
ABSTRACT
Introduction: Duchenne muscular dystrophy (DMD) is a lethal, X-linked muscle-wasting disease caused by the lack of dystrophin. A few years after the identification of the dystrophin gene, a ubiquitously expressed transcript with high homology to dystrophin was identified leading to the postulate that utrophin might be an effective surrogate to compensate for the lack of dystrophin in dystrophic muscles.
Areas covered: We review the utrophin gene, its regulation, the organisation of the corresponding protein and its dynamic expression pattern in comparison with dystrophin. In view of the evidence that utrophin acts as an efficient substitute to dystrophin to prevent the pathology, we describe the therapeutic approaches to modulate utrophin expression. We review the current status in the development of ezutromid, the first small utrophin modulator drug currently tested in DMD patients.
Expert opinion: A utrophin based strategy is a highly promising approach applicable to all DMD patients irrespective of their genetic defect. Whereas accurate utrophin and regeneration quantification tools are urgently needed, the foundation of utrophin based therapies, defined 20 years ago, recently delivered first promising results in DMD patients with ezutromid. The field is at an exciting stage on the road of a universal treatment for all DMD patients.
1. Introduction – utrophin: Discovery, expression, and molecular basis
1.1. Duchenne muscular dystrophy and the lack of dystrophin
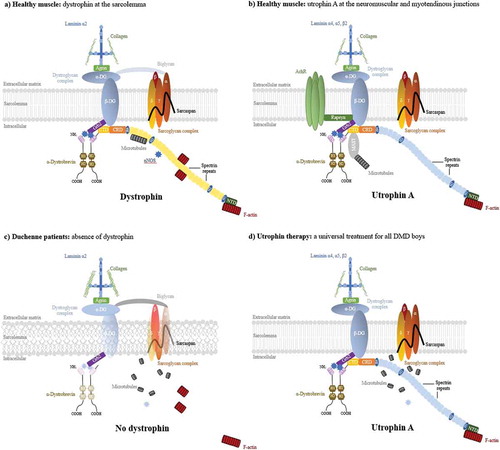
The gene for Duchenne muscular dystrophy (DMD, MIM no. 310200), an X-linked progressive muscle wasting disorder affecting 1:5000 boys [Citation1,Citation2], was identified 30 years ago [Citation3]. Patients have progressive muscle weakness from late childhood and lose the ability to walk around the age of 12 and die in their late 20s [Citation4]. The gene is the largest in the human genome (2.3 megabases) and the disease shows one of the highest new mutation rates which results predominantly in deletions of the gene. The 14-kb RNA transcript is encoded by 79 exons and is expressed predominantly in muscle and at lower levels in brain. The corresponding protein, dystrophin, is a 427-kDa cytoplasmic protein which provides an essential structural link between the dystrophin-associated protein complex at the sarcolemma and the costameric actin which maintains the biomechanical properties of fiber strength, flexibility, and stability in skeletal muscle ()) [Citation5,Citation6]. Absence in DMD or reduction of dystrophin in the milder Becker Muscular dystrophy (BMD, MIM no. 300376) [Citation7] leads to costamere perturbation, sarcolemma fragility, and subsequent chronic inflammation associated with a failed regeneration process leading to necrotic and fibrotic events and ultimately loss of muscle fibers and reduction in muscle mass and function muscle ()) [Citation8,Citation9].
Figure 1. The dystrophin and utrophin-associated protein complexes and utrophin based therapies. (a) In healthy muscle, the dystrophin and its associated proteins (DAPC) protect the sarcolemma from contraction-induced injury, maintaining the biomechanical properties of fibre strength, flexibility and stability in skeletal muscle. Dystrophin contains N-terminal (NTD), central rod, cysteine-rich (CRD), and C-terminal (CTD) domains. The NTD and spectrin-like repeat 11–15 bind the costameric actin filament to aid in shock absorbance during muscle contraction. The central rod domain contains 24 spectrin-like repeats and 4 putative hinge modules. Spectrin repeat 11–17 interact with a second independent actin binding domain and spectrin repeats 16/17 in dystrophin are able to recruit nNOS. Dystrophin interacts indirectly with microtubules through ankyrin-B and directly via spectrin repeats 20–23. The CRD links dystophin to the sarcolemmal-bound β-dystroglycan which binds to α-dystroglycan to form the dystroglycan complex. β-dystroglycan in association with δ-sarcoglycan participates in the stabilization of the (α, β, δ and γ) sarcoglycan-sarcospan complex at the sarcolemma and the α-dystroglycan links laminin-α2 at the extracellular matrix which both strengthen the DPAC. The CTD binds several cytosolic proteins such as α-dystrobrevin 2, α- and β-syntrophins and recruits signalling molecules such as nNOS via PDZ domains. (b) At the neuromuscular junction, utrophin forms the UAPC complex with similar protective functions as the DAPC. Acting as a receptor for agrin and laminin, the UAPC links the actin cytoskeleton through the NTD to the extracellular matrix via laminin-α4, -α5, and -β2. Utrophin binds to MAST and is associated with microtubules by the CTD and nNOS can only be recruited indirectly through the syntrophins. In addition, the UAPC binds to rapsyn and is involved in the clustering of acetylcholine receptors. (c) In DMD, the absence of dystrophin results in loss of the DAPC w hich lead to costamere perturbation and sarcolemma fragility. Subsequent sarcolemmal lesions trigger muscle degeneration and chronic inflammation associated with failed regeneration leading to necrotic and fibrotic events. (d) As a structural and functional surrogate to dystrophin, utrophin can replace and compensate for the lack of dystrophin in dystrophic muscles. A large majority of the elements of the DAPC and in consequence sarcolemma stability is rescued leading to prevention of the dystrophic phenotype despite the lack of restoration of the microtubules and nNOS.
The unmet clinical need has resulted in many translational efforts which either target downstream effects (inflammation, fibrosis) [Citation10,Citation11] or attempt to correct the patient mutation through mutation-specific strategies such as stop codon readthrough [Citation12] or exon skipping [Citation13]. Gene therapy is also being developed [Citation14] but there are challenges in systemic delivery and virus production. This review is focused on therapy of DMD through the modulation of expression of the dystrophin-related protein, utrophin, which will be applicable to all patient mutations and mediated through small molecules.
1.2. Utrophin: genetic basis, protein structure, and localization
The utrophin gene encoding a 13-kb transcript is located on human chromosome 6q24 and is ubiquitously expressed [Citation15]. The 900-kb gene exhibits 65% nucleic-acid identity and an intron-exon structure very similar to the gene encoding dystrophin [Citation16], suggesting that the two genes arose through an ancient duplication event ()). Despite their high level of structural identity and relative conservation, utrophin and dystrophin present different spatiotemporal expression [Citation17]. Driven by at least seven tissue-specific promoters, expression of dystrophin is mainly confined to adult skeletal, smooth, and cardiac muscles and brain [Citation18] whereas the utrophin is more ubiquitously expressed. In early human developing muscles [Citation19], utrophin is found at the sarcolemma and is progressively replaced by dystrophin toward birth [Citation17]. In adult tissues, utrophin is expressed in a wide range of tissues such as lung, kidney, liver, spleen and is limited not only to neuromuscular and myotendinous junctions in muscles but also the sarcolemma in regenerating myofibers and blood vessels ()) [Citation20]. To date, two utrophin promoters, A and B, differentially regulated, have been reported to drive two distinct mRNA isoforms translated into full length utrophin proteins with unique N-termini and different expression patterns ( and (b)) [Citation20,Citation21]. Unlike the dystrophin tissue-specific promoters, utrophin A promoter encoding the full-length protein is associated with a CpG island at the 5ʹ-end of the gene [Citation22]. Expressed in many tissues, Utrophin-A is the isoform that is expressed in muscle at the neuromuscular and myotendinous junctions, choroid plexus, pia mater, and renal glomerulus and found at the sarcolemma in regenerating myofibers. Utrophin B, which differs from A by a slightly different N-terminal acting binding site, is confined to endothelial cells and blood vessels. Both dystrophin and utrophin have smaller transcripts driven by similar internal promoters (Dp71, Dp140, Up71, Up140) [Citation23,Citation24] (,b)). Another short isoform G-utrophin, expressed in sensory ganglia and brain, presented as the autosomal encoded counterpart to Dp116 [Citation25], emphasizing the high degree of structural conservation between the utrophin and dystrophin genes (,b)). The function of these smaller transcripts is not fully understood.
Figure 2. Schematic organisation of the dystrophin (A), utrophin (B) genes with associated promoters and utrophin A and B transcripts (c). (a). Dystrophin expression is driven by at least 7 tissue-specific promoters. The full-length dystrophin transcription is controlled by 3 promoters (B, brain; M, muscle; P, Purkinje) localised upstream to the first exon and which only differs in its NH2-terminal sequences. 4 internal promoters, dp260, dp140, dp116 and dp71 were reported. Dp427m is the main variant produced in muscle and is involved in DMD and BMD. (b). In contrast to dystrophin, the utrophin gene has a long 5ʹprime untranslated region composed of 2 exons and a cluster of unmethylated, rare-cutting restriction enzyme sites. Expression of utrophin transcripts is controlled by two independently regulated promoters, A and B. Whereas each promoter results in a transcript with unique 5ʹ exons that splice into a common mRNA at exon 3 (black box), utrophin A promoter is CpG rich at the 5ʹ-end and contains an N-box motif controlling the synaptic expression. Different elements of the utrophin A promoter are specified. Promoter B, devoid of the N-box motif lies within the second exon of utrophin. 3 shorter isoforms Up140, G-Utrophin and Up71 were reported similar to the smaller transcripts of the dystrophin gene. Exons (grey boxes with exon numbers) and intronic regions (black line), intronic enhancer (DME-1 for dystrophin, DUE for utrophin, green boxes), untranslated first exon 1A of utrophin (yellow) are specified. Arrows indicate transcription start sites. (c) Schematic representation of the utrophin-A and B transcripts. The unique translated exon of the utrophin A transcript is 2A. Utrophin B transcript starts at exon 1B. Promoters A and B produce proteins that differ at their N-termini with, respectively, unique sections of 31 and 26 amino acids. Adapted from [Citation11]. Full color available online.
![Figure 2. Schematic organisation of the dystrophin (A), utrophin (B) genes with associated promoters and utrophin A and B transcripts (c). (a). Dystrophin expression is driven by at least 7 tissue-specific promoters. The full-length dystrophin transcription is controlled by 3 promoters (B, brain; M, muscle; P, Purkinje) localised upstream to the first exon and which only differs in its NH2-terminal sequences. 4 internal promoters, dp260, dp140, dp116 and dp71 were reported. Dp427m is the main variant produced in muscle and is involved in DMD and BMD. (b). In contrast to dystrophin, the utrophin gene has a long 5ʹprime untranslated region composed of 2 exons and a cluster of unmethylated, rare-cutting restriction enzyme sites. Expression of utrophin transcripts is controlled by two independently regulated promoters, A and B. Whereas each promoter results in a transcript with unique 5ʹ exons that splice into a common mRNA at exon 3 (black box), utrophin A promoter is CpG rich at the 5ʹ-end and contains an N-box motif controlling the synaptic expression. Different elements of the utrophin A promoter are specified. Promoter B, devoid of the N-box motif lies within the second exon of utrophin. 3 shorter isoforms Up140, G-Utrophin and Up71 were reported similar to the smaller transcripts of the dystrophin gene. Exons (grey boxes with exon numbers) and intronic regions (black line), intronic enhancer (DME-1 for dystrophin, DUE for utrophin, green boxes), untranslated first exon 1A of utrophin (yellow) are specified. Arrows indicate transcription start sites. (c) Schematic representation of the utrophin-A and B transcripts. The unique translated exon of the utrophin A transcript is 2A. Utrophin B transcript starts at exon 1B. Promoters A and B produce proteins that differ at their N-termini with, respectively, unique sections of 31 and 26 amino acids. Adapted from [Citation11]. Full color available online.](/cms/asset/5987e9c3-fa2e-40bb-abf3-71b2f0278f75/ieod_a_1438261_f0002_oc.jpg)
Figure 3. The domain composition of the full-length dystrophin protein (A), full-length utrophin A and B and various shorter isoforms (B). (a) Dystrophin contains an amino-terminal domain (NTD) followed by 24 spectrin like central rod domain, the cysteine rich (CRD) and the carboxy-terminal (CTD) domain. The full-length dystrophin proteins Dp427m, Dp427b and Dp427p are respectively expressed in skeletal, smooth and cardiac muscle, brain and Purkinje neuron. Dystrophin interacts with actin-F via two low affinity sites via the amino-terminal CH domain and spectrin repeats 11–17. Other binding sites are indicated. (b) The full-length utrophin A and B proteins present the same overall structure as the dystrophin full-length protein but have a shorter central rod domain and a 22 spectrin like domain. Utrophin is interacting with actin filaments through a unique continuous binding domain via the N-terminal domain and the spectrin 1–10. Utrophin A and B differ at their N-termini with unique sections of 31 and 26 amino acids, respectively. Various shorter utrophin isoform as Up140 and Up71 ubiquitously expressed were reporter as G-utrophin expressed in brain and sensory ganglia. Identity and similarity score per domain and expression patterns are specified. Data taken from [Citation6,Citation27].
![Figure 3. The domain composition of the full-length dystrophin protein (A), full-length utrophin A and B and various shorter isoforms (B). (a) Dystrophin contains an amino-terminal domain (NTD) followed by 24 spectrin like central rod domain, the cysteine rich (CRD) and the carboxy-terminal (CTD) domain. The full-length dystrophin proteins Dp427m, Dp427b and Dp427p are respectively expressed in skeletal, smooth and cardiac muscle, brain and Purkinje neuron. Dystrophin interacts with actin-F via two low affinity sites via the amino-terminal CH domain and spectrin repeats 11–17. Other binding sites are indicated. (b) The full-length utrophin A and B proteins present the same overall structure as the dystrophin full-length protein but have a shorter central rod domain and a 22 spectrin like domain. Utrophin is interacting with actin filaments through a unique continuous binding domain via the N-terminal domain and the spectrin 1–10. Utrophin A and B differ at their N-termini with unique sections of 31 and 26 amino acids, respectively. Various shorter utrophin isoform as Up140 and Up71 ubiquitously expressed were reporter as G-utrophin expressed in brain and sensory ganglia. Identity and similarity score per domain and expression patterns are specified. Data taken from [Citation6,Citation27].](/cms/asset/2e46230e-7810-4470-9e09-18013512d644/ieod_a_1438261_f0003_oc.jpg)
The full length 74 exons of the utrophin gene encode a protein of 3433 amino acids with a predicted molecular weight of 395 kDa and 80% amino acid identity to dystrophin () [Citation26]. Both proteins contain an actin-binding N-terminus, a triple coiled-coil spectrin repeat central region, and a C-terminus that consists of protein–protein interaction motifs which shares many binding partners, such as α-dystrobrevin-1, β-dystroglycan, and F-actin [Citation27]. However, the two proteins show some differences. Both proteins bind actin filaments with similar affinity. However, molecular contact with actin filaments is operated through a unique continuous binding domain for utrophin. By contrast, dystrophin utilizes two low-affinity distinct and spatially separated actin-binding modules [Citation28]. Utrophin also differs from dystrophin in its mode of interaction with microtubules [Citation29]. Furthermore, utrophin does not recruit neuronal nitrogen synthase (nNOS) to the sarcolemma to regulate blood flow to muscles [Citation30]. The significance of this observation is unclear; a recent study reported no relationship between the expression of nNOS at the sarcolemma and the disease severity in BMD patients [Citation31] who remain mildly affected and ambulant without nNOS membrane localization, suggesting compensatory nNOS pathways.
In summary, despite subtle differences, the structural similarities and relative conservation of dystrophin and utrophin led to the postulate that utrophin might be an effective surrogate to compensate for the lack of dystrophin in dystrophic muscles (.
2. Evidence for utrophin as an efficient surrogate for dystrophin
2.1. Utrophin expression in dystrophic muscles
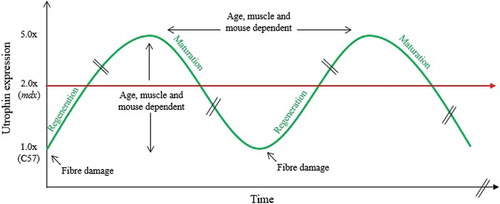
In the widely used mdx mouse model for DMD [Citation32], utrophin is increased by ~1.8/2.2-fold in muscle and localized at the sarcolemma of regenerating fibers [Citation33]. This repair mechanism also occurs in BMD and DMD patients with a respective ~2.5 and ~4-fold upregulation of utrophin protein [Citation34,Citation35]. Some studies reported that a small increase in utrophin delays the age when boys are wheel chair bound [Citation36] while others described no relationship [Citation37], or a counterintuitive negative correlation of utrophin expression with clinical severity [Citation38]. Often described as a regeneration-associated protein, utrophin levels need to be correlated with regeneration markers such as developmental mysosin and fiber size. The level of regeneration and utrophin vary between muscle types and are age dependent in both animals and DMD boys, emphasizing the complexity of quantifying utrophin in dynamic and dystrophic muscles (). Thus, the development of accurate utrophin and regeneration quantification are urgently needed to understand this dynamic and to study utrophin expression at the individual fiber level [Citation39,Citation40]. In addition, utrophin levels are also often complicated by the presence of mutant dystrophin in BMD patients and other mechanisms such as the stabilization of the protein complex at the muscle membrane might be involved.
Figure 4. Dynamic utrophin protein levels in mdx dystrophic muscles. Between the age of 3 and 10 weeks, dystrophin deficient muscles of the mdx mouse undergo many degeneration and regeneration cycles which contribute to the progressive muscle wasting. The dynamic utrophin level is variable and dependent on the muscle, the age and the animal and associated with high regeneration and repair processes emphasizing the complexity of quantifying utrophin in dynamic and dystrophic muscles.
Further evidence of the interrelationship between utrophin and dystrophin is provided by the fact that the mdx model presents with a mild dystrophic phenotype and a normal lifespan, whereas the absence of utrophin and dystrophin in doubly deficient dko animals worsen the skeletal and cardiac pathology. Whereas the utrophin-null mutant mice seem normal, the dko mouse displays many of the characteristics of DMD including severe muscle wasting, kyphosis, and a 4–20-week reduced lifespan [Citation41]. Absence of utrophin in a DMD boy also exacerbates the condition [Citation42].
2.2. Utrophin overexpression compensates the dystrophin loss
Evidence that utrophin can compensate for the lack of dystrophin was obtained through the generation of transgenic mdx mice expressing a truncated [Citation43] or full length utrophin gene [Citation44]. These studies established that a highly expressed truncated utrophin minigene only partially prevents the pathology, whereas expression of the full length utrophin gene suppresses functional signs of dystrophinopathy in a dose dependent way. This work also shows that the continuous localization of utrophin along the muscle membrane is essential and that low increases of utrophin (1.5×) can be beneficial. High levels of utrophin were recently shown to also rescue a large majority of serological mdx biomarkers [Citation45] and mitochondria defects in dystrophic muscles [Citation46]. Importantly, ubiquitous overexpression of utrophin in the mdx mouse is not toxic [Citation47] and utrophin therapy initiated after birth also prevents pathology. As with dystrophin, the earlier the administration of utrophin, the better the outcome is [Citation48]. These mouse studies strongly support utrophin as a functional surrogate for dystrophin and emphasize the potential of full length utrophin modulation for the therapy of DMD. Such an approach would be applicable to all DMD boys irrespective of their mutation.
3. Utrophin based strategies: preclinical development
Utrophin presents a complex pattern of transcriptional regulation and is subject to posttranscriptional modifications offering several avenues to develop therapeutic interventions. This section will describe the different pharmacological approaches, gene therapy solutions, and potential new avenues of interest to upregulate utrophin expression ().
Table 1. Status of the main and most promising utrophin therapeutic strategies for DMD.
3.1. Pharmacological utrophin activators
3.1.1. Utrophin modulation using small drugs
Transcriptional modulation of the utrophin A promoter using an orally bioavailable small molecule offering a systemic solution targeting skeletal and cardiac muscles for all DMD patients irrespective of their dystrophin mutation is an attractive strategy. In vitro screening using an immortalized mdx mouse H2K-mdx cell line carrying an 8.9-kb fragment of the human utrophin promoter linked to a luciferase reporter gene led to the discovery of the first orally bioavailable small molecule utrophin upregulator: ezutromid (formally SMT C1100), the active ingredient 5(ethylsulfonyl)-2-(naphthalen-2-yl) benzo[d]oxazole formulated for oral administration [Citation49,Citation50]. In vitro upregulation of utrophin in human cells dosed with ezutromid was confirmed at the mRNA and protein levels with, respectively, a significant 25% and 50% increase. In mdx animals, daily oral administration of ezutromid increased utrophin expression in skeletal, respiratory, and cardiac muscles, resulting in improved sarcolemmal stability. This expression resulted in a significant reduction in the key hallmarks of the disease such as regeneration, necrosis, and fibrosis which translated into physiological and muscle function improvements. Importantly, ezutromid treatment was shown to be efficient in exercised mdx mice and to act in combination and synergy with the approved gold standard steroid prednisolone treatment [Citation50]. As a first new pharmacological class of small molecules that act to modulate transcription of utrophin, ezutromid is currently in a Phase 2 efficacy trial in DMD boys (see Section 4). Other compounds, structurally related to ezutromid, have been developed and show efficacy in mdx animals validating the ezutromid drug series and strongly endorsing the therapeutic potential of utrophin modulation as a disease-modifying therapeutic strategy for all DMD patients irrespective of their dystrophin mutation [Citation51]. Whereas important developments in synthetic chemistry allow generation of thousands of new drug-like chemical entities in an automatic fashion, high-throughput screening (HTS) and subsequent medicinal chemical lead optimization offer great potential to identify novel pharmacological agents for utrophin transcriptional upregulation [Citation52]. The discovery of novel utrophin modulators depends on appropriate primary assays that reflect the in vivo situation as closely as possible. Robust secondary screens are also essential. HTSs have now been developed where the reporter gene is knocked into the utrophin locus allowing screening for small molecules which modulate the utrophin promoter in its genomic context. These new HTSs are providing new chemical leads which should deliver a new generation of utrophin modulators with the objective to define a best in class utrophin small molecule for DMD [Citation53].
With the characterization of several transcriptional regulatory elements in the utrophin A promoter – including MyoD/myogenin and E-box; N-Box and GA-binding protein; calcineurin/nuclear factor of activated T cells (NFAT), Ets-2 repressor factor, and E2F1; peroxisome proliferator-activated receptor-β/δ, peroxisome proliferator-activated receptor ϒ coactivator 1α (PGC1α); and sirtuin 1 (Sirt1) – a range of other agents has been also reported to modulate utrophin expression and to be efficient in mdx mice. Heregulin acts via the N-box motif of the utrophin A promoter and GAPB via an epigenetic regulation through activation of MSK1/2 and phosphorylation of histone H3 in an ERK dependent manner [Citation54–Citation56]. Recombinant interleukin-6 induces overexpression of utrophin the extrasynaptic sarcolemma via the activation of NRG-1/ErbB [Citation57]. The active form RhoAV14 of the GTPase RhoA also increases utrophin expression and stability and leads to an extrasynaptic distribution and re-localization of utrophin protein along the sarcolemma in mice [Citation58]. Pharmacological activators of the NO pathway such as l-arginine and hydroxyurea [Citation59] represent other pathways which induce utrophin levels. However, safety concerns and lack of cardiac benefits were reported after l-arginine long treatment.
3.1.2. Promotion of the slow and oxidative phenotype
Fiber-type composition and muscle structure influence utrophin expression [Citation60] independently from muscle regeneration [Citation61]. In slow oxidative type I myofibers, utrophin levels are elevated [Citation62] and may contribute to the highest resistance of slow muscle to contraction-induced damage [Citation63]. Concomitant with elevated utrophin A expression, a shift of fast and glycolytic myofibers toward a slow and oxidative myofibers program may be obtained with different pharmacological agents such as GW501516 which target the PPARδ-signaling pathways [Citation64], and 5-amino-4-imidazolecarboxamide riboside (AICAR) [Citation65], metformin [Citation66], quercetin [Citation67], and resveratrol [Citation68] which respectively act through the 5′ adenosine monophosphate-activated protein kinase, and Sirt1 and the PGC1α axis. In addition to transcriptional regulatory pathways, promotion of a slower and more oxidative phenotype can be also obtained via post-transcriptional events occurring at the 3′UTR and 5′UTR of utrophin A. Heparin treatment activates p38 MAP kinase and mediates a sequestration of the RNA-binding protein KSRP by the regulatory protein 14-3-3 resulting in ∼1.5–3-fold increase of the utrophin protein following posttranscriptional stabilization of the utrophin mRNA [Citation69]. However, excessive fiber-type switching may affect muscle function and could be inefficient in some human muscles with high content of slow type I fibers such as the diaphragm muscle. Interestingly, using different mechanisms of action, heparin and AICAR treatment act in combination and result in an additive effect on utrophin A expression. Molecular factors controlling fiber-type specification remain to be defined in detail and could offer new targets to increase utrophin expression by promotion of the slow and oxidative phenotype. For instance, transgenic mice overexpressing FHL1 show an increase of utrophin expression by promotion of the slow oxidative phenotype due to the activation of the calcineurin/NFATc1 signaling [Citation70]. Ablation of the heterotrimeric G proteins α13 (Gα13) [Citation71] or overexpression of some member of the Ephrin family are able to reprogram myofibers to an oxidative phenotype [Citation72] and could lead to new therapeutic targets.
3.2. Utrophin protein-based strategies
Direct protein replacement, inhibition of the protein degradation, and stabilization of the utrophin protein complex are alternative attractive strategies. Direct utrophin protein replacement using chimeric proteins encoding the protein transduction domain of the HIV-1 TAT protein fused to full-length TAT-Utr and a truncated TAT-μUtr are effective in transducing skeletal and cardiac muscles, brain, and liver [Citation73]. Cell-penetrating recombinant TAT-μUtr expressed using the baculovirus system restores a proper membrane, stabilizes muscle membrane integrity and mitigates the pathophysiology of dystrophin and utrophin double-knockout mice [Citation74]. Nevertheless, despite promising preclinical results, no further progress has been reported. Another interesting avenue is the use of calpain inhibitors to prevent utrophin protein cleavage [Citation75]. Nonetheless, despite some reports describing delay of muscle degeneration and necrosis in mdx mice after calpain inhibition [Citation76], a leupeptin-derived calpain inhibitor fails to ameliorate the severe muscle pathology in the DMD GRMD dog model [Citation77]. Finally, biglycan, a small leucine-rich proteoglycan, involved in the maintenance of muscle cell integrity, directs the assembly of a functional utrophin-associated complex and regulates the stabilization of utrophin and nNOs at the sarcolemma [Citation78]. As the recombinant human biglycan results in utrophin protein increase without transcriptional change and is inefficient in dko mice, benefits arise from posttranscriptional effects on utrophin protein. Following encouraging preclinical results with systemic dose of recombinant human biglycan, Tivorsan Pharamceutics manufactured a highly purified formulation of TVN-102 and is on track to file an IND and enter the clinic in 2018.
3.3. Utrophin gene therapies
In 1990, the description of a 61-year-old Becker patient still ambulant and presenting with a very mild muscular dystrophy associated with the deletion of 46% of dystrophin [Citation7] paved the way of gene-therapy development to treat DMD using a gene coding for a truncated dystrophin or utrophin delivered by adeno-associated virus (AAV) or recombinant adeno-associated viral (rAAV) [Citation79]. Initial proof of principle of adenovirus-mediated utrophin transfer presented a viable and nontoxic viral strategy in mouse able to mitigate the dystrophic phenotype of mdx muscles [Citation80]. Using an optimized (DeltaR4-R21/DeltaCT) microutrophin systemic delivery through a rAAV2/6, Odom and colleagues showed that gene therapy using an utrophin mini-gene could also alleviate most of the pathophysiological abnormalities associated with muscular dystrophy in the more severe dko mouse [Citation81]. The dystrophic phenotype of GRMD dogs is also mitigated by adenovirus-mediated utrophin gene transfer [Citation82]. However, despite great promise, the major challenges facing AAV gene delivery are the high titer necessary to achieve systemic transfection in human and the immune reaction against the AAV vector preventing readministration with the same muscle-tropic serotype. An immune response against the exogenously administered transgene potentially perceived as a neoantigen may lead to destruction of transduced myofibers by immune effector cells. Furthermore, limitation of the effective packaging capacity with rAAV (−4.5 kb) only offers the possibility to deliver a highly truncated utrophin, previously described as less efficient than the full-length gene. A direct, head-to-head comparison of a 6.3-kb dystrophin minigene (AdV-Dys) with a 6-kb utrophin minigene (AdVUtr) gene transfer in immunocompetent mdx mice concluded that dystrophin and utrophin are largely equivalent in their intrinsic abilities to prevent the development of the disease [Citation83]. Nevertheless, the efficacy of dystrophin gene replacement may be impaired by immunity against the exogenous dystrophin, whereas utrophin, expressed in dystrophic tissues, offers a larger margin of efficacy with higher production of force as well as safety. Whereas subcellular localization of dystrophin subdomains was recently described as different in skeletal and cardiac muscles offering critical insights to develop new optimized mini-gene versions [Citation84], clinical trials from Solid Biosciences [Citation85] and Pfizer [Citation86] with optimized micro-dystrophin are about to start. With the novel rationally designed AAV microutrophin vector able to recruit nNOS to the sarcolemma [Citation87], utrophin gene therapy may offer great promise to treat DMD.
A number of other utrophin-based viral strategies using alternative mechanisms recently opened new avenues to obtain a therapeutic strategy for treatment of DMD. This includes the artificial zinc finger transcription factor ‘Jazz’ and subsequent ‘UtroUp’ [Citation88] able to recognize specific domains of the utrophin A promoter to transcriptionally drive utrophin expression. After treatment with a mAAV8-Jazz, a 1.5–2.0-fold increase of utrophin protein in skeletal and cardiac muscles results in significant histological and functional recovery of the dystrophic phenotype [Citation89]. Another interesting viral strategy is the overexpression of the cytotoxic T-cell GalNAc transferase (GALGT2). Increased level Galgt2 leads to the α-dystroglycan glycosylation resulting in cell-surface expression of utrophin by increasing transportation of utrophin-α-DG from endoplasmic reticulum/Golgi membranes and the stabilization of the utrophin protein complex at the sarcolemma [Citation90]. Similarly, it was noted that overexpression of sarcospan increases GalNAc modifications and enhances transportation of utrophin-DG at the sarcolemma [Citation91]. Despite some utrophin-independent mechanisms, Galgt2 gene therapy inhibits muscular dystrophy in mdx mice [Citation92]. Following positive reviews, complete toxicology safety studies, and submission of an IND to the FDA, a Phase 1/2a gene transfer clinical trial to deliver rAAVrh74.MCK.GALGT2 for DMD is enrolling participants by invitation only (NCT03333590).
3.4. New therapeutic avenues
An alternative novel and promising mechanism is the repressive function of some microRNAs (let-7c, miR-150, miR-196b, miR-296-5p, miR-133b) acting on the 3ʹUTR of utrophin mRNA [Citation93]. Using a 2ʹ-o-methyl oligonucleotide to disrupt the site of interaction of Let-7c on the utrophin 3ʹUTR, Mishra and colleagues reported a two to threefold increase of utrophin protein expression in mdx skeletal muscles associated with histological benefits and specific force improvement [Citation94]. Of importance, another recent publication noted that miR-206 is acting as an important transacting factor capable of stimulating skeletal muscle regeneration and that overexpression of this miR results in increased utrophin levels [Citation95]. This study highlights the pleiotropic and complex regulation of miRs and their effects on utrophin mRNA.
Other alternative and exploratory avenues to consider are the use of skeletal muscle resident protease inhibitors such as Serpina3n which stabilizes the sarcolemma [Citation96] and the use of modulators of the ER stress occurring in dystrophic muscles. Overexpression of thrombospondin-4 induces ATF6α, a part of the unfolded protein response, which selectively enhances vesicular trafficking of dystrophin–glycoprotein and integrin attachment complexes to stabilize sarcolemmal protein such as utrophin [Citation97].
4. Clinical trials in DMD patients
4.1. Utrophin modulation: ezutromid
Summit Therapeutics is developing the small molecule utrophin modulator, ezutromid, for the treatment of DMD. The potential of ezutromid as a universal treatment approach for DMD has been demonstrated in preclinical studies (see Section 3) and these data have led to evaluation in human clinical trials. The clinical program to date has comprised Phase 1 studies in healthy volunteers [Citation98] and patients with DMD [Citation99] and an ongoing Phase 2 proof-of-concept study called PhaseOut DMD.
4.1.1. Pharmacokinetics, safety, and tolerability of ezutromid
Ezutromid has a complex pharmacokinetic profile. Overcoming the challenge of reaching systemic exposure levels anticipated to be sufficient for the modulation of utrophin was achieved though formulation development and gaining a greater understanding the impact of diet has on drug absorption as part of a rigorous Phase 1 clinical program.
Two formulations of ezutromid have emerged: A microfluidized aqueous oral suspension formulation called F3 and a powder for suspension formulation called F6.
Evaluation of the F3 formulation in Phase 1 trials revealed a consistent theme of variable exposure. This was observed in all subjects, with this lower in pediatric DMD patients than those in healthy volunteers. It was noted that some of the DMD patients were on a low-fat diet purportedly to control weight gain whilst on concomitant corticosteroid treatment. The impact of administering ezutromid with 100 mL of whole (full fat) milk combined with a balanced diet was duly examined; exposure was substantially improved and it was determined to advance F3 into Phase 2 testing.
In parallel formulation, development work was undertaken with the aim to further optimize the pharmaceutical characteristics. This resulted in the identification of formulation F6. Phase 1 testing of F6 showed that administration was undertaken at lower doses compared to those of F3 from prior Phase 1 trials. Despite these lower doses, F6 achieved a greater than sixfold increase in maximum plasma levels in DMD patients compared to that seen with F3. Consequently, F6 was also taken forward to be investigated in Phase 2 trials.
It is expected that utrophin modulation will be a life-long treatment for patients with DMD. In all Phase 1 studies, ezutromid was shown to be safe and well tolerated. With formulation F3, no serious or severe adverse events were reported. Adverse events were primarily gastrointestinal, with pale feces accounting for the majority of events, and most were considered mild by the investigator.
Formulation F6 was generally safe and well tolerated in both healthy volunteers and patients. The majority of the treatment emergent adverse events (TEAE) were gastrointestinal in nature, with feces discolored most frequently reported in healthy volunteers and diarrhea the most frequently reported in patients. All but one of the AEs were mild or moderate. In patients there was one SAE (considered severe and possibly related) of elevated liver function test abnormalities with the 250-mg BID dose. The patient was clinically well but discontinued from the study and his liver function tests returned to normal 10 days after drug discontinuation. Overall, the safety findings were consistent with those from preclinical studies and showed that ezutromid had an unremarkable safety profile.
4.1.2. PhaseOut DMD, a Phase 2 clinical trial of ezutromid in patients with DMD
PhaseOut DMD is a Phase 2, open-label, multicenter study of the activity and safety of the F3 and F6 formulations of ezutromid. A total of 40 ambulatory boys with DMD aged between their fifth and tenth birthday have been enrolled. The inclusion criteria are shown in as in the overall study design ().
Table 2. Inclusion and exclusion criteria in PhaseOut DMD trial with ezutromid.
Figure 5. Overview of the PhaseOut DMD, a Phase 2 Clinical Trial of Ezutromid in Patients with DMD.
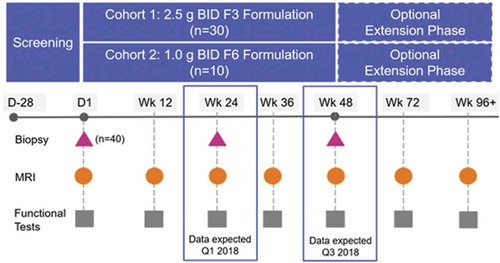
The 24-week interim analysis of this study recently revealed that treatment with ezutromid increases utrophin protein expression and significantly reduced muscle damage in DMD patients with a reduction in developmental myosin, a biomarker of muscle damage found in repairing fibers [Citation100]. These promising interim data suggest that ezutromid may slow the relentless cycle of muscle fiber degeneration and regeneration and provide the first evidence of utrophin modulation working in DMD patients. Longer term treatment with ezutromid could achieve meaningful functional benefits for patients living with DMD and the final 48-week readout is expected in the third quarter of 2018. An external data monitoring committee has supported the progress of this study. Following completion of the 48 weeks, patients are eligible for an extension study and can receive ezutromid until approval or study drug discontinuation.
Clinical trials in DMD are challenging for the following reasons: (a) disease progression does not follow a linear course with rapid declines in functional measures seen at key milestones such as loss of ambulation; (b) the DMD population, including all genotypes, is heterogeneous leading to high variability; (c) DMD is a rare disease making clinical trials challenging to recruit from both a patient and site perspective; and (d) clinical function tests are dependent on patient collaboration, motivation, and attention. The number of validated endpoints in clinical studies is currently limited, and EMA and FDA guidance encourages the development of new and suitable biomarkers that could serve as primary or key secondary end points in clinical studies.
The primary end point in PhaseOut DMD is using quantitative MRI (qMRI) to measure fat fraction, the percentage of fat within muscle tissue (% fat fraction), as this has been studied as a marker of DMD severity and progression. In contrast to functional tests, qMRI is less dependent on patient collaboration and is thought to show good sensitivity and reproducibility in order to detect disease progression [Citation101]. In addition to qMRI measures of fat fraction, PhaseOut DMD will also analyze imaging-based T2 values which are directly related to muscle damage and inflammation.
All patients in PhaseOut DMD will have a muscle biopsy taken at baseline and a second after either 24 or 48 weeks of dosing. The muscle biopsies will be assessed by an automated imaging method to quantify the intensity of utrophin staining at the muscle fiber membrane, the staining of developmental heavy chain myosin (MHCd) in the cytoplasm, and myofiber diameter within a muscle biopsy section. The number and intensity of MHCd positive fibers and the fiber size distribution within a biopsy sample are reflective of levels of muscle fiber regeneration and are postulated as a surrogate for disease pathology in muscular dystrophy.
A variety of functional end points including 6-min walk distance and North Star Ambulatory Assessment are measured at regular intervals throughout the study, including the extension phase.
5. Expert opinion
Nearly 30 years after the discovery of the utrophin gene, the characterization of the gene and protein organization revealed a high homology to dystrophin, disrupted in DMD. Utrophin is an autosomal, structural, and functional paralog of dystrophin able to compensate for the lack of dystrophin in different animal models of DMD. Utrophin modulation approach has the potential to offer a universal treatment to all DMD patients regardless of their genetic defect. Despite a high level of structural identity, utrophin and dystrophin differ in their spatiotemporal expression. Utrophin is ubiquitously expressed mainly in embryonic tissue and is primarily restricted to the neuromuscular junction in adult muscle. Utrophin is also expressed in cardiac fibers and in regenerating skeletal muscle fibers. Dystrophin is expressed in the skeletal, cardiac, and smooth muscles in adult. Some sequence differences between the two proteins underlie functional differences in binding partners and explain that utrophin may not be able to recapitulate all the functions of dystrophin. Thus, utrophin modulation strategy may lead to a mild BMD phenotype. The ongoing clinical trials of utrophin modulation will test this hypothesis. This approach to therapy is still very attractive since both exon skipping and AAV-mediated gene therapy with dystrophin minigenes will also lead to a BMD phenotype.
The better understanding of the regulation of the utrophin gene is opening up new strategies for therapeutic intervention. Utrophin transcription can be activated at multiple levels via transcription factors, intracellular signaling cascades, or through extracellular growth factors. Utrophin levels can also be increased by mRNA and protein stabilization. Other strategies which modulate utrophin stabilization such as promotion of the slow and oxidative phenotype are also being explored. Drugs promoting utrophin modulation via different mechanisms could be used in synergy and in combination with other strategies targeting primary or secondary defects of the disease as their effects would be predicted to be additive.
High-throughput screens have been successfully developed to identify utrophin modulators and the first chemical leads are emerging. The first of these, ezutromid, has shown efficacy in the mdx model and shown to be safe in healthy volunteers and DMD patients by Summit Therapeutics plc. This first in class utrophin modulator is currently in a Phase 2 trial in DMD boys and interim 24-week data showed that ezutromid increases utrophin and significantly reduces muscle damage in patients living with DMD. With the development and improvements in synthetic chemistry, new HTS mimicking the in vivo chromatin context of utrophin gene, and more robust secondary assays, new generation of small modulators drugs are under development to define a best in class small utrophin modulator.
Despite clear advances and high expectations in the field, some challenges remain. First, the absolute level of utrophin levels is complicated to interpret because of regeneration and others mechanisms such as stabilization, which differ according to muscle type, as well as the timing due to specific regeneration/degeneration cycles in dystrophic muscle. As utrophin is increased in dystrophic muscles in part due to the higher regeneration process, it is essential to correlate the utrophin levels after drug treatment with a panel of regeneration markers. An efficient small utrophin modulator will lead to a reduction in regeneration, lowering the initial increase of utrophin and markers of regeneration. Thus, the development of accurate tools to quantify utrophin levels in correlation with regeneration at the individual fiber level rather than in the muscle tissue as a whole will be essential. Second, as a clinically effective utrophin modulator would promote the expression of utrophin continuously along the muscle fibers, these tools will help to define homogeneity of the utrophin signal across the muscle.
Foundation of utrophin-based therapies defined 20 years ago recently delivered the first promising results in DMD patients with ezutromid. We are at an exciting stage on the road of a universal treatment for all DMD patients.
Article highlights
Utrophin is an autosomal and functional paralogue of dystrophin able to compensate for the primary defect in Duchenne Muscular dystrophy.
A utrophin based strategy targeting all muscles has the potential to offer a universal treatment to all DMD patients irrespective of their genetic defect.
The control of utrophin expression at both the transcriptional and post-transcriptional levels provides several avenues to develop therapeutic interventions which are potentially complementary.
Utrophin levels are dynamic in adult muscle where they are muscle type and time dependent. The development of accurate utrophin and regeneration quantification is urgently needed.
Ezutromid is the first in class small molecule utrophin modulator currently in Phase 2 clinical trial. After 24 weeks of treatment, utrophin is increased and muscle damage significantly reduce in DMD patients.
Novel chemical series of small utrophin modulators are currently under development.
This box summarizes key points contained in the article.
Declaration of interest
K Davies is a founder and shareholder of Summit Therapeutics plc. D Roblin is employed by and is a shareholder of Summit Therapeutics plc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Acknowledgments
The authors apologize to all the authors whose work has not been cited due to space restrictions and thank all the collaborators who made possible the completion of the work reviewed here.
Additional information
Funding
Related Research Data
References
- Guiraud S, Aartsma-Rus A, Vieira NM, et al. The pathogenesis and therapy of muscular dystrophies. Annu Rev Genomics Hum Genet. 2015;16:281–308.
- Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015 Apr;36(4):395–402.
- Monaco AP, Neve RL, Colletti-Feener C, et al. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986 Oct 16–22;323(6089):646–650.
- Bach JR, O’Brien J, Krotenberg R, et al. Management of end stage respiratory failure in Duchenne muscular dystrophy. Muscle Nerve. 1987 Feb;10(2):177–182.
- Hoffman EP, Brown RH Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987 Dec 24;51(6):919–928.
- Blake DJ, Weir A, Newey SE, et al. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002 Apr;82(2):291–329.
- England SB, Nicholson LVB, Johnson MA, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343(6254):180–182.
- Emery AE. Dystrophin function. Lancet. 1990 May 26;335(8700):1289.
- Guiraud S, Davies KE. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr Opin Pharmacol. 2017 May;6(34):36–48.
- Guiraud S, Chen H, Burns DT, et al. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp Physiol. 2015 Dec;100(12):1458–1467.
- Khurana TS, Davies KE. Pharmacological strategies for muscular dystrophy. Nat Rev Drug Discov. 2003 May;2(5):379–390.
- Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477–487.
- Niks EH, Aartsma-Rus A. Exon skipping: a first in class strategy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2017 Feb;17(2):225–236.
- Robinson-Hamm JN, Gersbach CA. Gene therapies that restore dystrophin expression for the treatment of Duchenne muscular dystrophy. Hum Genet. 2016 Sep;135(9):1029–1040.
- Love DR, Hill DF, Dickson G, et al. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. 1989 May 4;339(6219):55–58.
- Pearce M, Blake DJ, Tinsley JM, et al. The utrophin and dystrophin genes share similarities in genomic structure. Hum Mol Genet. 1993 Nov;2(11):1765–1772.
- Tome FM, Matsumura K, Chevallay M, et al. Expression of dystrophin-associated glycoproteins during human fetal muscle development: a preliminary immunocytochemical study. Neuromuscular Disorders. 1994 Jul;4(4):343–348.
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003 Dec;2(12):731–740.
- Clerk A, Morris GE, Dubowitz V, et al. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J. 1993 Aug;25(8):554–561.
- Weir AP, Burton EA, Harrod G, et al. A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J Biol Chem. 2002 Nov 22;277(47):45285–45290.
- Burton EA, Tinsley JM, Holzfeind PJ, et al. A second promoter provides an alternative target for therapeutic up-regulation of utrophin in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 1999 Nov 23;96(24):14025–14030.
- Dennis CL, Tinsley JM, Deconinck AE, et al. Molecular and functional analysis of the utrophin promoter. Nucleic Acids Res. 1996 May 1;24(9):1646–1652.
- Wilson J, Putt W, Jimenez C, et al. Up71 and up140, two novel transcripts of utrophin that are homologues of short forms of dystrophin. Hum Mol Genet. 1999 Jul;8(7):1271–1278.
- Sadoulet-Puccio HM, Kunkel LM. Dystrophin and its isoforms. Brain Pathology. 1996 Jan;6(1):25–35.
- Blake DJ, Schofield JN, Zuellig RA, et al. G-utrophin, the autosomal homologue of dystrophin Dp116, is expressed in sensory ganglia and brain. Proc Natl Acad Sci USA. 1995 Apr 25;92(9):3697–3701.
- Tinsley JM, Blake DJ, Roche A, et al. Primary structure of dystrophin-related protein. Nature. 1992 Dec 10;360(6404):591–593.
- Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007 Feb;1772(2):108–117.
- Rybakova IN, Humston JL, Sonnemann KJ, et al. Dystrophin and utrophin bind actin through distinct modes of contact. J Biol Chem. 2006 Apr 14;281(15):9996–10001.
- Belanto JJ, Mader TL, Eckhoff MD, et al. Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci USA. 2014 Apr 15;111(15):5723–5728.
- Li D, Bareja A, Judge L, et al. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci. 2010 Jun 15;123(Pt 12):2008–2013.
- van den Bergen JC, Wokke BH, Hulsker MA, et al. Studying the role of dystrophin-associated proteins in influencing Becker muscular dystrophy disease severity. Neuromuscular Disorders. 2015 Mar;25(3):231–237.
- Bulfield G, Siller WG, Wight PA, et al. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984 Feb;81(4):1189–1192.
- Schofield JN, Gorecki DC, Blake DJ, et al. Dystroglycan mRNA expression during normal and mdx mouse embryogenesis: a comparison with utrophin and the apo-dystrophins. Dev Dyn. 1995 Oct;204(2):178–185.
- Helliwell TR, Man NT, Morris GE, et al. The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromuscular Disorders. 1992;2(3):177–184.
- Arechavala-Gomeza V, Kinali M, Feng L, et al. Immunohistological intensity measurements as a tool to assess sarcolemma-associated protein expression. Neuropathol Appl Neurobiol. 2010 Jun;36(4):265–274.
- Kleopa KA, Drousiotou A, Mavrikiou E, et al. Naturally occurring utrophin correlates with disease severity in Duchenne muscular dystrophy. Hum Mol Genet. 2006 May 15;15(10):1623–1628.
- Vainzof M, Passos-Bueno MR, Man N, et al. Absence of correlation between utrophin localization and quantity and the clinical severity in Duchenne/Becker dystrophies. Am J Med Genet. 1995 Sep 25;58(4):305–309.
- van den Bergen JC. Schade van Westrum SM, Dekker L, et al. Clinical characterisation of Becker muscular dystrophy patients predicts favourable outcome in exon-skipping therapy. J Neurol Neurosurg Psychiatry. 2014 Jan;85(1):92–98.
- Janghra N, Morgan JE, Sewry CA, et al. Correlation of utrophin levels with the dystrophin protein complex and muscle fibre regeneration in duchenne and becker muscular dystrophy muscle biopsies. PloS One. 2016;11(3):e0150818.
- Wilson K, Faelan C, Patterson-Kane JC, et al. Duchenne and becker muscular dystrophies: a review of animal models, clinical end points, and biomarker quantification. Toxicol Pathol. 2017 Oct;45(7):961–976.
- Deconinck AE, Rafael JA, Skinner JA, et al. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997 Aug 22;90(4):717–727.
- Chevron MP, Echenne B, Demaille J. Absence of dystrophin and utrophin in a boy with severe muscular dystrophy. N Engl J Med. 1994 Oct 27;331(17):1162–1163.
- Deconinck N, Tinsley J, De Backer F, et al. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med. 1997 Nov;3(11):1216–1221.
- Tinsley J, Deconinck N, Fisher R, et al. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998 Dec;4(12):1441–1444.
- Guiraud S, Edwards B, Squire SE, et al. Identification of serum protein biomarkers for utrophin based DMD therapy. Sci Rep. 2017 Mar;2(7):43697.
- Kennedy TL, Moir L, Hemming S, et al. Utrophin influences mitochondrial pathology and oxidative stress in dystrophic muscle. Skeletal Muscle. 2017 Oct 24;7(1):22.
- Fisher R, Tinsley JM, Phelps SR, et al. Non-toxic ubiquitous over-expression of utrophin in the mdx mouse. Neuromuscul Disord. 2001;11(8):713–721.
- Squire S, Raymackers JM, Vandebrouck C, et al. Prevention of pathology in mdx mice by expression of utrophin: analysis using an inducible transgenic expression system. Hum Mol Genet. 2002 Dec 15;11(26):3333–3344.
- Chancellor DR, Davies KE, De Moor O, et al. Discovery of 2-arylbenzoxazoles as upregulators of utrophin production for the treatment of Duchenne muscular dystrophy. J Med Chem. 2011 May 12;54(9):3241–3250.
- Tinsley JM, Fairclough RJ, Storer R, et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PloS One. 2011;6(5):e19189.
- Guiraud S, Squire SE, Edwards B, et al. Second-generation compound for the modulation of utrophin in the therapy of DMD. Hum Mol Genet. 2015 Aug 1; 24(15):4212–4224.
- Moorwood C, Lozynska O, Suri N, et al. Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. PloS One. 2011;6(10):e26169.
- Wynne G, Uorinen A, Emer E, et al. Discovery of small molecule utrophin modulators for the therapy of Duchenne muscular dystrophy. World Muscle Society Conference, Saint Malo France. 2017.
- Gramolini AO, Angus LM, Schaeffer L, et al. Induction of utrophin gene expression by heregulin in skeletal muscle cells: role of the N-box motif and GA binding protein. Proc Natl Acad Sci USA. 1999 Mar 16;96(6):3223–3227.
- Basu U, Gyrd-Hansen M, Baby SM, et al. Heregulin-induced epigenetic regulation of the utrophin-A promoter. FEBS Lett. 2007 Sep 4;581(22):4153–4158.
- Krag TO, Bogdanovich S, Jensen CJ, et al. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc Natl Acad Sci USA. 2004 Sep 21;101(38):13856–13860.
- Fujimori K, Itoh Y, Yamamoto K, et al. Interleukin 6 induces overexpression of the sarcolemmal utrophin in neonatal mdx skeletal muscle. Hum Gene Ther. 2002 Mar 1;13(4):509–518.
- Gauthier-Rouviere C, Bonet-Kerrache A. RhoA leads to up-regulation and relocalization of utrophin in muscle fibers. Biochem Biophys Res Commun. 2009 Jul 3;384(3):322–328.
- Chaubourt E, Fossier P, Baux G, et al. Nitric oxide and l-arginine cause an accumulation of utrophin at the sarcolemma: a possible compensation for dystrophin loss in Duchenne muscular dystrophy. Neurobiol Dis. 1999 Dec;6(6):499–507.
- Banks GB, Combs AC, Odom GL, et al. Muscle structure influences utrophin expression in mdx mice. PLoS Genet. 2014 Jun;10(6):e1004431.
- Weir AP, Morgan JE, Davies KE. A-utrophin up-regulation in mdx skeletal muscle is independent of regeneration. Neuromuscular Disorders. 2004 Jan;14(1):19–23.
- Chakkalakal JV, Stocksley MA, Harrison MA, et al. Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc Natl Acad Sci USA. 2003 Jun 24;100(13):7791–7796.
- Consolino CM, Brooks SV. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J Appl Physiol. 2004 Feb;96(2):633–638.
- Miura P, Chakkalakal JV, Boudreault L, et al. Pharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum Mol Genet. 2009 Dec 1;18(23):4640–4649.
- Ljubicic V, Miura P, Burt M, et al. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet. 2011 Sep 01;20(17):3478–3493.
- Ljubicic V, Jasmin BJ. Metformin increases peroxisome proliferator-activated receptor gamma Co-activator-1alpha and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve. 2015 Jul;52(1):139–142.
- Ballmann C, Hollinger K, Selsby JT, et al. Histological and biochemical outcomes of cardiac pathology in mdx mice with dietary quercetin enrichment. Exp Physiol. 2015 Jan;100(1):12–22.
- Gordon BS, Delgado Diaz DC, Kostek MC. Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of Duchenne muscular dystrophy. Clin Nutrition. 2013 Feb;32(1):104–111.
- Peladeau C, Ahmed A, Amirouche A, et al. Combinatorial therapeutic activation with heparin and AICAR stimulates additive effects on utrophin A expression in dystrophic muscles. Hum Mol Genet. 2016 Jan 01;25(1):24–43.
- D’Arcy CE, Feeney SJ, McLean CA, et al. Identification of FHL1 as a therapeutic target for Duchenne muscular dystrophy. Hum Mol Genet. 2014 Feb 1;23(3):618–636.
- Koo JH, Kim TH, Park SY, et al. Galpha13 ablation reprograms myofibers to oxidative phenotype and enhances whole-body metabolism. J Clin Invest. 2017 Oct 2;127(10):3845–3860.
- Stark DA, Coffey NJ, Pancoast HR, et al. Ephrin-A3 promotes and maintains slow muscle fiber identity during postnatal development and reinnervation. J Cell Biol. 2015 Dec 7;211(5):1077–1091.
- Sonnemann KJ, Heun-Johnson H, Turner AJ, et al. Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS Med. 2009 May 26;6(5):e1000083.
- Call JA, Ervasti JM, Lowe DA. TAT-muUtrophin mitigates the pathophysiology of dystrophin and utrophin double-knockout mice. J Appl Physiol. 2011 Jul;111(1):200–205.
- Courdier-Fruh I, Briguet A. Utrophin is a calpain substrate in muscle cells. Muscle Nerve. 2006 Jun;33(6):753–759.
- Badalamente MA, Stracher A. Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve. 2000 Jan;23(1):106–111.
- Childers MK, Bogan JR, Bogan DJ, et al. Chronic administration of a leupeptin-derived calpain inhibitor fails to ameliorate severe muscle pathology in a canine model of duchenne muscular dystrophy. Front Pharmacol. 2011;2:89.
- Amenta AR, Yilmaz A, Bogdanovich S, et al. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci USA. 2011 Jan 11;108(2):762–767.
- Chamberlain JR, Chamberlain JS. Progress toward gene therapy for duchenne muscular dystrophy. Mol Therapy. 2017 May 3;25(5):1125–1131.
- Gilbert R, Nalbantoglu J, Petrof BJ, et al. Adenovirus-mediated utrophin gene transfer mitigates the dystrophic phenotype of mdx mouse muscles. Hum Gene Ther. 1999 May 20;10(8):1299–1310.
- Odom GL, Gregorevic P, Allen JM, et al. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol Therapy. 2008 Sep;16(9):1539–1545.
- Cerletti M, Negri T, Cozzi F, et al. Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther. 2003 May;10(9):750–757.
- Ebihara S, Guibinga GH, Gilbert R, et al. Differential effects of dystrophin and utrophin gene transfer in immunocompetent muscular dystrophy (mdx) mice. Physiol Genomics. 2000 Sep 8;3(3):133–144.
- Zhao J, Kodippili K, Yue Y, et al. Dystrophin contains multiple independent membrane-binding domains. Hum Mol Genet. 2016 Sep 1;25(17):3647–3653.
- Biosciences S. Letter to duchenne community about solid’s first clinical trial. 2017. Available from: https://solidbiocom/content/letter-duchennecommunity-about-solid-s-first-clinical-trial
- Pfizer. A study to evaluate the safety and tolerability of PF-06939926 gene therapy in duchenne muscular dystrophy. 2017. Available from: https://clinicaltrialsgov/ct2/show/NCT03362502
- Lai Y, Zhao J, Yue Y, et al. A novel rationally designed AAV micro-utrophin vector recruits nNOS to the sarcolemma. 15th Annual Meeting of the American-Society-of-Gene-and-Cell-Therapy 2012;20.
- Onori A, Pisani C, Strimpakos G, et al. UtroUp is a novel six zinc finger artificial transcription factor that recognises 18 base pairs of the utrophin promoter and efficiently drives utrophin upregulation. BMC Mol Biol. 2013 Jan 30;14:3.
- Strimpakos G, Corbi N, Pisani C, et al. Novel adeno-associated viral vector delivering the utrophin gene regulator jazz counteracts dystrophic pathology in mdx mice. J Cell Physiol. 2014 Sep;229(9):1283–1291.
- Durko M, Allen C, Nalbantoglu J, et al. CT-GalNAc transferase overexpression in adult mice is associated with extrasynaptic utrophin in skeletal muscle fibres. J Muscle Res Cell Motil. 2010 Sep;31(3):181–193.
- Gibbs EM, Marshall JL, Ma E, et al. High levels of sarcospan are well tolerated and act as a sarcolemmal stabilizer to address skeletal muscle and pulmonary dysfunction in DMD. Hum Mol Genet. 2016 Oct 24.
- Xu R, Camboni M, Martin PT. Postnatal overexpression of the CT GalNAc transferase inhibits muscular dystrophy in mdx mice without altering muscle growth or neuromuscular development: evidence for a utrophin-independent mechanism. Neuromuscular Disord. 2007 Mar;17(3):209–220.
- Basu U, Lozynska O, Moorwood C, et al. Translational regulation of utrophin by miRNAs. PloS One. 2011;6(12):e29376.
- Mishra MK, Loro E, Sengupta K, et al. Functional improvement of dystrophic muscle by repression of utrophin: let-7c interaction. PloS One. 2017;12(10):e0182676.
- Amirouche A, Jahnke VE, Lunde JA, et al. Muscle-specific microRNA-206 targets multiple components in dystrophic skeletal muscle representing beneficial adaptations. Am J Physiol Cell Physiol. 2017 Mar 1;312(3):C209–C21.
- Tjondrokoesoemo A, Schips T, Kanisicak O, et al. Genetic overexpression of Serpina3n attenuates muscular dystrophy in mice. Hum Mol Genet. 2016 Mar 15;25(6):1192–1202.
- Vanhoutte D, Schips TG, Kwong JQ, et al. Thrombospondin expression in myofibers stabilizes muscle membranes. eLife. 2016 Sep 26;5.
- Tinsley J, Robinson N, Davies KE. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to healthy male adult volunteers. J Clin Pharmacol. 2015 Jun;55(6):698–707.
- Ricotti V, Spinty S, Roper H, et al. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with duchenne muscular dystrophy. PloS One. 2016;11(4):e0152840.
- Summit therapeutic plc. Ezutromid significantly reduced muscle damage in DMD patients ini 24-week interim data from Summit’s PhaseOut DMD clinical trial. 2018. Available from: http://otpinvestiscom/clients/uk/summit_corporation_plc/rns/regulatory-storyaspx?cid=1575&newsid=970514.
- Forbes SC, Walter GA, Rooney WD, et al. Skeletal muscles of ambulant children with Duchenne muscular dystrophy: validation of multicenter study of evaluation with MR imaging and MR spectroscopy. Radiology. 2013 Oct;269(1):198–207.