
ABSTRACT
Introduction: Urea cycle disorders are rare inborn errors of metabolism resulting in the accumulation of ammonia. Over the last 3 decades, the use of alternative nitrogen excretion pathways has been exploited to improve survival and outcomes for urea cycle disorder patients.
Areas covered: Early discovered nitrogen scavengers (sodium benzoate, phenylacetate, and phenylbutyrate) are effective in lowering ammonia. However, they have side effects and administration issues that can reduce compliance to therapy and limit optimal outcomes. Glycerol phenylbutyrate, a pro-drug of phenylbutyrate, was developed to improve therapy for patients with urea cycle disorders. Research with glycerol phenylbutyrate has produced the largest body of controlled study data in urea cycle disorder patients to date.
Expert opinion: Glycerol phenylbutyrate is better tolerated than sodium phenylbutyrate and enables patients with urea cycle disorders to reach ammonia and glutamine targets. Maintenance of target ammonia levels allows better long-term control in patients with urea cycle defects with reduced neurological sequelae.
1. Introduction
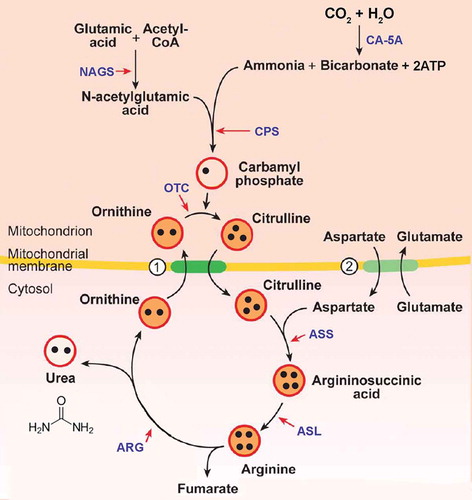
The urea cycle is a biochemical pathway in the mammalian liver and to a lesser extent in the kidneys, with the primary purpose of converting highly toxic ammonia (NH3) to urea ((NH2)2CO) for renal excretion () [1]. Urea cycle disorders (UCDs) are rare inborn errors of metabolism caused by a defect in any one of the enzymes or transporters required for the normal function of the urea cycle (). A deficiency in any one of the enzymes or transporters typically results in hyperammonemia with a variable clinical presentation depending on the severity of the mutation and the residual enzyme activity [Citation2]. UCD presentation is typically more severe among the proximal versus distal enzyme defects [Citation3]. An estimated prevalence of the different subtypes is presented in . The overall incidence of UCD is estimated to be 1/35,000, with the majority of patients presenting after the newborn period [Citation5].
Table 1. Estimated prevalence of the different UCD subtypes [Citation4].
Figure 1. Urea cycle.
Circles 1. ornithine/citrulline exchanger and 2. aspartate/glutamate transporterBlack dots figuratively show the number of nitrogen molecules moving through the urea cycle.NAGS, N-acetylglutamate synthetase; CA-5A, carbonic anhydrase 5A; CPS, carbamoyl phosphate synthetase; OTC, ornithine transcarbamylase; ASS, argininosuccinate synthetase; ASL, argininosuccinate lyase; ARG, arginase.
2. Clinical presentation and outcomes
Ornithine transcarbamylase (OTC) deficiency is the only X-linked and the most common subtype of UCD [Citation6]. Males with an X-linked OTC deficiency tend to present earlier. Females who carry two X-chromosomes can inactivate either the normal allele or the mutant one causing a wide variability in clinical presentations ranging from asymptomatic to neonatal onset [Citation7].
Only a minority of UCD patients are diagnosed with newborn screening (i.e. ASS, ASL, and ARG deficiency subtypes), while most are diagnosed after presenting with symptoms of hyperammonemia. Because UCD patients typically present with nonspecific symptoms, long delays in diagnosis are common [Citation8,Citation9]. Some critically ill infants are misdiagnosed, resulting in delayed treatment or death [Citation10]. UCDs typically manifest in the neonatal period with lethargy progressing to coma and death if left untreated [Citation11]. Approximately one-third of patients present in the neonatal period with an estimated mortality of 24% [Citation6]. A majority of patients present with late onset disease with diverse manifestations such as neurological, hepatogastric, or psychiatric symptoms [Citation8,Citation12]. Although late onset disease is thought to be less severe, it is associated with a 28% mortality rate and long-term neurological disability [Citation8]. Early treatment of UCDs decreases the risk for morbidity and mortality and may reduce some neurologic sequelae [Citation13].
Hyperammonemia has direct toxic effects on the brain and is also associated with an increase in glutamine levels leading to astrocyte swelling, brain edema, increased intracranial pressure, and can lead ultimately to brain damage and death [Citation14]. Neurological outcome has been correlated with the magnitude and duration of hyperammonemia [Citation15–Citation17]. Neurologic dysfunction tends to be most severe in patients with neonatal onset disease, but even episodic hyperammonemia increases the risk of neurocognitive deficits [Citation15,Citation18–Citation20]. Interestingly, executive dysfunction and working memory deficits (associated with brain white matter damage) have been demonstrated in asymptomatic females with OTC deficiency who have average IQ scores [Citation21]. Peak plasma ammonia concentration during the initial hyperammonemic crisis (HAC) (ranging from 350 to >1000 µmol/L) and the age of onset of disease (neonatal onset) may be predictive of a poor neurological outcome [Citation16,Citation22]. Seventy-five percent of patients with a diagnostic delay of >1 year have been reported to experience cognitive impairment compared to 46% if the delay was <1 year [Citation9]. Neuroimaging suggests that UCDs are associated with white matter abnormalities of the brain even in OTC females [Citation21,Citation23,Citation24]. Neuropsychological testing and new neuroimaging capabilities may assist in diagnosis and assessment of UCD patients by allowing the identification of microstructural abnormalities in the white matter of the brain [Citation25–Citation27].
3. Treatment
UCD patients should be treated by metabolic specialists given their complexity [Citation28]. Early diagnosis and treatment with a low protein diet combined with essential amino acids and nitrogen scavengers (sodium benzoate, sodium and glycerol phenylbutyrate (GPB)) when indicated for maintenance therapy have been shown to improve long-term neurological outcomes [Citation13,Citation20]. In the event of an acute HAC, protein intake should be stopped and intravenous (IV) calories, such as glucose and intralipids, should be commenced together with initiation of first-line medications such as IV l-arginine and IV nitrogen scavengers [Citation28]. Hemodialysis may be necessary if ammonia levels increase to very high levels. The goal of long-term management should be preventing hyperammonemia and HACs, promoting growth and development while maintaining a good quality of life. The ultimate long-term ‘cure’ for most UCDs is liver transplantation [Citation29]. Exceptions exist, since some urea cycle defects might also affect nitric oxide metabolism and/or guanidinoacetate/creatine biosynthesis resulting in neurological impairment independently from hyperammonemia and, in some cases, liver transplant [Citation29]. This has prompted some to recommend that liver transplantation be considered only in ASL deficient patients with recurrent hyperammonemia or those resistant to conventional medical therapy (diet/arginine/oral scavengers) [Citation30]. Another exception is N-acetylglutamate synthetase deficiency in which therapy with carglumic acid can prevent hyperammonemia while allowing for a normal diet [Citation31].
3.1. Nitrogen scavengers
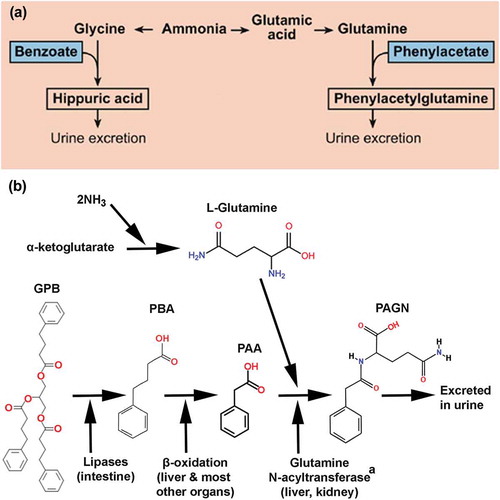
Alternative nitrogen excretion pathways were pioneered by Brusilow and Batshaw in the early 1980s with the use of exogenously administered sodium benzoate (NaBz) and sodium phenylacetate () [Citation3,Citation32–Citation34]. Based on stoichiometric calculations, each mole of NaBz administered can remove 1 mol of nitrogen by conjugating with glycine to form hippuric acid, which is then renally excreted. Phenylacetic acid (PAA), on the other hand, conjugates with glutamine to form phenylacetylglutamine (PAGN) providing for the excretion of 2 mol of nitrogen for each mole of PAA administered [Citation3]. In the US, NaBz is not approved for oral use, but an oral prodrug of PAA, sodium phenylbutyrate (NaPBA) was approved in 1996 as Buphenyl® for chronic management and the IV combination of PAA and NaBz in 2005 as Ammonul® for acute management [Citation35]. NaPBA requires large volumes of either granules or tablets to be consumed, has a foul smell, contains a high sodium load, and produces an unpleasant body odor, which is the number one patient reported complaint [Citation36]. Eighty percent of patients have been reported to experience ‘some’ or ‘a lot’ of side effects from NaPBA with 64% reporting that it is difficult to take because of its taste and strong odor. Care givers also reported body odor (45%) and burning sensation in the mouth or throat (23%) as major side effects [Citation37]. Adverse events (AEs) like gastrointestinal disturbances and severe reflux render the medication unappealing to many patients with a net result of reducing adherence and compliance to treatment [Citation37]. To improve on existing therapies, GPB was developed and approved in the US in 2013 as Ravicti®.
Figure 2. (a) Alternate pathways for nitrogen removal. (b) Biotransformation of glycerol phenylbutyrate (GPB).
This conversion likely involves more than 1 enzyme in the liver and kidney.NH3, ammonia; GPB, glycerol phenylbutyrate; PAA, phenylacetic acid; PAGN, phenylacetylglutamine; PBA, phenylbutyric acid.
4. Glycerol phenylbutyrate
4.1. Description
GPB is a liquid triglyceride formulation consisting of three molecules of phenylbutyrate (PBA) joined to a glycerol backbone () [Citation38]. It is a nearly tasteless and odorless liquid, contains no sugar or sodium, and is administered in low volume doses [Citation39]. As shown in , GPB is hydrolyzed by pancreatic lipases to glycerol and PBA, absorbed and then converted via β-oxidation to its active metabolite, PAA, which is conjugated with glutamine to form renally excreted PAGN. GPB is a prodrug of PBA and a pre-prodrug of PAA, the active moiety of the compound. Because hydrolysis begins after GPB exits the stomach and enters the proximal small intestine where it is exposed to pancreatic lipases, PBA delivered orally as GPB enters the circulation about 75% more slowly than NaPBA and demonstrates presystemic conversion of PBA to PAA and ultimately to PAGN. This first-pass metabolism produces a more metered PBA release into the splanchnic circulation as compared with the ‘bolus-like’ release from NaPBA [Citation38]. This slow-release of PBA is associated with clinically important advantages of GPB in comparison with NaPBA resulting in more sustained ammonia control throughout the day [Citation38].
Box 1. Drug summary box.
4.2. Clinical studies
GPB is currently approved in the US and EU for the chronic management of patients with UCDs aged 2 months and older and in Canada for those aged 2 years and older, who cannot be managed by dietary protein restriction and/or amino acid supplementation alone [Citation40]. These approvals were based on individual and pooled results from five short-term studies () and three long-term follow-on and pooled study results (). The protocols and informed consent for each of these studies were reviewed by the respective Institutional Review Board of the participating study centers.
Table 2. Short-term clinical studies.a,b,c
Table 3. Pooled analyses of short- and long-term clinical studies.
A total of 100 patients contributed data for these original approvals with 10 additional patients less than 2 years of age reported recently. Furthermore, 88 subjects who had completed the three long-term follow-on studies continued an additional long-term safety assessment (HPN100-011), with ammonia and neurocognitive outcomes also monitored. The median duration of treatment among the patients was 2.84 years. In the analysis of GPB in UCD patients aged 2 months to 2 years, GPB was found to be safe and effective with short- and long-term ammonia control and no evidence of previously unknown toxicity [Citation45]. Finally, a UCD registry (Study HPN100-014, named THRIVE) was established to allow both previous study patients and those newly prescribed therapy to be monitored on an ongoing basis. A total of 149 patients were entered in the THRIVE Registry as of June 2017. These studies collectively constitute the largest UCD treatment population studied in a controlled manner to-date.
4.3. Pooled clinical trial summary data
Patients in the study program mirrored those seen in registry databases with female OTC patient predominance. There were equal numbers of pediatric and adult patients ().
Table 4. Selected demographic from pooled trial data.
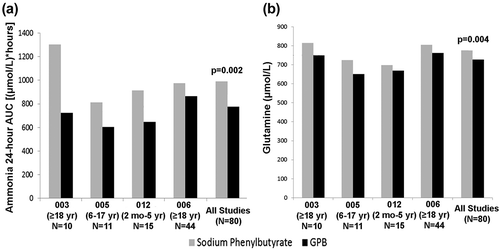
A clinically meaningful and statistically significant difference in favor of GPB versus NaPBA was seen in controlling blood ammonia levels among those who completed the short-term studies, as measured by area under the curve for 24 h (AUC0–24, 774.1 versus 991.2 μmol∙h/L, respectively) (). The mean difference between GPB and NaPBA (n = 78) was −179 μmol∙h/L (p = 0.004, paired t-test; p = 0.002, Wilcoxon signed-rank test). The upper bound of the 95% confidence interval (CI) was 0.949. Similar results were seen in mean short-term glutamine levels which were significantly lower after GPB treatment compared with NaPBA treatment by 50.4 ± 154.8 μmol/L (p = 0.006, paired t-test; p = 0.004, Wilcoxon signed-rank test ()). Mean glutamine levels with GPB over time were similar at baseline (763 μmol/L) and Month 12 (748 μmol/L) and remained within normal range throughout the 12-month period.
Figure 3. (a) Pooled analysis: blood ammonia AUC0-24, (b) Mean blood glutamine levels across studies.
p-values obtained with Wilcoxon signed-rank test.003 = UP1204-003; 005 = HPN100-005; 012 = HPN100-012; 006 = HPN100-006; AUC, area under the curve; GPB, glycerol phenylbutyrate; mo, month; yr, year.
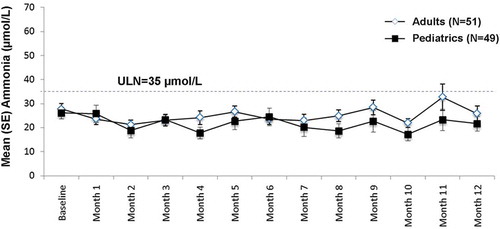
Long-term mean ammonia levels were within the normal range over 12 months of analysis in both pediatric and adult patients ().
Figure 4. Pooled long-term mean ammonia levels.
Ammonia levels normalized to reference range of 9–35 μmol/L.ULN, upper limit of normal; SE, standard error.
The rate of HACs per patient decreased while on GPB versus preenrollment () [Citation46]. HACs were most frequent in patients aged 2–5 years. Overall, patients preenrollment had on average 0.58 events per patient per year. While on GPB, patients experienced on average 0.29 events per patient per year [Citation46,Citation47].
Figure 5. Number of HACs pre-study and long-term follow-up. Adapted with permission from [Citation46].
p-values obtained with Wilcoxon signed rank test.
![Figure 5. Number of HACs pre-study and long-term follow-up. Adapted with permission from [Citation46].p-values obtained with Wilcoxon signed rank test.](/cms/asset/444758cc-95fd-4f2d-bb8e-40db385928cf/ieod_a_1405807_f0005_b.gif)
4.4. Growth and neurocognitive outcomes
Dietary data were pooled from four studies in 45 adult and 49 pediatric UCD patients treated over 12 months. Actual protein intake as evidenced by patient diary inspection was very similar to that prescribed [Citation50]. Prescribed protein was lower in the UCD subjects compared to that recommended for healthy subjects; however, protein intake was greater than traditional recommendations for UCD patients. This was not anticipated due to previous reports of dietary protein aversion in UCD patients. Pediatric patients demonstrated normal Z-scores for height, weight, and body mass index during the studies. Additionally, patients 2 months to <2 years showed normal growth with 71% having Z-scores higher than typical for their age and gender [Citation45].
Patients enrolled in the 12-month long-term follow-up received several neurocognitive tests at baseline and at the end of study. Parent questionnaires were used for pediatric patients and examined internalizing/externalizing behaviors and executive function among others [Citation13]. All neurological tests remained stable in adults over the course of the study. Twenty-two pediatric patients who completed the long-term studies demonstrated statistically significantly improved executive function, as measured by the Behavior Rating Inventory of Executive Function scales () [Citation13]. These results may indicate that certain neurocognitive outcomes may improve with effective therapy, especially when started early.
4.5. Pharmacokinetic (PK) and metabolite studies of importance to dosing and safety
In an effort to understand the effect of GPB dosing on plasma PBA, PAA, and PAGN, PK analyses were conducted during the clinical trials and population PK modeling completed where appropriate. Initial studies with single dose exposure of GPB and NaPBA in 24 healthy adults found that GPB resulted in lower levels of plasma PBA, PAA, and PAGN (likely due to the slower absorption of GPB), with a prolonged and similar urinary excretion of PAGN compared to NaPBA [Citation38,Citation48]. Further studies have found that the dose of GPB strongly correlated with urinary PAGN (UPAGN) rather than with the plasma metabolites, suggesting that pre-systemic conversion of GPB to PAA and PAGN likely occurs [Citation49]. Population PK modeling and dosing simulations found that the rate of PAA metabolism to PAGN correlates most strongly with body surface area, such that smaller children metabolize PAA to PAGN more slowly and show higher plasma PAA levels [Citation38]. At the upper end of the dose range, median PAA exposure for GPB was generally ≤200 µg/mL for all age groups and the upper 95% CI for Cmax does not extend above 500 µg/mL in the pediatric population, a threshold previously identified as potentially neurotoxic [Citation51,Citation52]. In contrast, for NaPBA, median PAA exposure was similar to GPB but the upper 95% CI frequently extended above 500 µg/mL in the pediatric population [Citation38]. PAA levels were examined in 100 UCD patients enrolled in 12-month studies and no relationship was observed between PAA levels and specific AEs known to be associated with PAA toxicity [Citation53].
4.6. Safety and AEs
GPB was generally well tolerated in the 114 pooled patients in the safety analysis. The most frequently reported organ symptom AEs were gastrointestinal disorders [32 (28.2%)], metabolism and nutrition disorders [12 (12.3%)], and nervous system disorders [14 (12.3%)]. When pooled, no individual AE met the criteria of very common. Of the AEs meeting the common criteria (≥2 patients), the most frequently (≥5% (6 patients)) reported included the following: diarrhea, flatulence, and headache (10, 8.8% each); vomiting (7, 6.1%); and fatigue, nausea and skin odor abnormalities (6, 5.3% each). The most common AEs are presented in .
Table 5. Adverse events occurring in ≥10% of patients as evaluated in clinical trials [Citation40].
Twenty-six of the 100 patients studied long-term reported a serious AE, but none of these were deemed related to the GPB [Citation54].
4.7. Patient reported UCD and treatment symptoms
Overall, 69% of patients reported at least one symptom commonly associated with nitrogen-scavenging medications at baseline while taking NaPBA. The mean (standard deviation) number of symptoms per patient was approximately 2.5 (3.0) for all patient groups while on NaPBA baseline therapy and was significantly reduced in each age category while on GPB treatment over 3 months ().
Figure 6. (a) Mean number of symptoms per patient. (b) Common Symptoms in all UCD Patients Before and After 3 Months of GPB Treatment.
Abbreviations: NaPBA, sodium phenylbutyrate; GPB, glycerol phenylbutyrate.
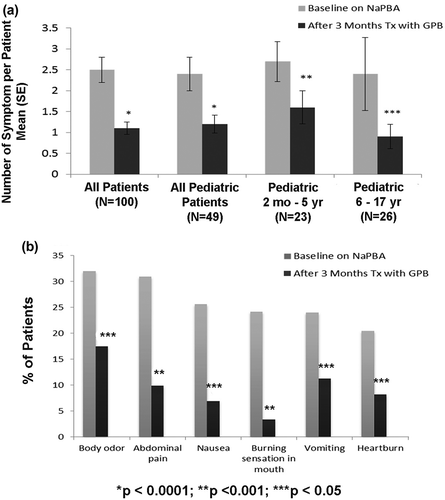
For patients <6 years of age, parents or caregivers reported treatment symptoms quarterly for 12 months. In this subgroup, the percentage of patients reporting no treatment-associated symptoms increased from 21.7% at baseline to 43.5%, 40.9%, 52.4%, and 61.9% at Months 3, 6, 9, and 12, respectively with a decrease in the mean number of symptoms per patient from 2.6 at baseline to 1.6, 1.1, 1.1, and 1.1, respectively.
After 3 months of treatment with GPB, the proportion of patients with no UCD-related symptoms increased from 31.0% to 53.6% for all patients and from 30.6% to 47.9% for pediatric patients. Increases in the proportion of patients with no symptoms were also observed for patients aged 2 months to 5 years, 6–17 years, and adult patients.
The most common UCD-related symptoms at baseline while on NaPBA were body odor, abdominal pain, and nausea for all patients and body odor, abdominal pain/distress, and vomiting for pediatric patients. After 3 months of treatment with GPB, there were decreases in the proportions of patients with each symptom and these decreases were statistically significant for all symptoms for all patients () and for body odor, abdominal pain, nausea, and burning sensation in the mouth for pediatric patients [Citation36].
5. Conclusion
GPB is at least similar to NaPBA in terms of efficacy and safety in patients with UCD 2 months and older, with pooled data indicating improved ammonia control and lower rates of HACs. It is a major addition to our armamentarium of therapies that help UCD patients have the best chance to grow and develop normally while having a good quality of life.
6. Expert opinion
UCD patients are being diagnosed earlier and patients are living longer than ever before due to earlier diagnosis and better management and understanding of UCD treatment approaches. There are still challenges such as the lack of data on the appropriate management of patients below 2 months of age as well as those on higher protein diets, particularly with respect to initiating and titrating nitrogen scavenging therapy and improving adherence and compliance to treatment. A study evaluating the use of GPB in neonates from birth to 2 months of age is ongoing and will provide new data on efficacy and safety in this population.
GPB may offer the opportunity to more finely tune doses in infants allowing for optimal protein intake for proper growth and development. However, we have limited data on modeling protein intake, assessing residual enzyme activity, and calculating an effective and safe dose. For example, the theoretical calculations on dosing that appear in the prescribing information for nitrogen scavengers in the US do not take into account the heterogeneity of UCD patients. Conversion rates of PBA/PAA to PAGN vary and on average are only about 70% complete, without good models to account for the appropriate amount of drug to bind nitrogen from ingested protein. Estimates are based on the pioneering work of Brusilow in the late 1980s and early 1990s but not updated since then [Citation54]. Analysis of the plasma metabolites and ratios of PAA to PAGN may help us optimize dose adjustment decisions in the future, if we can get them in a timely manner. Unlike plasma PAA, the PAA-to-PAGN ratio is relatively constant over 24 h and is not affected by timing of the blood draw as that of ammonia, which is best taken upon fasting [Citation53].
Another important consideration in monitoring patients is adherence to the prescribed diet and medication [Citation56,Citation57]. Nonadherence to medication and diet is the second most frequent precipitant of a HAC. In a survey, physicians reported concerns with noncompliance to NaPBA and NaBz therapy (42% and 36% noncompliance, respectively), due to the amount of medication taken each time, difficulty in swallowing medication, the number of times medication is taken each day, patient forgetting to take, side effects from medication, and difficulty in keeping medication down [Citation37]. The net result of this has been that many patients take less than the required amounts of these nitrogen scavengers. In fact, approximately one quarter and one-third of pediatric and adult UCD patients, respectively, received NaPBA doses recognized by their physicians as suboptimal. Therefore, when patients switch from NaPBA to GPB they are likely put on a lower than optimal dose of GPB. In this context, GPB, which has a lower incidence of body odor, abdominal pain, vomiting, nausea, burning sensation in mouth, and heartburn, is expected to result in improved compliance in patients and ultimately in a reduction in the number of HACs as reviewed previously [Citation36]. To this end, it has been proposed that UPAGN concentration can serve as a biomarker for adherence to therapy in UCD patients treated with GPB [Citation45,Citation58].
Regular ammonia monitoring of patients with UCD can prevent HACs, hospitalization, morbidity, and mortality. In the studies reported here, 35 hospitalizations occurred in the pre-GPB evaluation versus 25 while on GPB [Citation46]. Fasting ammonia, and to a lesser degree glutamine, has been shown to correlate with daily ammonia exposure and the rate of HACs [Citation47,Citation58]. UCD patients with a fasting ammonia above the upper limit of normal (ULN) have a higher risk of developing a HAC as compared to patients with a fasting ammonia <0.5 ULN [Citation47]. Hence patients are recommended to maintain a fasting ammonia <0.5 ULN.
Therapy with NaPBA in patients with UCD has been associated with a decrease in the branched-chain amino acids (BCAA) leucine, isoleucine, and valine, possibly due to inhibition of branched-chain α-ketoacid dehydrogenase kinase, an enzyme that inhibits branched-chain α-ketoacid dehydrogenase complex activity [Citation60–Citation62]. NaPBA does not adversely affect whole-body proteolysis or net protein catabolism despite lowering BCAA levels [Citation63]. Use of GPB up to 1 year in the clinical trials reviewed here has not demonstrated any significant changes in amino acid levels, including BCAA levels, which remain within normal limits [Citation44]. Additionally, this was confirmed in a study of infants 2 months to <2 years of age [Citation45]. Further investigations are necessary to determine whether prolonged exposure to GPB reduces BCAA levels as does NaPBA.
Nitrogen scavengers may improve or stabilize neurocognitive dysfunction in some patients with UCDs [Citation64,Citation65]. The mechanisms of neurologic damage due to hyperammonemia or elevated glutamine levels have not been clearly defined, but they may result in brain white matter damage and decrease in executive function [Citation21]. It is hypothesized that hyperammonemia may lead to high concentrations of glutamine in the astrocytes leading to an influx of water into the astrocytes, astrocyte swelling and brain edema [Citation14]. Long-term GPB trials involving 77 adult and pediatric patients suggest that GPB might mitigate, or at a minimum stabilize neurocognitive dysfunction if started early enough [Citation13]. However, this needs to be confirmed in prospective longer term clinical trials.
Declaration of interest
Dr. Longo’s institution, University of Utah, has received research funds from Horizon Pharma. He has served as a Horizon Pharma Advisory Board Member and has no ownership in Horizon Pharma. R.J. Holt is an employee of, and has stock in, Horizon Pharma USA, Inc. Editorial/Writing support was provided by Teresa M.Y. Kok RPh, MBA and Megan Francis-Sedlak PhD, Employees of Horizon Pharma, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. A reviewer on this manuscript has disclosed they are an employee of Sanofi Genzyme. The company does not own competing products for urea cycle defects.
Additional information
Funding
References
- Brusilow SW, Horwich AL. Urea cycle enzymes. In: Scriver CRBAL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8th ed. New York (NY): McGraw-Hill; 2001.
- Hoffmann GF, Kölker S. Defects in amino acid catabolism and the urea cycle. Handb Clin Neurol. 2013;113:1755–1773.
- Batshaw ML. Hyperammonemia. Curr Probl Pediatr. 1984 Nov;14(11):1–69.
- Seminara J, Tuchman M, Krivitzky L, et al. Establishing a consortium for the study of rare diseases: the urea cycle disorders consortium. Mol Genet Metab. 2010;100 Suppl 1:S97–105.
- Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013 Sep-Oct;110(1–2):179–180.
- Batshaw ML, Tuchman M, Summar M, et al. A longitudinal study of urea cycle disorders. Mol Genet Metab. 2014 Sep-Oct;113(1–2):127–130.
- Batshaw ML, Msall M, Beaudet AL, et al. Risk of serious illness in heterozygotes for ornithine transcarbamylase deficiency. J Pediatr. 1986 Feb;108(2):236–241.
- Nassogne MC, Héron B, Touati G, et al. Urea cycle defects: management and outcome. J Inherit Metab Dis. 2005;28(3):407–414.
- Ruegger CM, Lindner M, Ballhausen D, et al. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J Inherit Metab Dis. 2014 Jan;37(1):21–30.
- Leonard JV, Morris AA. Urea cycle disorders. Semin Neonatol. 2002 Feb;7(1):27–35.
- Burgard P, Kölker S, Haege G, et al. Neonatal mortality and outcome at the end of the first year of life in early onset urea cycle disorders–review and meta-analysis of observational studies published over more than 35 years. J Inherit Metab Dis. 2016 Mar;39(2):219–229.
- Summar ML, Dobbelaere D, Brusilow S, et al. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr (Oslo, Norway: 1992). 2008 Oct;97(10):1420–1425.
- Diaz GA, Krivitzky LS, Mokhtarani M, et al. Ammonia control and neurocognitive outcome among urea cycle disorder patients treated with glycerol phenylbutyrate. Hepatology (Baltimore, MD). 2013 Jun;57(6):2171–2179.
- Norenberg MD, Rao KV, Jayakumar AR. Mechanisms of ammonia-induced astrocyte swelling. Metab Brain Dis. 2005 Dec;20(4):303–318.
- Msall M, Monahan PS, Chapanis N, et al. Cognitive development in children with inborn errors of urea synthesis. Acta Paediatr Japon. 1988 Aug;30(4):435–441.
- Uchino T, Endo F, Matsuda I. Neurodevelopmental outcome of long-term therapy of urea cycle disorders in Japan. J Inherit Metab Dis. 1998;21 Suppl 1:151–159.
- Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003 Jun;162(6):410–416.
- Batshaw ML, Roan Y, Jung AL, et al. Cerebral dysfunction in asymptomatic carriers of ornithine transcarbamylase deficiency. N Engl J Med. 1980;302(9):482–485.
- Gyato K, Wray J, Huang ZJ, et al. Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann Neurol. 2004 Jan;55(1):80–86.
- Krivitzky L, Babikian T, Lee H-S, et al. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009 Jul;66(1):96–101.
- Gropman A. Brain imaging in urea cycle disorders. Mol Genet Metab. 2010;100 Suppl 1:S20–30.
- Posset R, Garcia-Cazorla A, Valayannopoulos V, et al. Age at disease onset and peak ammonium level rather than interventional variables predict the neurological outcome in urea cycle disorders. J Inherit Metab Dis. 2016 Sep;39(5):661–672.
- Gropman AL, Prust M, Breeden A, et al. Urea cycle defects and hyperammonemia: effects on functional imaging. Metab Brain Dis. 2013 Jun;28(2):269–275.
- Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis. 2007 Nov;30(6):865–879.
- Pacheco-Colón I, Fricke S, VanMeter J, et al. Advances in urea cycle neuroimaging: proceedings from the 4th International Symposium on urea cycle disorders, Barcelona, Spain, September 2013. Mol Genet Metab. 2014 Sep-Oct;113(1–2):118–126.
- Gioia GA, Isquith PK, Guy SC. Behavior Rating Inventory of Executive Function. Odessa (FL): Psychological Assessment Resources; 2000.
- Harrison P, Oakland T. Adaptive behavior assessment system® - second edition (ABAS® — second edition). San Antonio (TX): Pearson; 2003.
- Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012 May;7(1):32.
- Erez A, Nagamani SC, Lee B. Argininosuccinate lyase deficiency-argininosuccinic aciduria and beyond. Am J Med Genet C Semin Med Genet. 2011 Feb 15;157C(1):45–53.
- Nagamani SCS, Erez A, Lee B, et al. Argininosuccinate lyase deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R). Seattle (WA): University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle; 1993. All rights reserved.
- Gessler P, Buchal P, Schwenk HU, et al. Favourable long-term outcome after immediate treatment of neonatal hyperammonemia due to N-acetylglutamate synthase deficiency. Eur J Pediatr. 2010 Feb;169(2):197–199.
- Batshaw ML, Brusilow S, Waber L, et al. Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med. 1982 Jun 10;306(23):1387–1392.
- Brusilow S, Tinker J, Batshaw ML. Amino acid acylation: a mechanism of nitrogen excretion in inborn errors of urea synthesis. Science. 1980 Feb 08;207(4431):659–661.
- Brusilow SW, Valle DL, Batshaw M. New pathways of nitrogen excretion in inborn errors of urea synthesis. Lancet. 1979 Sep 01;2(8140):452–454.
- Mistry PK. Rare disease clinical research network’s urea cycle consortium delivers a successful clinical trial to improve alternate pathway therapy. Hepatology (Baltimore, Md). 2013 Jun;57(6):2100–2102.
- Nagamani SC, Diaz GA, Rhead W, et al. Self-reported treatment-associated symptoms among patients with urea cycle disorders participating in glycerol phenylbutyrate clinical trials. Mol Genet Metab. 2015 Sep-Oct;116(1–2):29–34.
- Shchelochkov OA, Dickinson K, Scharschmidt BF, et al. Barriers to drug adherence in the treatment of urea cycle disorders: assessment of patient, caregiver and provider perspectives. Mol Genetics Metab Rep. 2016 Sep;8:43–47.
- Monteleone JP, Mokhtarani M, Diaz GA, et al. Population pharmacokinetic modeling and dosing simulations of nitrogen-scavenging compounds: disposition of glycerol phenylbutyrate and sodium phenylbutyrate in adult and pediatric patients with urea cycle disorders. J Clin Pharmacol. 2013 Jul;53(7):699–710.
- Cederbaum S, Lemons C, Batshaw ML. Alternative pathway or diversion therapy for urea cycle disorders now and in the future. Mol Genet Metab. 2010 Jul;100(3):219–220.
- Ravicti [package insert]. Lake Forest (IL): Horizon Therapeutics; 2017 April.
- Lee B, Rhead W, Diaz GA, et al. Phase 2 comparison of a novel ammonia scavenging agent with sodium phenylbutyrate in patients with urea cycle disorders: safety, pharmacokinetics and ammonia control. Mol Genet Metab. 2010 Jul;100(3):221–228.
- Lichter-Konecki U, Diaz GA, Merritt JL 2nd, et al. Ammonia control in children with urea cycle disorders (UCDs); phase 2 comparison of sodium phenylbutyrate and glycerol phenylbutyrate. Mol Genet Metab. 2011 Aug;103(4):323–329.
- Smith W, Ga D, Lichter-Konecki U, et al. Ammonia control in children ages 2 months through 5 years with urea cycle disorders: comparison of sodium phenylbutyrate and glycerol phenylbutyrate. J Pediatr. 2013 Jun;162(6):1228–1234, 34.e1.
- Berry SA, Lichter-Konecki U, Diaz GA, et al. Glycerol phenylbutyrate treatment in children with urea cycle disorders: pooled analysis of short and long-term ammonia control and outcomes. Mol Genet Metab. 2014 May;112(1):17–24.
- Berry SA, Longo N, Diaz GA, et al. Safety and efficacy of glycerol phenylbutyrate for management of urea cycle disorders in patients aged 2 months to 2 years. Mol Genet Metab. 2017 Sep 8. pii: S1096-7192(17)30491-2. doi: 10.1016/j.ymgme.2017.09.002. [Epub ahead of print].
- Kent JD, Holt RJ. Hyperammonemic crises in patients with urea cycle disorders on chronic nitrogen scavenger therapy with either sodium phenylbutyrate or glycerol phenylbutyrate. Neuropsychiatry (London). 2017;7(2):131–136.
- Lee B, Diaz GA, Rhead W, et al. Blood ammonia and glutamine as predictors of hyperammonemic crises in patients with urea cycle disorder. Genet Med. 2015 Jul;17(7):561–568.
- McGuire BM, Zupanets IA, Lowe ME, et al. Pharmacology and safety of glycerol phenylbutyrate in healthy adults and adults with cirrhosis. Hepatology (Baltimore, Md). 2010 Jun;51(6):2077–2085.
- Mokhtarani M, Diaz GA, Rhead W, et al. Urinary phenylacetylglutamine as dosing biomarker for patients with urea cycle disorders. Mol Genet Metab. 2012 Nov;107(3):308–314.
- Hook D, Diaz GA, Lee B, et al. Protein and calorie intakes in adult and pediatric subjects with urea cycle disorders participating in clinical trials of glycerol phenylbutyrate. Mol Genetics Metab Rep. 2016 Mar;6:34–40.
- Thibault A, Cooper MR, Figg WD, et al. A phase I and pharmacokinetic study of intravenous phenylacetate in patients with cancer. Cancer Res. 1994 Apr 01;54(7):1690–1694.
- Thibault A, Samid D, Cooper MR, et al. Phase I study of phenylacetate administered twice daily to patients with cancer. Cancer. 1995 Jun 15;75(12):2932–2938.
- Mokhtarani M, Diaz GA, Rhead W, et al. Elevated phenylacetic acid levels do not correlate with adverse events in patients with urea cycle disorders or hepatic encephalopathy and can be predicted based on the plasma PAA to PAGN ratio. Mol Genet Metab. 2013 Dec;110(4):446–453.
- European Medicines Agency Ravicti Assessment Report. 2015. [cited 2017 Nov 14]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003822/WC500199159.pdf.
- Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res. 1991 Feb;29(2):147–150.
- McGuire PJ, Lee H-S; Members of the Urea Cycle Disorders Consortium, Summar ML. Infectious precipitants of acute hyperammonemia are associated with indicators of increased morbidity in patients with urea cycle disorders. J Pediatr. 2013 Dec;163(6):1705–1710.e1.
- Wilson D, Bressani R, Scrimshaw NS. Infection and nutritional status. I. The effect of chicken pox on nitrogen metabolism in children. Am J Clin Nutr. 1961 Mar-Apr;9:154–158.
- Mokhtarani M, Diaz GA, Lichter-Konecki U, et al. Urinary phenylacetylglutamine (U-PAGN) concentration as biomarker for adherence in patients with urea cycle disorders (UCD) treated with glycerol phenylbutyrate. Mol Genetics Metab Rep. 2015 Dec;5:12–14.
- Lee B, Diaz GA, Rhead W, et al. Glutamine and hyperammonemic crises in patients with urea cycle disorders. Mol Genet Metab. 2016 Jan;117(1):27–32.
- Burrage LC, Jain M, Gandolfo L, et al. Sodium phenylbutyrate decreases plasma branched-chain amino acids in patients with urea cycle disorders. Mol Genet Metab. 2014 Sep-Oct;113(1–2):131–135.
- Scaglia F, Carter S, O’Brien WE, et al. Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Mol Genet Metab. 2004 Apr;81(Suppl 1):S79–85.
- Paxton R, Harris RA. Clofibric acid, phenylpyruvate, and dichloroacetate inhibition of branched-chain alpha-ketoacid dehydrogenase kinase in vitro and in perfused rat heart. Arch Biochem Biophys. 1984 May 15;231(1):58–66.
- Marini JC, Lanpher BC, Scaglia F, et al. Phenylbutyrate improves nitrogen disposal via an alternative pathway without eliciting an increase in protein breakdown and catabolism in control and ornithine transcarbamylase-deficient patients. Am J Clin Nutr. 2011 Jun;93(6):1248–1254.
- Maestri NE, Brusilow SW, Clissold DB, et al. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med. 1996 Sep 19;335(12):855–859.
- Maestri NE, Hauser ER, Bartholomew D, et al. Prospective treatment of urea cycle disorders. J Pediatr. 1991 Dec;119(6):923–928.