
Abstract
Background: Amyotrophic lateral sclerosis (ALS) shows considerable clinical heterogeneity, which affects clinical trials. A clinical staging system has been proposed for ALS with potential applications in patient care, research, trial design and health economic analyses. The King’s system consists of five stages. We have previously shown that progressive clinical stages were reached at predictable proportions through the disease course, but this needs to be validated in other independent samples. Objectives: We aimed to compare King’s clinical staging in ALS in four patient groups, located in different regions and countries and using different health care systems from the original study population in South London. Methods: Clinical data were extracted from two European phase 3 randomized controlled trials (MitoTarget and LiCALS) and from two databases predominately from the United States: the PRO-ACT Consortium Database and a database of patients from the PatientsLikeMe website. Clinical stage was estimated using an algorithm, and standardized time to each clinical stage was calculated in deceased patients. Results: 8,796 patients were included, of whom 1,959 had died by the end of follow-up. Stages occurred in the same order as in the original study for all cohorts. Median standardized times to stages (interquartile range) were Stage 2: 0.61 (0.47–0.75), Stage 3: 0.68 (0.56–0.81), Stage 4A: 0.82 (0.71–0.91), Stage 4B: 0.82 (0.69–0.92) and Stage 4 0.80 (0.67–0.91). Discussion: Timings for all stages were similar to those reported in the original study, except Stage 2 which occurred later in the clinical trial databases due to recruitment occurring after diagnosis.
Introduction
On average, 50% of people with amyotrophic lateral sclerosis (ALS) die within 30 months of symptom onset, but 15–20% are alive at 5 years (Citation1). These survival differences make clinical trial design difficult and need to be taken into account when planning services and interventions. As a potential solution to these problems, we previously proposed a staging system for ALS consisting of five clinical milestones occurring in a specified order and at predictable points through the disease course. The system has been partially validated in a UK ALS population of 1459 patients and staging has been used in multiple other studies (Citation2–12). Stage 4A represents nutritional failure, defined by the requirement for gastrostomy, and Stage 4B represents respiratory failure, defined by the requirement for noninvasive ventilation (NIV), and these two stages have now been combined into a single Stage 4 (Citation13). Each milestone is reached at a standardized time, which is the proportion elapsed through the disease course. This staging system can be used as a clinical trial endpoint, and retrospective analysis of Riluzole and Edaravone clinical trial data demonstrates the specific stages which are prolonged with these drugs, and shows an increase in the number of events reached during the trial compared with conventional analyses, improving statistical power (Citation14,Citation15).
We aimed to investigate use of the King’s staging system in four new patient groups. The Lithium Carbonate in ALS (LiCALS) and the MitoTarget clinical trials together consist of 725 patients, from 23 ALS centers from five European countries, providing useful cohorts in which to validate the staging system (Citation16–18). PatientsLikeMe is an online global community of people living with ALS, the majority from the United States. People with ALS using the website are a useful cohort to study, as they are unlike the original study population in being self-selected and using different health care systems, yet likely represent a significantly biased group compared with an unselected population. The Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) Consortium Database contains the largest ALS clinical trial dataset, with records from 18 clinical trials (Citation19). Together these four international databases consist of 8,796 patients. We aimed to assess whether staging milestones occur in the same order as the previous study in these databases (Citation3).
Materials and methods
Data sources
Databases of ALS patients who had participated in the MitoTarget clinical trial (Eudract number: HEALTH F2-2008-223388), a double-blind randomized placebo-controlled parallel group trial of olesoxime in ALS (Citation18), and the LiCALS clinical trial (Eudract number: 2008-006891-31), a double-blind randomized placebo-controlled parallel group trial of lithium carbonate in ALS (Citation16,Citation17), were analyzed. A database of people living with ALS from PatientsLikeMe, an online patient community, was analyzed. Quality control was performed on the PatientsLikeMe database so that only patients with adult-onset ALS were included and any people with invalid data (age of onset of disease in negative years, diagnosis date before or on date of symptom onset, ALSFRS-R report before date of symptom onset or after day of death) were removed from subsequent analysis. In addition, data used in the preparation of this article were obtained from the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) Database. In 2011, Prize4Life, in collaboration with the Northeast ALS Consortium, and with funding from the ALS Therapy Alliance, formed the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) Consortium. The data available in the PRO-ACT Database has been volunteered by PRO-ACT Consortium members. The data from the original published study on ALS staging, comprising patients seen at a tertiary UK ALS referral center, was also used for comparisons to be made to the other databases (Citation3).
Standard protocol approvals, registrations, and patient consents
The LiCALS study was ethically approved by the South East Research Ethics Committee in the UK, with the reference number 09/H1102/15 (Eudract number: 2008-006891-31). The MitoTarget study was ethically approved by the Comité de Protection des Personnes Ile de France VI—GH Pitié Salpétrière with the reference number 122-08 (Eudract 2008-007320-25). All patient identifying and experimental arm data from the five datasets were anonymised. This study was classified as a secondary analysis of fully anonymised preexisting clinical trial data by the King’s College London Psychiatry, Nursing and Midwifery Research and Ethics Subcommittee and therefore did not require ethical approval.
Patients were classified as having limb, bulbar or respiratory onset ALS. For the purposes of calculating standardized times to clinical milestones and analyzing survival, those with respiratory onset were classified with those with limb onset due to the common spinal basis of lower motor neuron degeneration.
In line with previous studies (Citation3), milestones were defined as Stage 1, symptom onset i.e. clinical involvement of the first CNS region, Stage 2, clinical involvement of a second CNS region and Stage 3, clinical involvement of a third CNS region. We also calculated milestones for Stage 4A, the need for gastrostomy and Stage 4B, the need for noninvasive ventilation, and Stage 4, representing the earliest milestone reached of Stage 4A and 4B.
For the MitoTarget trial, visits occurred at recruitment, at 1, 2 and 3 months after recruitment and then at 3-monthly intervals until exit from the trial through death or withdrawal. For the LiCALS trial, visits occurred at recruitment, then at 3-monthly intervals until exit from the trial through death or withdrawal. The Revised ALS Functional Rating Scale (ALSFRS-R) scores were recorded at each trial visit. On the PatientsLikeMe website patients or their caregivers enter data onto their online profile as frequently as they wish. The data inputted includes self-assessed ALSFRS-R scores. In the PRO-ACT database ALSFRS or ALSFRS-R scores were recorded during the course of the trials. As there had been no prospective staging at each clinical trial visit or during data collection on the PatientsLikeMe website, an algorithm based on ALSFRS-R score breakdown was used to retrospectively estimate disease staging at each trial visit and at each point of data entry onto the PatientsLikeMe website for every individual (Citation20). For the LiCALS and MitoTarget clinical trial data, actual dates of gastrostomy insertion and of commencement of noninvasive ventilation were available, and these were used as a proxy for timing of Stage 4 in trial patients.
Milestone timings were standardized as proportions of time elapsed though the disease course using information from patients who had died, by dividing time to a milestone by disease duration. Therefore, the time to each milestone was a value between 0 and 1, with 0 being symptom onset and 1 being death. All recorded milestones were used in the analysis.
We compared standardized times to each milestone calculated from the LiCALS, MitoTarget, PatientsLikeMe and PRO-ACT databases to each other and to standardized times calculated from the original King’s College Hospital database (Citation3).
Statistical analysis
Data that were not parametrically distributed were transformed and if transformation did not result in normality, non-parametric statistical tests were used. Baseline characteristics of the databases were compared using one-way ANOVA with post-hoc Games-Howell tests, Pearson’s Chi squared and Student’s t-test. Standardized times were expressed as medians with interquartile ranges. Kruskal-Wallis ANOVA was used to compare standardized times between databases and a Mann-Whitney U test was used to compare standardized times between limb and bulbar onset patients.
Analyses were performed in SPSS v27 (SPSS Inc, Illinois).
Results
Patient characteristics
There were 511 patients in the MitoTarget trial database, 214 patients in the LiCALS trial database, 3,620 patients in the PatientsLikeMe database and 4,841 patients with ALSFRS(-R) scores in the PRO-ACT database. Since the trial data had been through rigorous quality control but the PatientsLikeMe data had not, we applied exclusion criteria to ensure standardized data (Consort diagram in ).
Figure 1 Participant flow diagram for the PatientsLikeMe database.
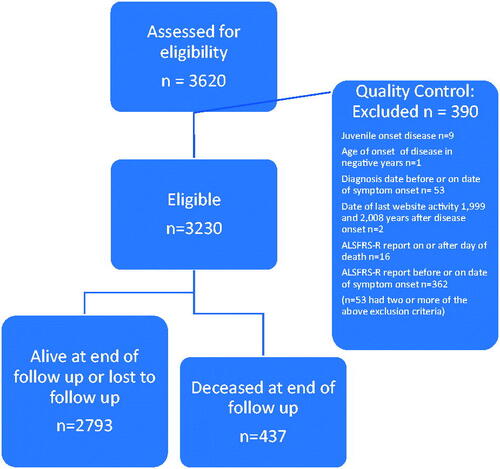
After exclusions and quality control there were 3230 patients from the PatientsLikeMe database included in the subsequent analysis (89.2% of the initial PatientsLikeMe cohort). In total there were 8,796 patients included in the analysis (95.8% of initial cohort).
Baseline characteristics between the four databases of ALS patients were different (). There were statistically significant differences between groups by one-way ANOVA for age of onset (Welch F(3,791)=132.6, p = 1.3 × 10−69), duration from disease onset to diagnosis (Welch F(3,938)=80.8, p = 1.7 × 10−46) and duration from disease onset to the end of follow up (Welch F(3,895)=228.0, p < 7.1 × 10−110). Age of onset was lower in the PatientsLikeMe database compared to the LiCALS (p = 3.6 × 10−13), MitoTarget (p = 2.8 × 10−13) and PRO-ACT (p = 2.6 × 10−12) databases. Age of onset was higher in the LiCALS compared to the MitoTarget (p = 0.036) and PRO-ACT (p = 0.03) databases. Diagnostic delay was longer in the PatientsLikeMe database compared to the MitoTarget (p < 1 × 10−36), LiCALS (p = 1.7 × 10−13) and PRO-ACT (p < 1 × 10−36) databases. Diagnostic delay was longer in the PRO-ACT database compared to the MitoTarget database (p = 1.3 × 10−7). Duration to end of follow up was longer in the PatientsLikeMe database compared to the MitoTarget (p = 9.9 × 10−13), PRO-ACT (p = 2.4 × 10−13) and LiCALS (p = 6.7 × 10−13) databases. Duration to end of follow up was longer in the PRO-ACT database compared to the MitoTarget database (p = 0.002). There was a larger proportion of male patients in the LiCALS compared to the PatientsLikeMe database (Pearson χ2(3) = 13.1, p = 0.045). There were no differences in the proportions of limb and bulbar onset patients between the four databases (Pearson χ2(3) = 2.6, p = 0.45). There was a larger proportion of deceased patients in the LiCALS compared to the PatientsLikeMe database (Pearson χ2(3) = 282.8, p = 5.2 × 10−61).
Table 1 Baseline characteristics King’s College Hospital (KCH), LiCALS and MitoTarget, PatientsLikeMe and PRO-ACT databases with values expressed as Mean (95% Confidence Interval) or Number (% of total).
Standardized times to each milestone
By the end of follow up, 101 patients from the LiCALS database, 159 patients from the MitoTarget database, 437 patients from the PatientsLikeMe database and 1262 from the PRO-ACT database had died. Patients who were still alive at the end of follow up could not have standardized times to milestones calculated, because we were unable to standardize stages to time of death.
Mean age of onset was significantly older in patients who had died (58.1 years, 95% CI 57.6–58.6) compared to those with no deceased date at the end of follow up (52.4 years, 95% CI 52.1–52.6, p = 1.8 × 10−88). Diagnostic delay was significantly shorter in patients who had died (11.8 months, 95% CI 11.1–12.5) compared to those with no deceased date at the end of follow up (14.8 months, 95% CI 14.3–15.3, p = 9.6 × 10−11).
Duration and standardized times from onset to every clinical milestone are shown in . Standardized milestone timings occurred at progressive proportions through the disease course in all databases. The stages occurred in the same order as in the previous study in all databases. Median standardized times (interquartile range) for all 3,026 deceased patients from the five databases (King’s College Hospital (KCH), PatientsLikeMe, LiCALS, MitoTarget and PRO-ACT) were: Stage 2 0.50 (0.28–0.68), Stage 3 0.66 (0.51–0.81), Stage 4A 0.81 (0.69–0.91), Stage 4B 0.82 (0.67–0.92) and Stage 4 0.80 (0.67–0.91) (, ). In addition, standardized times excluding the original KCH database are presented (, ). Need for gastrostomy (Stage 4A) and need for NIV (Stage 4B) occurred at similar standardized times through the disease.
Figure 2 Box plot of standardized times to each stage in all 3,026 deceased patients from the five databases (KCH, PatientsLikeMe, LiCALS, MitoTarget and PRO-ACT) using every milestone reached.
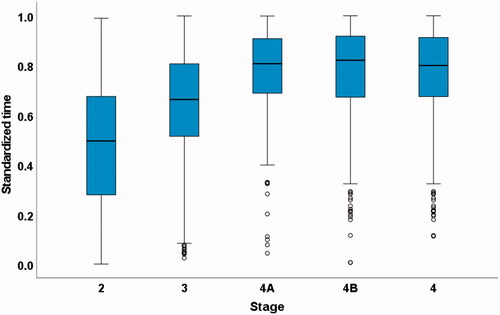
Figure 3 Box plot of standardized times to each stage in the 1,959 deceased patients from PatientsLikeMe, LiCALS, MitoTarget and PRO-ACT databases using every milestone reached.
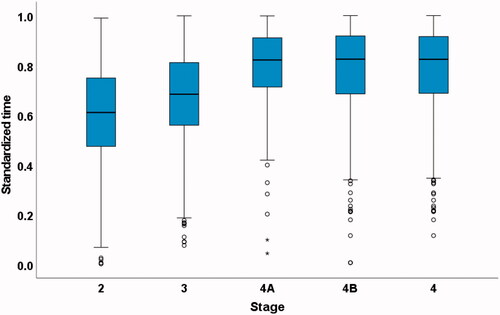
Table 2 Standardized times to every milestone reached and milestone timings for King’s College Hospital (KCH), PatientsLikeMe, LiCALS, MitoTarget and PRO-ACT databases.
Standardized times to Stages 2, 3, 4A, 4B and 4 were significantly different between databases. Standardized times to Stage 2 were no different between the LiCALS, MitoTarget and PRO-ACT databases but for all these trial databases Stage 2 occurred later than the KCH database (p < 0.001) and PatientsLikeMe database (p < 0.001). Stage 2 occurred earlier in the KCH compared to the PatientsLikeMe database (p = 0.014). Standardized times to Stage 3 were no different between the LiCALS, MitoTarget and PRO-ACT databases. Stage 3 was later in the PRO-ACT database compared to both the PatientsLikeMe database (p < 0.001) and the KCH database (p < 0.001) and later in the LiCALS database compared to the PatientsLikeMe database (p = 0.028) but between the other groups were no different. Stage 4A was later in the PRO-ACT database compared to the PatientsLikeMe database (p = 0.004) and the KCH database (p < 0.001). Standardized time to Stage 4B was earlier in the PatientsLikeMe database compared to the KCH database (p = 0.026), the LiCALS database (p < 0.001), the MitoTarget database (p < 0.001) and the PRO-ACT database (p < 0.001). Stage 4B was earlier in the KCH database compared to the PRO-ACT database (p < 0.001) and the LiCALS database (p = 0.047). Stage 4 was earlier in the PatientsLikeMe database compared to the LiCALS database (p = 0.035), MitoTarget database (p = 0.005) and PRO-ACT database (p < 0.001). Stage 4 was earlier in the KCH database compared to the PRO-ACT database (p < 0.001).
We examined whether the timing of milestones was affected by site of disease onset in the PatientsLikeMe, LiCALS, MitoTarget and PRO-ACT databases (). As in the previous study (Citation3), we found that noninvasive ventilation was usually needed before gastrostomy in patients with limb onset ALS but after gastrostomy in those with bulbar onset ALS. In bulbar onset, Stage 3 (p < 0.001), Stage 4A (p < 0.001) and Stage 4 (p < 0.01) were reached earlier compared to limb onset disease. There were no significant differences between standardized times to the other milestones between limb versus bulbar onset disease.
Table 3 Standardized times in PatientsLikeMe, LiCALS, MitoTarget and PRO-ACT databases in limb onset versus bulbar onset patients.
Discussion
This ALS staging system had been partially validated in a population of ALS patients from King’s College Hospital, and we have now found that the staging milestones occur in the same order in four separate international populations of patients. The approximate proportions through the disease course at each Stage in these four databases are Stage 2: 60%, Stage 3: 70% and Stage 4: 80% which are similar to the original study, but they do differ between datasets (Citation3).
The baseline characteristics of each population are not identical. In the PatientsLikeMe database, age of disease onset was younger than for the other cohorts, likely reflecting the online nature of the platform. Age of onset in this database also occurs earlier than typically in ALS populations worldwide (Citation21–23) including in population-based studies in the US (Citation24,Citation25). The PatientsLikeMe database had a longer diagnostic delay than the other databases, which is known to associate with slower disease progression. In addition, follow-up data were available for a longer duration than the two trial databases, which is likely to be due to clinical trials having a defined trial duration after which patients are no longer followed up, whereas patients can continue to enter their data onto the PatientsLikeMe website for as long as they wish. Despite the longer duration of follow up in the PatientsLikeMe database, there was a lower proportion of deceased patients than the other databases. As these patient data are collected online with patients and their caregivers inputting their own data, it is likely that not all patients who have died are captured in this database, therefore this proportion reflects only the people who have deceased dates available.
People with ALS with a younger age of onset tend to be those with limb onset disease, while those with an older age of onset tend to have bulbar onset disease (Citation26–28). Although the proportions of limb and bulbar onset patients were not different between the databases, in the PatientsLikeMe database, site of disease onset was unknown for almost a quarter of patients. Therefore, we cannot be certain if the proportions in the patients with unknown onset reflected those of the rest of the database, and with a younger age of onset we might expect a higher proportion of limb-onset disease. We have shown Stage 4A occurs earlier in patients with bulbar onset than with limb onset disease. If indeed the PatientsLikeMe cohort had a greater proportion of bulbar onset disease with the patients with unknown onset included, then this may partly explain the reason for the standardized time to Stage 4A occurring earlier in the PatientsLikeMe database compared to the PRO-ACT database. To test this further we examined median standardized times to Stage 4A and 4B in the limb and bulbar onset patients in the PatientsLikeMe database but we found no difference between the groups.
Stage 2 and Stage 3 occurred later in both clinical trial databases compared to the KCH and PatientsLikeMe databases. The clinical trial databases only capture data from time of trial recruitment, not from disease onset, and also for a limited window for the duration of the trial. Since people can only be recruited after diagnosis, which in our previous work corresponded closely with Stage 2 times (Citation3), we would expect that this retrospective allocation to clinical stage would result in an apparent delay to Stage 2 since it is artificially left-censored at trial recruitment. The stage in this case is calculated retrospectively from ALSFRS-R data captured at recruitment, so the date of recruitment becomes the date of Stage 2 or the date of Stage 3, if a patient is recruited further through their disease progression, and the resulting timing is therefore shifted later. Future work assessing patients’ progression through the clinical milestones prospectively would help to overcome this bias.
We also compared standardized times to each milestone in patients with bulbar and limb onset disease, showing that Stage 3, Stage 4A and Stage 4 are reached earlier in bulbar onset disease. This reflects the fact that patients with bulbar onset progress more quickly than those with limb onset and that patients with bulbar onset disease tend to require gastrostomy earlier due to progressive dysphagia (Citation1,Citation29–33).
Limitations
A limitation of this study is that staging was calculated retrospectively using the ALSFRS-R, from clinical trial visits or at variable intervals according to when reports were entered onto the PatientsLikeMe website. The use of this retrospective ALSFRS-R data can lead to biases. Ideally staging information would be collected prospectively. However, the breakdown of the ALSFRS-R domain scores provides a sensitive tool for detecting functional involvement of CNS regions and we have shown that the algorithm used to convert ALSFRS-R to stage is strongly correlated with actual clinical stage (Citation20). However, ALSFRS-R detects only whether a patient has a gastrostomy or is using NIV, not whether there was in fact a need for these interventions determined by the multidisciplinary team. Therefore, there may be additional patients who have reached Stage 4 who are not accounted for in the data. This may be somewhat balanced by those patients who are recommended to consider gastrostomy or use NIV and who decline. Furthermore, it is likely that the need for NIV or gastrostomy occurs much earlier than when the intervention commences, as detected by the ALSFRS-R. Therefore, Stage 4 is likely to occur earlier than detected in these datasets. It is also important to note that the ALSFRS-R detects a functional deficit, whereas ALS staging assesses the involvement of a bulbar/spinal region in the disease; the presence of signs of motor neuron damage in a bulbar/spinal region will not necessarily lead to a functional deficit in that region, therefore ALSFRS-R will be less sensitive in detecting involvement of a region than prospectively assessing the disease stage.
The PatientsLikeMe database is an internet-based database, therefore this data may be compromised by inaccurate or false information being supplied. It is difficult to assess whether data are incorrect; however, we employed stringent quality control methods to maximize the validity of the cases included in the subsequent analysis. In addition ALSFRS-R in this database is self-administered by the patient, but self-administration of ALSFRS-R has been shown to have good reliability compared to standard administration of this tool which was also true over a three month follow-up time period (Citation34,Citation35). Furthermore, it has been shown that online self-administration or online caregiver administration of the ALSFRS-R has good reliability compared to administration by a health professional in clinic (Citation36).
Conclusions
Despite these limitations and the heterogenous baseline characteristics of each database, we have shown that the staging milestones occur in exactly the same order in the PatientsLikeMe database and the clinical trial databases compared to the original study, and that the standardized times to each milestone are similar in these databases. Stage 2 occurred later than the original study, which is likely to be due to the bias of Stage 2 appearing artefactually later in clinical trial data.
We also observed that Stage 4A and Stage 4B occur at very similar standardized times through the disease course in these groups of patients and in the KCH cohort. In the UK, need for noninvasive ventilation is defined by National Institute for Clinical Excellence (NICE) guidelines (Citation37) and there is evidence that patients are increasingly being referred for NIV by UK neurologists (Citation38). Worldwide, however, there is a wide variation across treatment centers as to when NIV is administered (Citation39–43). The earlier timing of Stage 4B in the PatientsLikeMe database may also reflect earlier use of noninvasive ventilation in the US compared to in Europe. There is also some variability as to whether gastrostomy insertion occurs as part of patient care (Citation42,Citation44). In an Italian study, frequency of gastrostomy in a population with ALS depended upon whether a patient had attended an ALS center or not (Citation45). Exactly which parameters to use when starting NIV are not clearly defined worldwide (Citation46–48) and in the UK, neurologists frequently do not assess parameters of respiratory muscle function regularly (Citation38). The need for gastrostomy at Stage 4A reflects progression of bulbar symptoms to the extent that alternative feeding is required (Citation49–51). The need for noninvasive ventilation at Stage 4B reflects respiratory insufficiency, predominantly caused by involvement of the cervical and thoracic lower motor neurons supplying the diaphragm and intercostal muscles, leading to impaired respiratory function (Citation52). A standard operating procedure for the application of the King’s staging system defines how to apply Stages 4A and 4B and furthermore combines Stage 4A and 4B (Citation13). Stage 4 is reached if there is evidence of feeding failure or respiratory failure secondary to ALS. The use of the standard operating procedure enables reliable and simple calculation of staging, so that stages can be determined objectively by clinicians and researchers universally. A further important use of a clinical staging system is in health economics analysis for assessing utility and socioeconomic costs in ALS (Citation53–55).
We have shown that King’s ALS stages occur in the same order as the original study population in international samples of ALS patients enrolled in clinical trials. Staging has benefits for clinical work and in resource allocation, and can now be applied more widely toward this remit. Patients at stage 1 need diagnosis, those at Stage 2 need therapy to maintain functional ability, and by stages 3 and 4 gastrostomy or ventilatory support may be required (Citation56).
Acknowledgments
We thank all the participants involved in the study.
Declaration of interest
AAC reports consultancies or advisory boards for Amylyx, Apellis, Biogen, Brainstorm, Cytokinetics, GenieUs, GSK, Lilly, Mitsubishi Tanabe Pharma, Novartis, OrionPharma, Quralis, and Wave Pharmaceuticals. At the time of study, PW was an employee of PatientsLikeMe. PW is employed by Wicks Digital Health Ltd, which has received funding from Ada Health, AstraZeneca, Baillie Gifford, Bold Health, Camoni, Compass Pathways, Coronna, EIT, Happify, HealthUnlocked, Inbeeo, Kheiron Medical, Sano Genetics, Self Care Catalysts, The Learning Corp, The Wellcome Trust, VeraSci, and Woebot. CAY is a Principal Investigator for Cytokinetics and receives speaker and consultancy fees from Biogen, Celgene, Cytokinetics, Genzyme, Janssen, Merck, Novartis, Orion, Roche and Teva. PJS is a consultant/Advisory Board member for Biogen, Benevolent AI, Quell Therapeutics, Aclipse Therapeutics and QurAlis.
Additional information
Funding
References
- Talbot K. Motor neuron disease: the bare essentials. Pract Neurol. 2009;9:303–9.
- Canosa A, Calvo A, Moglia C, Manera U, Vasta R, Di Pede F, et al. Brain metabolic changes across King's stages in amyotrophic lateral sclerosis: a (18)F-2-fluoro-2-deoxy-D-glucose-positron emission tomography study. Eur J Nucl Med Mol Imaging. 2021;48:1124–33.
- Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135:847–52.
- Balendra R, Jones A, Jivraj N, Steen IN, Young CA, Shaw J, et al. Use of clinical staging in amyotrophic lateral sclerosis for phase 3 clinical trials. J Neurol Neurosurg Psychiatry 2014;86:45–49.
- Consonni M, Dalla Bella E, Contarino VE, Bersano E, Lauria G. Cortical thinning trajectories across disease stages and cognitive impairment in amyotrophic lateral sclerosis. Cortex. 2020;131:284–94.
- Sugimoto K, Han Y, Song Y, Gao Y. Correlational analysis of als progression and serum nfl measured by simoa assay in chinese patients. Front Neurol 2020;11:579094.
- Abdul Aziz NA, Toh T-H, Loh E-C, Capelle DP, Goh K-J, Abdul Latif L, et al. The utility of ALS staging systems in a multi-ethnic patient cohort. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22:341–9.
- Sugimoto K, Mori M, Liu J, Shibuya K, Isose S, Koide M, et al. Novel serum autoantibodies against ss-actin (ACTB) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener.2021;22:388–94.
- Luna J, Couratier P, Lahmadi S, Lautrette G, Fontana A, Tortelli R, et al. Comparison of the ability of the King's and MiToS staging systems to predict disease progression and survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22:478–85.
- Wei Q-Q, Hou Y, Chen Y, Ou R, Cao B, Zhang L, et al. Health-related quality of life in amyotrophic lateral sclerosis using EQ-5D-5L. Health Qual Life Outcomes. 2021;19:181.
- Devenney EM, McErlean K, Tse NY, Caga J, Dharmadasa T, Huynh W, et al. Factors that influence non-motor impairment across the ALS-FTD spectrum: impact of phenotype, sex, age, onset and disease stage. Front Neurol 2021;12:743688.
- Floeter MK, Danielian LE, Braun LE, Wu T. Longitudinal diffusion imaging across the C9orf72 clinical spectrum. J Neurol Neurosurg Psychiatry. 2018;89:53–60.
- Balendra R, Al Khleifat A, Fang T, Al-Chalabi A. A standard operating procedure for King’s ALS clinical staging. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:159–64.
- Fang T, Al Khleifat A, Meurgey J-H, Jones A, Leigh PN, Bensimon G, et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018;17:416–22.
- Al-Chalabi A, Chiò A, Merrill C, Oster G, Bornheimer R, Agnese W, et al. Clinical staging in amyotrophic lateral sclerosis: analysis of Edaravone Study 19. J Neurol Neurosurg Psychiatry. 2021;92:165–71.
- UKMND-LiCALS Study Group, Morrison KE, Dhariwal S, Hornabrook R, Savage L, Burn DJ, et al. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013;12:339–45.
- Al-Chalabi A, Shaw PJ, Young CA, Morrison KE, Murphy C, Thornhill M, et al. Protocol for a double-blind randomised placebo-controlled trial of lithium carbonate in patients with amyotrophic lateral sclerosis (LiCALS) [Eudract number: 2008-006891-31]. BMC Neurol. 2011;11:111.
- Phase A. 2–3 trial of olesoxime in subjects with amyotrophic lateral sclerosis, 23rd International Symposium on ALS/MND Abstracts. Amyotrophic Lateral Sclerosis 2012;13:1–58.
- Gomeni R, Fava M. Amyotrophic lateral sclerosis disease progression model. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:119–29.
- Balendra R, Jones A, Jivraj N, Knights C, Ellis CM, Burman R, et al. Estimating clinical stage of amyotrophic lateral sclerosis from the ALS Functional Rating Scale. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:279–84.
- Gordon PH, Salachas F, Lacomblez L, Forestier NL, Pradat PF, Bruneteau G, et al. Predicting survival of patients with amyotrophic lateral sclerosis at presentation: a 15-year experience. Neuro-Degenerative Diseases 2012;12:81–90.
- Bae JS, Hong Y-H, Baek W, Sohn EH, Cho J-Y, Kim B-J, et al. Current status of the diagnosis and management of amyotrophic lateral sclerosis in Korea: a multi-center cross-sectional study. J Clin Neurol. 2012;8:293–300.
- Pradas J, Puig T, Rojas-García R, Viguera ML, Gich I, Logroscino G, et al. Amyotrophic lateral sclerosis in Catalonia: a population based study. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:278–83.
- Khishchenko N, Allen KD, Coffman CJ, Kasarskis EJ, Lindquist JH, Morgenlander JC, et al. Time to diagnosis in the National Registry of Veterans with Amyotrophic Lateral Sclerosis. Amyotroph Lateral Scler. 2010;11:125–32.
- Sorenson EJ, Stalker AP, Kurland LT, Windebank AJ. Amyotrophic lateral sclerosis in Olmsted County, Minnesota, 1925 to 1998. Neurology 2002;59:280–2.
- Bandettini di Poggio M, Sormani MP, Truffelli R, Mandich P, Origone P, Verdiani S, et al. Clinical epidemiology of ALS in Liguria, Italy. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:52–7.
- Forbes RB, Colville S, Swingler RJ. The epidemiology of amyotrophic lateral sclerosis (ALS/MND) in people aged 80 or over. Age Ageing. 2004;33:131–4.
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:639–49.
- Ganesalingam J, Stahl D, Wijesekera L, Galtrey C, Shaw CE, Leigh PN, et al. Latent cluster analysis of ALS phenotypes identifies prognostically differing groups. PloS One. 2009;4:e7107.
- Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10:310–23.
- Preux PM, Couratier P, Boutros-Toni F, Salle JY, Tabaraud F, Bernet-Bernady P, et al. Survival prediction in sporadic amyotrophic lateral sclerosis. Age and clinical form at onset are independent risk factors. Neuroepidemiology 1996;15:153–60.
- Turner MR, Bakker M, Sham P, Shaw CE, Leigh PN, Al-Chalabi A, et al. Prognostic modelling of therapeutic interventions in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology. Research Group on Motor Neuron Diseases.2002;3:15–21.
- Higo R, Tayama N, Nito T. Longitudinal analysis of progression of dysphagia in amyotrophic lateral sclerosis. Auris Nasus Larynx. 2004;31:247–54. doi:
- Montes J, Levy G, Albert S, Kaufmann P, Buchsbaum R, Gordon PH, et al. Development and evaluation of a self-administered version of the ALSFRS-R. Neurology 2006;67:1294–6. doi:
- Montes J, Levy G, Richard B, Kaufmann P, Buchsbaum R, Gordon PH, et al. Development and evaluation of the ALSFRS-R as a self-administered tool in patients with ALS. Neurology 2006;66:A246–A46.
- Maier A, Holm T, Wicks P, Steinfurth L, Linke P, Münch C, et al. Online assessment of ALS functional rating scale compares well to in-clinic evaluation: a prospective trial. Amyotrophic Lateral Sclerosis.2012;13:210–6. doi:
- National Institute for Health and Clinical Excellence. Motor neurone disease: the use of non-invasive ventilation in the management of motor neurone disease. London: National Institute for Health and Clinical Excellence. 2010. Available at: www.nice.org.uk/guidance/NG42
- O’Neill CL, Williams TL, Peel ET, McDermott CJ, Shaw PJ, Gibson GJ, et al. Non-invasive ventilation in motor neuron disease: an update of current UK practice. J Neurol Neurosurg Psychiatry. 2012;83:371–6. doi:
- Albert SM, Whitaker A, Rabkin JG, del Bene M, Tider T, O’Sullivan I, et al. Medical and supportive care among people with ALS in the months before death or tracheostomy. J Pain Symptom Manag.2009;38:546–53. doi:
- Borasio CD, Gelinas DF, Yanagisawa N. Mechanical ventilation in amyotrophic lateral sclerosis: a cross-cultural perspective. J Neurol. 1998;245:S7–S12.
- Melo J, Homma A, Iturriaga E, Frierson L, Amato A, Anzueto A, et al. Pulmonary evaluation and prevalence of non-invasive ventilation in patients with amyotrophic lateral sclerosis: a multicenter survey and proposal of a pulmonary protocol. J Neurol Sci. 1999;169:114–7. doi:
- Borasio GD, Shaw PJ, Hardiman O, Ludolph AC, Sales Luis ML, Silani V, et al. Standards of palliative care for patients with amyotrophic lateral sclerosis: results of a European survey. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:159–64. doi:
- Cedarbaum JM, Stambler N, Grp BS. Disease status and use of ventilatory support by ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:19–22.
- Bradley WG, Anderson F, Gowda N, Miller RG. Changes in the management of ALS since the publication of the AAN ALS Practice Parameter 1999. Amyotroph Lateral Sc.2004;5:240–4. doi:
- Georgoulopoulou E, Fini N, Vinceti M, Monelli M, Vacondio P, Bianconi G, et al. The impact of clinical factors, riluzole and therapeutic interventions on ALS survival: a population based study in Modena, Italy. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration.2013;14:338–45.
- Vrijsen B, Testelmans D, Belge C, Robberecht W, Van Damme P, Buyse B, et al. Non-invasive ventilation in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration.2013;14:85–95.
- Gruis KL, Lechtzin N. Respiratory therapies for amyotrophic lateral sclerosis: a primer. Muscle Nerve. 2012;46:313–31.
- Sivak ED, Shefner JM, Mitsumoto H, Taft JM. The use of non-invasive positive pressure ventilation (NIPPV) in ALS patients. A need for improved determination of intervention timing. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:139–45. doi:
- Marin B, Desport JC, Kajeu P, Jesus P, Nicolaud B, Nicol M, et al. Alteration of nutritional status at diagnosis is a prognostic factor for survival of amyotrophic lateral sclerosis patients. J Neurol Neurosur Ps.2011;82:628–34. doi:
- Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73:1218–26.
- Pena MJ, Ravasco P, Machado M, Pinto A, Pinto S, Rocha L, et al. What is the relevance of percutaneous endoscopic gastrostomy on the survival of patients with amyotrophic lateral sclerosis? Amyotrophic Lateral Sclerosis.2012;13:550–4. doi:
- Hardiman O. Symptomatic treatment of respiratory and nutritional failure in amyotrophic lateral sclerosis. J Neurol. 2000;247:245–51. doi:
- Jones AR, Jivraj N, Balendra R, Murphy C, Kelly J, Thornhill M, et al. Health utility decreases with increasing clinical stage in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:285–91.
- Oh J, An JW, Oh S-I, Oh KW, Kim JA, Lee JS, et al. Socioeconomic costs of amyotrophic lateral sclerosis according to staging system. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:202–8.
- Moore A, Young CA, Hughes DA. Health utilities and costs for motor neurone disease. Value Health. 2019;22:1257–65.
- Leigh PN, Abrahams S, Al-Chalabi A, et al. The management of motor neurone disease. Journal of Neurology, Neurosurgery, and Psychiatry 2003;74:iv32–iv47.