycle
An investigation cycle was defined as a contiguous and uninterrupted chain of related health care contacts, including referrals and follow-up visits. For instance, an investigation cycle might begin with a first visit to a PCP which refers the patient to a different medical specialty clinic, which in turn refers the patient to another clinic and so on. The investigation cycle was considered terminated if the patient was referred to a specialist ALS clinic, if an alternate diagnosis was made, or if the patient or the health care provider chose to end the investigation for any other reason.
Occupation
Current or pre-retirement. Categorized as service sector (e.g. store employees, nurses, and teachers), manual labor (e.g. factory workers and craftsmen), managerial positions, creative/artistic, white-collar non-qualified (e.g. administrators and accountants), and white-collar qualified (e.g. engineers and academics).
Clinical phenotype at onset
Based on the predominant presenting symptoms at the initial health care visit: LE, UE, bulbar, or respiratory onset.
Time of onset, patients’ and doctors’ delay
Time of onset was defined as the date of first symptoms of ALS (extremity, bulbar, or respiratory weakness), as reported by the patient. The total time to diagnosis was reported as three components: (1) the time from symptom onset to initial health care visit, (2) the time from initial health care visit to referral to a specialist ALS clinic, and (3) the time from referral to ALS diagnosis. The first component being the patients’ delay and the second and third comprising the doctors’ delay.
Progression rate
The progression rate was calculated as 48 minus ALSFRS-R score divided by the time interval between time of symptom onset and the time of ALSFRS-R measurement (months). Thus, a full score of 48 was used for the time of symptom onset.
Statistics
Decimals were limited to one throughout the paper except for mean progression rate. Categorical variables were summarized as proportions (percent) and bivariate analyses were performed using the χ2 test to assess for significance of differences between groups. Continuous variables except progression rate were reported as medians with the 25th–75th percentile range (IQR) due to skewed distribution. Progression rate was reported as mean with a confidence interval (CI) of 95%. The one-way ANOVA test was performed to assess for differences in age at presentation.
A generalized linear model (GLM) (negative binomial with log link to account for overdispersion) was used to calculate relative risk (RR) for longer time to diagnosis. Separate analyses were performed for time from onset to first health care visit (patients’ delay) and for time from first health care visit to ALS specialist referral (doctors’ delay). The models included putative predictors based on plausibility and included 322 patients as there were missing data (for occupation) for 31 patients.
In all tests the significance level was set to 95%. All statistical analyses were conducted in IBM SPSS Statistics version 28 (IBM SPSS Statistics, Armonk, NY).
Results
Cohort characteristics
A total of 353 ALS patients were grouped based on clinical phenotype at onset: LE (n = 117 [33.1%]), UE (n = 85 [24.1%]), bulbar (n = 136 [38.5%]), and respiratory (n = 15 [4.2%]) onset ().
Table 1 Patient characteristics by clinical phenotype at onset.
Missing data was <1% for all baseline variables except for current or pre-retirement occupation (8.8% missing). Median age at the initial health care visit was 68 years (IQR 59–73). Sex distribution was equal in total (50.1% male) but differed between subgroups: more male patients in the UE and respiratory onset groups and more female patients in the bulbar onset group. Most patients, 72.5%, were living in their own home together with a partner and 63.2% were retired. Cardiovascular conditions were relatively common, particularly hypertension (35.1%) and ischemic heart disease/heart failure (9.9%). Both spinal degenerative disease and osteoarthritis were more common in the LE onset group, compared with other patients. The respiratory onset group had the highest prevalence of multimorbidity: 66.7% versus 28.6% in all patients, although they were only slightly older than the other groups.
Time to diagnosis
The median time from onset to diagnosis was 12.9 (IQR 8.3–18.8) months in all patients and was comprised of two main components: the patient’s and doctors’ delay (). The median time from onset until the initial health care visit, the patients’ delay, was 4.2 (2.0–8.6) months. From the initial heath care visit until referral to a specialist ALS clinic, there was a median delay of 3.7 (1.0–8.0) months. With an additional three months before the diagnosis was made, a median time of 6.8 (3.6–12.4) months was observed from initial health care visit to diagnosis, the doctors’ delay. The diagnostic delay varied between groups (). It was longest for the LE onset group while relatively short for those presenting with bulbar and respiratory symptoms. The total median time from onset to diagnosis was 16.0 (9.4–27.5) months in LE, 12.9 (8.8–17.8) months in UE, 11.7 (7.4–16.0) months in bulbar, and 8.3 (4.7–15.6) months in respiratory onset.
Figure 1 Median delay times grouped by clinical phenotype at onset. Displayed as three components: from onset to initial health care visit (blue), from initial health care visit until referral to a specialist ALS clinic (yellow), and from initial health care visit to ALS diagnosis (green). The first component being the patients’ delay and the second and third comprising the doctors’ delay. Reported as median time in months with the 25th–75th percentile in parentheses.
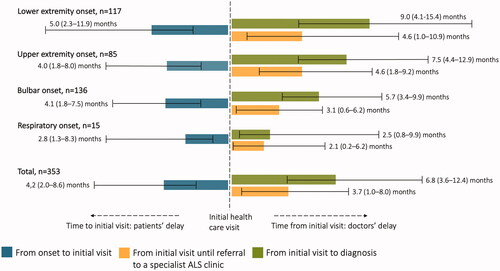
A multivariate analysis confirmed that UE and bulbar onset were associated with a shorter time to diagnosis: both for patients’ and doctors’ delay (). Additionally, respiratory onset was associated with a shorter patients’ delay, while male sex and higher Kings’ stage at the initial health care visit were associated with a shorter doctors’ delay. Other key variables such as age, occupation, living alone, family history of ALS, and multimorbidity showed no significant correlation to time to diagnosis.
Table 2 Predictors of longer time to diagnosis.
Referral patterns and alternate diagnoses
Sixty-four percent of all patients were referred to a specialist ALS clinic as a result of the first investigation cycle, 31.6% following a second cycle, 3.1% following a third cycle, and only one patient (with respiratory onset) had a fourth cycle (Supplementary Table 1). Differences between onset phenotype groups were minor.
specifically describes the first investigation cycle. In total, 74.5% of the initial health care visits took place at a PCPs’ office, 14.7% at the emergency department (ED), 2.8% at an orthopedics clinic, and 2.5% at an ear, nose, and throat (ENT) clinic. These proportions varied according to onset phenotype, e.g. those with LE or UE onset were more likely to first present at an orthopedics clinic (4.3% and 4.7%, respectively) while patients with bulbar of respiratory onset more often first presented at the ED (22.8% and 60.0%, respectively).
Figure 2 (A–D) Patient path during the first investigation cycle. Shown separately for each of the four onset phenotype groups. The yellow arrows indicate the proportion of patients where there was involvement of additional health care providers other than that of the first health care visit. Many patients were referred to more than one additional health care provider during the course of the investigation cycle and therefore the sum of percentages in the box might exceed 100%. A substantial proportion of patients were admitted to hospital for inpatient care as part of the investigation cycle. This proportionis included in the box showing additional health care providers. The proportion of patients referred directly or indirectly to a specialist ALS clinic is shown in green and the proportion receiving an alternate or no diagnosis is shown in red. Only additional health care providers and diagnoses >3% and n > 1 are shown, except in the respiratory onset group were all are shown due to low number of patients. COPD: chronic obstructive pulmonary disease; ED: emergency department; ENT: ear, nose, and throat; HCP: health care provider; PCP: primary care provider.
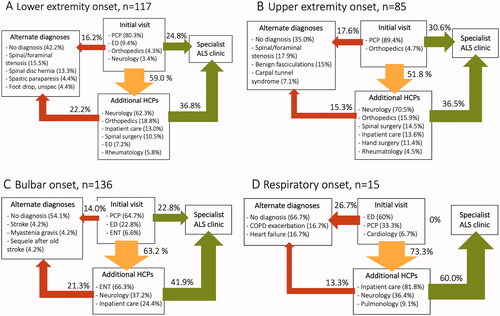
Only 24.4.% of all patients were directly referred to a specialist ALS clinic by the first health care provider whereas in 59.5%, other health care providers were involved. In patients with UE or LE onset this more commonly included an orthopedics or spinal/hand surgery clinic (29.3% for LE and 41.8% for UE), and for bulbar patients, ENT (66.3%). In all groups, a large proportion (52.4% in total) was referred to a neurologist other than a specialist ALS clinic during the investigation cycle. In a substantial proportion of cases, the patient was admitted to hospital for inpatient care as part of the investigation cycle: 13.0% for LE, 13.6% for UE, 24.4% for bulbar, and 81.8% for the respiratory onset group.
The proportion receiving an alternate or no diagnosis, effectively ending the first investigation cycle, was 36.0% in total (). For those with LE or UE onset, the most common alternate diagnosis was spinal/foraminal stenosis. For bulbar onset patients, the diagnoses myasthenia gravis and stroke were more common and for the respiratory group, cardiopulmonary diagnoses such as heart failure and chronic obstructive pulmonary disease (COPD) exacerbation predominated. In addition, there was a plethora of differentialdiagnoses considered during the investigation, such as stroke, brain tumor, peripheral neuropathy, and neuroborreliosis (Supplementary Table 2).
Clinical progression
Many patients experienced substantial clinical progression during the investigation (). King’s stage was estimated based on symptom spread at the initial health care visit and at diagnosis and was available for all patients. There was statistically significant clinical progression in all groups except in the respiratory onset group (all patients were in stage 4 per definition) (). The proportion in King’s stage 3 or 4 increased from 11.3% to 46.1% in all patients; 4.3% to 37.6% in LE onset; 1.2% to 31.8% in UE onset; 14.0% to 56.6% in bulbar onset and remained at 100% in the respiratory onset group.
Figure 3 (A–D) Symptom spread at the initial health care visit versus at diagnosis. Showing the percentage of patients with symptoms from a particular area (from top to bottom: speech, cognition, swallowing, breathing, right arm, left arm, right leg, left leg), separately for each of the four onset phenotype groups. Of note, the graphic presentation is a result of pooled data at group level and fails to illustrate the asymmetrical spread of ALS symptoms in individual patients.
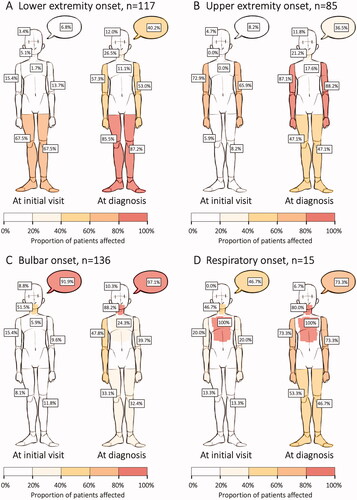
Table 3 King’s stage at the initial health care visit versus at diagnosis.
ALSFRS-R was available for 235 (66.6%) of the patients in conjunction with diagnosis (within two months) with a median (IQR) of 39 (34–42) for LE, 40 (36–44) for UE, 40 (32–43) for bulbar, and 31 (25–39) for respiratory onset. The mean (CI 95%) progression rate was estimated to 0.85 (0.63–1.07), 0.75 (0.59–0.91), 1.05 (0.82–1.27), and 1.65 (0.81–1.05) in these groups, respectively.
Discussion
The ALS CRC in Stockholm diagnoses about one hundred patients with ALS each year. A patient’s diagnosis marks the end of a long journey unique to each individual, mirroring the heterogeneity of the disease. The noted average time from onset to diagnosis is in line with previous reports: slightly longer than 1 year (Citation7,Citation9–12,Citation16), and we can show great variation in the path to diagnosis depending on the clinical phenotype at onset. For instance, patients with spinal onset (UE and LE) had longer delay times compared to bulbar and respiratory patients, consistent with previous research (Citation4,Citation7,Citation9). The reason for this discrepancy might be the result of a more flagrant presentation in bulbar and respiratory onset (difficulty speaking, swallowing, or breathing), prompting early health care contact and a thorough investigation to find the underlying cause. Also, for bulbar symptoms the differential is relatively limited. The respiratory onset group followed a different path compared to the other groups: most being admitted to hospital and many receiving pulmonary or cardiac diagnoses. By the time of presentation, many also displayed symptoms from other areas, most often bulbar weakness, which might partly explain the relatively short diagnostic delay. However, the number of patients was small (n = 15), limiting the reliability of these results.
Misdiagnosis was a major reason for diagnostic delay in all patients: 36.0% received an alternate or no diagnosis as a result of the first investigation cycle. Previous research report similar numbers: 27–44% of ALS patients misdiagnosed at some point during the investigation (Citation7–9).
Additional factors associated with a longer delay were lower Kings’ stage at presentation and female sex, while other factors such as age, occupation, living alone, family history of ALS, and multimorbidity were not.
Importantly, we show a substantial clinical progression from the initial health care visit to diagnosis, e.g., an increase from 11.3% to 46.1% of patients in King’s stage 3 or 4. These results are particularly concerning considering that King’s stage is strongly correlated with shorter survival. One study demonstrated a median survival of 28, 13, and 6–8 months at King’s stage 2, 3, and 4, respectively (Citation20). Thus, the time from initial health care visit until diagnosis is a critical period. Even though therapeutic options for ALS are limited, there are many benefits of shortening the diagnostic delay. The first is to provide answers in a timely manner to mitigate the uncertainty and anxiety of patients (Citation13). Second, admission to a multidisciplinary team with a special interest in ALS might increase survival and quality of life by working preventively, anticipating the progression and the difficulties arising (Citation21). Third, patients identified at an early stage have a better chance of being eligible for enrollment in clinical trials (Citation15). Also, diagnostic delay has been associated with increased health care costs (Citation14).
The time from initial health care visit to referral to a specialist ALS clinic is perhaps most amenable to improvement. This is particularly true for patients presenting with spinal symptoms (LE and UE) where the delay is longer. However, making the final diagnosis of ALS in the primary care setting is often impossible and should not be expected. Instead, the role of the physician initially seeing the patient should be to recognize red flags and refer the patient forward to a neurologist (Citation22). An open dialogue between PCPs and the neurological community needs to be encouraged in order to support decision-making and clinical reasoning. Further research exploring the interaction between primary care and the neurological community is warranted.
Strengths and weaknesses
This is, to our knowledge, the largest and most comprehensive analysis of the path of ALS patients through the health care system until diagnosis. The study has collected information through review of medical records from many health care providers. However, in around 15% of the patients, medical records from some part of the investigation were unavailable. Even though the information most often could be retrieved from secondary sources, some data had to be inferred. Also, the extent and quality of data entered in medical records by different providers varied.
ALS care in Stockholm is highly centralized and coordinated. However, the diagnostic process is likely different in other parts of Sweden, and in other countries, depending on local circumstances and resources. For instance, the Swedish health care system is largely publicly funded, universal for all citizens, and regionally administered. Comparative data from other regions and countries are needed for further verification and contextualization of our results. Specifically, research focused on the early investigation and care of ALS patients in regions of limited resources is lacking.
Conclusion
This is a comprehensive description of the investigation leading up to an ALS diagnosis in Stockholm patients. The initial investigation was most often conducted from primary care clinics and as a result of misdiagnosis and clinical uncertainty, diagnostic delay was prolonged, particularly in those presenting with upper or lower extremity weakness. During the investigation, there was substantial clinical progression in most patients.
Supplemental Material
Download MS Word (18.8 KB)Acknowledgments
The authors are grateful for the help of statistician Aniko Lovik at the Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden, in constructing the general linear model ().
Declaration of interest
The authors report that there are no competing interests to declare.
Data availability statement
The study is partly based on data from the Swedish MND Quality Registry. Having obtained the appropriate approval from a research ethics board, these data can be accessed from the registry office upon request.
Additional information
Funding
References
- Brown RH, Jr., Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602.
- Grad LI, Rouleau GA, Ravits J, Cashman NR. Clinical spectrum of amyotrophic lateral sclerosis (ALS). Cold Spring Harb Perspect Med. 2017;7:a024117.
- Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005;65:586–90.
- del Aguila MA, Longstreth WT, Jr., McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology 2003;60:813–9.
- Belsh JM. Diagnostic challenges in ALS. Neurology 1999;53:S26–S30.
- Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2013;41:118–30.
- Belsh JM, Schiffman PL. Misdiagnosis in patients with amyotrophic lateral sclerosis. Arch Intern Med. 1990;150:2301–5.
- Belsh JM, Schiffman PL. The amyotrophic lateral sclerosis (ALS) patient perspective on misdiagnosis and its repercussions. J Neurol Sci. 1996;139:110–6.
- Kraemer M, Buerger M, Berlit P. Diagnostic problems and delay of diagnosis in amyotrophic lateral sclerosis. Clin Neurol Neurosurg. 2010;112:103–5.
- Paganoni S, Macklin EA, Lee A, Murphy A, Chang J, Zipf A, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:453–6.
- Palese F, Sartori A, Logroscino G, Pisa FE. Predictors of diagnostic delay in amyotrophic lateral sclerosis: a cohort study based on administrative and electronic medical records data. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:176–85.
- Nzwalo H, de Abreu D, Swash M, Pinto S, de Carvalho M. Delayed diagnosis in ALS: the problem continues. J Neurol Sci. 2014;343:173–5.
- O’Brien MR, Whitehead B, Jack BA, Mitchell JD. From symptom onset to a diagnosis of amyotrophic lateral sclerosis/motor neuron disease (ALS/MND): experiences of people with ALS/MND and family carers - a qualitative study. Amyotroph Lateral Scler. 2011;12:97–104.
- Galvin M, Ryan P, Maguire S, Heverin M, Madden C, Vajda A, et al. The path to specialist multidisciplinary care in amyotrophic lateral sclerosis: a population- based study of consultations, interventions and costs. PLoS One. 2017;12:e0179796.
- van Eijk RPA, Westeneng H-J, Nikolakopoulos S, Verhagen IE, van Es MA, Eijkemans MJC, et al. Refining eligibility criteria for amyotrophic lateral sclerosis clinical trials. Neurology 2019;92:e451–60.
- Galvin M, Madden C, Maguire S, Heverin M, Vajda A, Staines A, et al. Patient journey to a specialist amyotrophic lateral sclerosis multidisciplinary clinic: an exploratory study. BMC Health Serv Res. 2015;15:571.
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron DiseasesWorld Federation of Neurology Research Group on Motor Neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Longinetti E, Regodón Wallin A, Samuelsson K, Press R, Zachau A, Ronnevi L-O, et al. The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:528–37.
- Cedarbaum JM, Stambler N. Performance of the amyotrophic lateral sclerosis functional rating scale (ALSFRS) in multicenter clinical trials. J Neurol Sci 1997;152:S1–S9.
- Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135:847–52.
- Hogden A, Foley G, Henderson RD, James N, Aoun SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. J Multidiscip Healthc. 2017;10:205–15.
- Baxter S, McDermott CJ. Decision-making and referral processes for patients with motor neurone disease: a qualitative study of GP experiences and evaluation of a new decision-support tool. BMC Health Serv Res. 2017;17:339.