
ABSTRACT
Introduction: X-linked retinitis pigmentosa caused by mutations in the retinitis pigmentosa GTPase regulator (RPGR) gene is the most common form of recessive RP. The phenotype is characterised by its severity and rapid disease progression. Gene therapy using adeno-associated viral vectors is currently the most promising therapeutic approach. However, the construction of a stable vector encoding the full-length RPGR transcript has previously proven to be a limiting step towards gene therapy clinical trials. Recently however, a codon optimised version of RPGR has been shown to increase the stability and fidelity of the sequence, conferring a therapeutic effect in murine and canine animal models.
Areas covered: This manuscript reviews the natural history of X-linked retinitis pigmentosa and the research performed from the discovery of the causative gene, RPGR, to the preclinical testing of potential therapies that have led to the initiation of three clinical trials.
Expert opinion: X-linked retinitis pigmentosa is an amenable disease to be treated by gene therapy. Codon optimisation has overcome the challenge of designing an RPGR vector without mutations, and with a therapeutic effect in different animal models. With the RPGR gene therapy clinical trials still in the early stages, the confirmation of the safety, tolerability and potency of the therapy is still ongoing.
1. Introduction
Retinitis pigmentosa (RP) is an heterogeneous group of inherited retinal dystrophies that affects 1 in 3000–4000 people worldwide. Although there is no ethnic specificity, mutations in particular genes are more frequent in some populations due to founder effects [Citation1].
RP presents clinical variability in age of onset, progression of the disease, and secondary clinical manifestations. Generally, primary degeneration affects photoreceptor rods, causing night blindness as the first clinical manifestation. Degeneration advances from the periphery toward the fovea, progressively decreasing the visual field and leading to tunnel vision. In the end stages of a typical rod-cone dystrophy, cones are also affected, leading to the loss of visual acuity [Citation2]. In some cases where the causative gene is expressed in cones and rods, patients can present with a cone rod dystrophy phenotype, in which visual acuity loss and peripheral rod loss occur synchronously. RP can also be syndromic, in which other organs are affected. The most common forms of syndromic RP are Usher syndrome, characterized by RP and hearing loss with or without vestibular dysfunction, and Bardet-Biedl syndrome, where other major signs are obesity, intellectual disability, polydactyly, and kidney abnormalities [Citation3,Citation4].
More than 3000 mutations in over 200 genes have been associated with RP. The genetic trait can be inherited as autosomal dominant (30–40% of cases), autosomal recessive (50–60%), or X-linked (5–15%) [Citation5,Citation6]. X-linked RP (XLRP) is particularly severe in males, characterized by early onset and rapid progression of vision loss, resulting in legal blindness by the end of the third decade. Most female carriers do not report symptoms or present with clinically relevant changes. However, the carrier phenotype can vary with the ratio of X-inactivation and ranges from asymptomatic to severe retinal disease with significant visual disability [Citation7]. The most common form of XLRP is that caused by mutations in the retinitis pigmentosa GTPase regulator (RPGR) gene, accounting for over 70% of XLRP-affected families and 10–20% of all RP cases [Citation8]. Less frequent forms of XLRP are caused by mutations in RP2 gene and OFD1 gene [Citation9].
2. Clinical spectrum of X-linked RP
Mutations in the RPGR gene can be associated with a rod-cone or cone-rod dystrophy phenotype. The most common presentation is as a rod-cone dystrophy. It is one of the most severe forms of RP with nyctalopia in most affected males before 10 years of age and progression to legal blindness by the third to fourth decade [Citation10]. It is initially identified with difficulties in scotopic visual function, where there is a predominant loss of rod photoreceptors. Simultaneously, peripheral vision deteriorates, resulting in visual field constriction on perimetry findings. The majority of cases present with a rod-cone dystrophy-type disease progression, where central visual acuity is initially less impaired than the peripheral field loss. However, some also present with early cone involvement and correspondingly impaired central visual acuity during early stages of the disease. The fovea is ultimately affected in all cases during the late stages of the disease by subsequent cone photoreceptor degeneration [Citation11].
There is a great variability in the clinical phenotype of these patients. The classical description of RP with peripheral bone spicules, waxy pallor of the optic disc, and attenuation of the retinal blood vessels is not always present. Bone spicules arise from the photoreceptor death that triggers the migration of retinal pigment epithelium (RPE) cells along the path of the inner retinal vessels [Citation12]. Bone spicules are primarily found in the rod-rich peripheral retina until progression of disease leads to pigmentation encroaching on the posterior pole of the retina (). In some patients, it is common to see a parafoveal hyper-autofluorescent ring on the blue autofluorescence (AF) imaging [Citation13]. The increased AF represents abnormal parafoveal accumulation of lipofuscin which usually encircles preserved areas. There is good correlation between ring radius and pattern P50 amplitude that is a measure of macular function. Larger rings may indicate a greater preservation of function, with progressive constriction over time [Citation14]. The rate of progression of the ring radius reduction is estimated between 0.8 and 15.8% per year [Citation15–Citation17]. An optical coherence tomography (OCT)-based model of disease progression has shown that there is an outer segment shortening early on, followed by a decrease of the outer nuclear layer thickness and a final disappearance of the inner segment ellipsoid zone (EZ) band [Citation18]. In a group of 28 patients affected by XLRP, the annual width decrease of the EZ band has been estimated at 248 μm/year over an observation time of 3 years. The equivalent area of the EZ at this time point corresponds to a 13% mean annual rate of change [Citation19]. There appears to be a functional correlation between the EZ area and static perimetry, as assessed in 48 patients [Citation19]. In this study, Hood and colleagues suggested that the EZ width might be considered a structural surrogate for the visual field in RP with high resolution and reproducibility. The figure of 13% annual change is also consistent with the 13% visual field and ERG loss/year reported in the clinical trial assessing docosahexaenoic acid supplementation [Citation20].
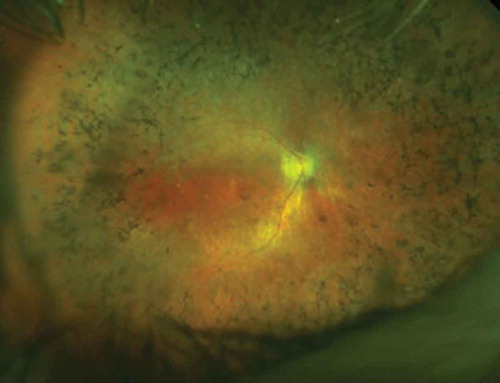
Figure 1. OPTOS image of a male RPGR patient showing the typical peripheral ‘bone spicule’ like pigmentary changes and signs of foveal atrophy. The inner retinal vessels are also attenuated due to the loss of outer retinal cells (photoreceptors) through physiological autoregulation of blood supply – this is not believed to be pathogenic.
In X-linked disease, males are almost always affected and females are carriers of the mutation, but still have degeneration and symptoms, albeit much milder and often subclinical. RPGR-related RP carriers, may however have highly variable phenotypes, with some demonstrating clinical signs similar to males [Citation21]. Female carriers that lie within this spectrum may show sparse patchy bone spicules in the extreme peripheral retina [Citation22]. A commonly described identification marker in female carriers is the tapetal-like reflex. This unique retinal feature in XLRP shows areas of high reflectance in a spoke-like fashion mainly visible on the blue AF and the red-free wavelength (). It is thought to represent an X-inactivation mosaic and speculated to be part of the early retinal degeneration process [Citation23,Citation24].
Figure 2. X-linked female carrier AF image showing the tapetal-like reflex centred on the fovea and patchy peripheral pigmentation. The mosaic picture is due to random X-inactivation generating clones of normal or degenerate photoreceptors. Since as with males, the degeneration takes time to manifest, a clearer X-linked carrier phenotype will generally be seen on the mothers of affected men rather than their daughters.
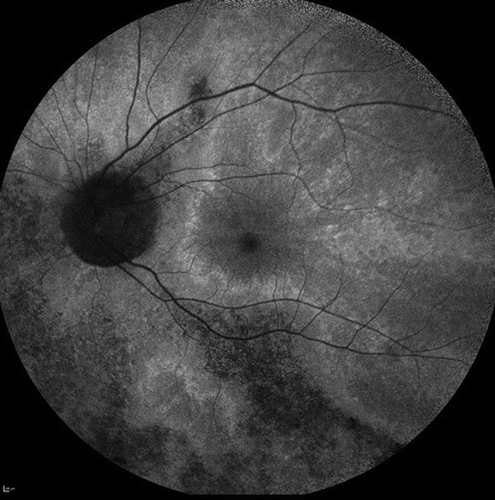
Multiple grading systems have been devised to categorize the variation of disease in X-linked carriers. These range from grade 0; tapetal reflex without any peripheral pigment retinal changes to grade 3; three-quadrants of bone spicule pigment or extensive peripheral atrophy [Citation21,Citation25].
Cone-rod dystrophies, a less commonly described form of XLRP, primarily affects cone function. Suffers complain of poor visual acuity, myopia, photophobia, and abnormal color vision. Cone-rich central regions are initially affected but significant variability exists in the involvement of rod photoreceptors [Citation26,Citation27]. Manifestations of cone-rod dystrophies in female carriers are unpredictable [Citation28].
RPGR is ubiquitously expressed in ciliated cells throughout the body. Although the findings of RP are confined to the eye, in some cases they can present in association with other non-ocular symptoms [Citation27]. Its role in cilia is still incompletely understood but defects in RPGR are thought to have a broader phenotypic variability than initially described. Extra-ocular tissues including the respiratory tract, auditory canal and sexual organs are shown to be dysfunctional in patients with mutations in RPGR [Citation29]. Therefore, it is most likely that RPGR-related ciliopathies exist within a larger clinical spectrum of disease, playing an important role in cilia function throughout the body [Citation30,Citation31].
3. Molecular genetics for X-linked RP
Six loci of genetic defects have been mapped to date in X-linked RP pathology: RP2 (MIM 312,600, Xp11.3), RP3 (MIM 312,610, Xp11.4), RP23 (MIM 300,424, Xp22.2), RP24 (MIM 300,155, Xq26-q27), RP6 (MIM 312,612, Xp21.3-p21.2), and RP34 (MIM 300,605, Xq28). However, only three genes have been identified so far: RP2, RPGR/RP3 and OFD1/RP23 [Citation6].
Mutations in RP2 are reported to cause 10–15% of XLRP. RP2 encodes a 350 amino acids protein, whose amino terminus shows homology to human cofactor C, involved in β-tubulin folding. RP2 protein acts as a GTPase-activating protein (GAP), probably controlling the protein trafficking between the Golgi apparatus and the ciliary membrane of photoreceptors [Citation32]. Mutations have been found along the RP2 sequence, with nonsense mutations and the consequent truncation of the protein as the most prevalent alteration [Citation33,Citation34]. Clinically, mutations in RP2 gene have been associated with an early onset of macular dystrophy and poor visual acuity combined with high myopia in XLRP male patients [Citation35]. A long-term dose efficacy study in Rp2-KO mice has shown a rescue of cone degeneration by delivering the RP2 gene with an adeno-associated viral (AAV) vector [Citation36].
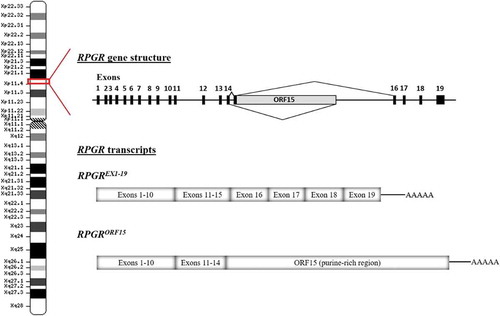
The RPGR gene was identified as cause of XLRP in 1996 by positional cloning [Citation37]. RPGR mutations account for 70–80% of XLRP, representing a major contribution to the RP disease. Its high contribution to X-linked RP and the severity of the disease caused by mutations in this gene makes the development of genetic therapies a high-impact approach for RP patients. The RPGR gene is located in the short arm of the X chromosome (Xp11.4) and encodes 10 alternatively-spliced transcripts, five of which are predicted to be protein coding. The different RPGR transcripts could be differentially spliced among tissues. The canonical sequence is composed of 19 exons that encodes a protein of 815 amino acids widely expressed in different tissues (RPGREx1−19 isoform, also termed the constitutive variant). Exons 1–14 harbor the majority of the disease-causing variants, although in total they account for less than 25% of XLRP cases. No disease-causing mutations have been reported in exons 16–19 [Citation1,Citation38]. The second major isoform is RPGRORF15, which is highly expressed in rod and cone photoreceptors of the retina. It shares exons 1–14 with RPGREx1−19, plus a large alternatively spliced exon, ORF15 (). Exon ORF15 contains a purine-rich repetitive region, and is considered a mutational hot spot since 80% of the RPGR mutations have been identified in this region [Citation39]. The high frequency of mutations in exon ORF15 may be explained by the repetitive nature of the sequence that can adopt unusual non-standard double helix DNA conformations, affecting DNA replication and transcription, and contributing to genome instability [Citation40]. The ORF15 mutations comprise mostly deletions/insertions, duplications, and nonsense mutations, thus resulting in the truncation of RPGRORF15 protein. Regardless the mutation type, it has been proposed that there may be a correlation between the location of the mutations within ORF15 and the severity of the disease; the longer the encoded wild-type amino acid sequence, the milder the disease and vice versa [Citation41,Citation42].
Figure 3. Schematic representation of the RPGR gene structure and the main alternatively spliced transcripts. Note that there is normally a splice donor site within the 5ʹ region of ORF15. In gene therapy, the cDNA encoding the gene is transcribed as a primary RNA transcript in the nucleus and may be subject to splicing. Hence disabling this splice donor site is also an essential part of codon optimisation.
4. Expression and function of RPGR protein
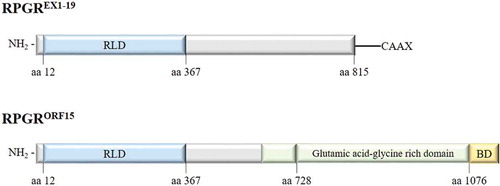
The RPGREx1−19 transcript encodes an 815 amino acid protein, expressed in the cilia in different tissues. As with all predicted isoforms, RPGREx1−19 protein includes an amino-terminal domain homologous to the regulator of chromosome condensation 1 (RCC1), known as the RCC1-like domain or RLD, encoded by exons 1–10 [Citation37]. RCC1 is a guanine nucleotide exchange factor (GEF) for Ran GTPase with a key role in the regulation of nucleo-cytoplasmic transport, mitosis and nuclear-envelope assembly [Citation43]. The carboxy-terminus of the constitutive isoform contains an isoprenylation motif (CAAX), a posttranslational modification that regulates the association to the membrane and the protein trafficking [Citation44].
The RPGRORF15 transcript encodes an 1152 amino acid protein that shows the highest expression in the retina where the protein product is located to the connecting cilia of photoreceptors. Exon ORF15 is translated into an acidic, repetitive glutamic acid-glycine rich domain plus a nonrepetitive, basic domain in the carboxy-terminus (). This basic domain is known to be evolutionary conserved with a high sequence identity across species [Citation45], suggesting that it constitutes a functional domain. Due to the repetitive nature of the glutamic acid-glycine-rich region and the fact that its length varies among species, it is challenging to decipher the function of this domain, however, the posttranslational poly-glutamylation near to the C-terminus is typical of the modification of tubulins that are essential for intracellular trafficking [Citation46]. In humans, a lack of glutamylation caused by TTLL5 enzyme deficiency causes RP, similar to RPGR deficiency, hence indicating that the glutamylated protein is essential for normal RPGRORF15 function [Citation47,Citation48].
Figure 4. Major RPGR protein isoforms (constitutive RPGREXON1-19 and RPGRORF15) schematic representation. RLD: RCC1-like domain, BD: basic domain.
RPGR protein interacts with other ciliary proteins to form protein complexes involved in cilia regulatory signaling pathways. The phosphodiesterase 6 delta subunit (PDE6δ) was the first RPGR interacting protein identified in a yeast two-hybrid screen using the RLD (amino acid 51–370) [Citation49]. Other interactions with the RCC1-like domain of RPGR include RPGR interacting protein 1 (RPGRIP1), SMC1 and SMC3, and RAB8A. RPGRIP1 may function as scaffolding protein facilitating the transport of molecules through the connecting cilium [Citation50]. The two chromosome-associated proteins, SMC1 and SMC3, are involved in microtubule-based movement of chromosomes and also associated with ciliary microtubules in the retina [Citation51]. RPGR not only interacts with RAB8A, a critical GTPase involved in photoreceptor protein trafficking, but also regulates its activity by exhibiting guanine exchange factor activity toward RAB8A [Citation52]. The basic domain of RPGRORF15 (amino acid 1079–1152) interacts with whirlin and with nucleophosmin. Whirlin protein is required for elongation and maintenance of inner and outer hair cell stereocilia in the organ of Corti in the inner ear. Mutations in this gene may cause Usher syndrome [Citation53]. Nucleophosmin is a multifunctional protein chaperone which functions in cell division [Citation45]. Co-immunoprecipitation revealed other ciliary proteins that interact with RPGR outside the mentioned domains (amino acid 370–585), such as IFT88, IQCB1 or CEP290 [Citation54].
Posttranslational modifications of proteins constitute an important mechanism for the regulation of protein–protein interactions. Recently, RPGRORF15 has been described as a novel glutamylation substrate [Citation48,Citation55]. Glutamylation is an evolutionarily conserved posttranslational modification that consists in the addition of glutamates to the C-terminal tail domain of target proteins. The ORF15 region in the C-terminus of RPGR protein contains 11 glutamate-rich consensus motifs. The addition of negatively charged glutamates to this region, is likely to affect the stabilization and the folding of RPGRORF15 protein and its interaction with other proteins in the connecting cilia, particularly with regard to trafficking.
The connecting cilia represents a critical junction between the inner and the outer segments, allowing the bidirectional transport of opsins and other phototransduction proteins and contributing to the viability of the photoreceptors. Although RPGRORF15 function is not well understood, evidence suggest that it plays an important role in microtubule-based transport to and from the basal bodies and within photoreceptor axonemes, contributing to the intracellular cargo movement between inner and outer segments [Citation54,Citation56]. The glutamylation of the ORF15 region appears to be critical for that function [Citation48].
5. Animal models to investigate the RPGR-associated pathology
Different animal models have helped not only to unravel the pathological mechanisms linked to RPGR-causing RP but also to shed light on the function of RPGR protein. In 1999, Aguirre and colleagues identified the canine X-linked progressive retinal atrophy (XLPRA) as the locus homologue of RP3 in the Siberian Husky, proposing this purebred dog as spontaneous model of XLRP [Citation57]. The XLPRA phenotype is similar to XLRP in most male human patients with mutations in RPGR. In this canine model, they observed a disruption and disintegration of the rod outer segments preceding inner segments abnormalities and, eventually the degeneration of cones triggered by the rod loss. These changes, observed in hemizygous males, were more severe than those seen in XLPRA carriers. Later in 2002, the same researchers identified the disease-associated mutation in RPGRORF15 in XLPRA canine models: XLPRA1 (Syberian Husky and Samoyed) and XLPRA2 (mongrel-derived) [Citation58]. XLPRA1 is caused by a five-nucleotide deletion (del1028-1032) that gives rise to a frameshift and immediate premature stop. The truncated protein is missing 230 amino acids in the C-terminus causing a slight decrease in the isoelectric point. In this model, characterized by a slowly progressive rod-cone dystrophy, the retinal function remains normal until 6 months of age. In contrast, XLPRA2 model is characterized by an anomalous retinal development and a rapid degeneration. It is caused by a two-nucleotide deletion (del1084-1085) that results in a frameshift. The mutated protein has an increased isoelectric point and 34 additional basic residues before a premature termination 71 amino acids downstream. Recently in 2016, Dekomien and colleagues identified the Weimaraner dog as the third naturally occurring model for XLRP [Citation59]. In this case, the mutation is a large deletion of the first four exons of RPGR gene. The opthalmoscopic examination confirmed the progressive retinal atrophy in males, although no further analysis has been done to evaluate the effect of the RPGR mutation at histological or molecular level. Despite the valuable information provided by the XLPRA canine models, the use of small animal models over the large models present several advantages such as the ease of care, the short life span and the generation of large and homogeneous samples.
In mice, Rpgr is the homologue of human RPGR, although the mouse protein is shorter. One natural and two genetically engineered mouse models with Rpgr mutations exist. The first Rpgr mouse model was created in 2000 by targeted knockout [Citation60]. In this model, exons 4–6 were replaced by a selectable marker, which implies the early truncation of both RPGR isoforms, constitutive and ORF15. Considered a moderate model in terms of progression of the disease, the early consequences of the functional RPGR absence in nullizygous mice are the mislocalization of opsins to the inner segments and proximal synaptic endings, and the upregulation of GFAP, indicative of the retinal degeneration. At 6 months of age, the photoreceptor cell loss is evident, and comparable to the reduction of the retinal function. Electroretinography (ERG) shows ~25% and ~30% reduction of scotopic and photopic amplitudes, respectively. In the second genetically engineered mouse model, termed Rpgr conditional knockout (Rpgr-cko), the proximal promoter and first exon were deleted ubiquitously [Citation61]. In this model, with also both RPGR isoforms affected, the rod and cone ERGs responses are reduced ~40% by 6 months of age, while the decrease of the outer nuclear layer (ONL) thickness is evident at 13 months.
The naturally occurring model, known as Rd9 mouse, has a 32 base pair duplication in ORF15 that produces a frameshift and a truncated protein where 108 amino acids in the C-terminal are missing [Citation62]. This mutation in ORF15 does not affect the constitutive isoform, Rpgr1−19 which is expressed in this model. However, it does not seem to compensate RpgrORF15 function. In this model, Rd9/Y males and Rd9/Rd9 females exhibit a uniform phenotype, with an early onset of the disease. The retinal function is reduced at early ages with a progressive loss of ERG amplitudes with aging, the cone opsins are mislocalized to the inner segments and a significant ONL thickness reduction is observed at 12 months of age.
Even though these mouse models have meant a useful tool to study the cellular and molecular mechanisms of different retinal diseases and also, in proof-of-concept studies to develop therapeutic strategies, one major limitation is the lack of a cone rich area in the central retina, such as the macula present in humans. Furthermore, and in contrast to the human disease, the photoreceptor loss in mice with Rpgr deficiency is very slow and mild.
6. Clinical and molecular diagnosis
Typical fundoscopy features of rod-cone dystrophy with bone spicule shaped pigmentary changes, begin in the rod-rich peripheral retina. These early signs coupled with symptoms of nyctalopia and tunnel vision field defects are diagnostic of RP. Features identified at a young age with rapid progression are characteristic of X-linked RP.
With the progression of disease, pigmentary changes may progress to the posterior pole of the retina and can be seen surrounding the macular area. Although perimetry findings show worsening peripheral visual field constriction, central visual acuity can remain relatively stable until the late stages of disease [Citation2].
X-linked carriers, however, can have an unpredictable spectrum of clinical features with the tapetal like reflex shown to be a unique characteristic [Citation23]. All patients require a comprehensive medical and family history to be elicited, especially if there is a suspicion of RPGR related ciliopathy that could affect multiple organs.
Imaging techniques can be employed to confirm the diagnosis and monitor progression. A detailed examination of retinal architecture using OCT can demonstrate the integrity of retinal structure. The photoreceptor inner/outer segment line, otherwise known as the ellipsoid layer, is thought to be important parameter in the monitoring of RP patients; more so, due to its recent correlation with visual acuity and microperimetry findings [Citation63]. Coupled with this, imaging techniques such as AF that uses a confocal scanning laser ophthalmoscope can also be useful in assessment of the RPE. Lipofuscin accumulates in the RPE cells as a by-product of photoreceptor outer segment degradation and the abnormal metabolism, as in RP, leads to hyper-fluorescence on AF imaging. In contrast, areas of RPE atrophy and photoreceptor dysfunction show an absence of fluorescence.
AF imaging has been identified as a useful tool in female carriers. Retinal abnormalities of the RPE are thought to be seen earlier on autofluorescence imaging than fundoscopy, with patchy areas of retina showing hyper-fluorescence. Additionally, the tapetal-like reflex can also be seen on AF but is best demonstrated on infrared reflectance imaging [Citation64,Citation65].
An enhanced autofluorescent para-foveal ring has also been recognized in XLRP patients and carriers. Hyper-fluorescence in this rod-dense region is hypothesized to be due to the high turnover rate of the photoreceptor outer segments and is therefore an accumulation of lipofuscin. It is suggested to be a marker of the border between the functional and nonfunctional retina, with the radius of the extra-foveal ring correlating linearly with the area of functioning macular [Citation66]. Cone-rod dystrophies, however, show an inversely proportional relationship; a large AF para-foveal ring indicates a worse P50 ERG amplitude and so poorer macular function [Citation16]. It is therefore a combination of symptoms, clinical features and imaging which can be used to aid the diagnosis of XLRP.
Besides the clinical phenotype, the study of the family history is important to identify the underlying genetic defect. However, in X-linked inheritance (no male-to-male transmission), female carriers may not show any symptoms. Importantly, families with apparent autosomal dominant RP, with affected females but no male-to-male transmission, may, in fact have X-linked RP caused by RPGR mutations combined with skewed X-inactivation in the female carriers [Citation67]. Thus, genetic testing is the best tool to diagnose the disease. The molecular diagnosis using next-generation sequencing (NGS) is a robust approach, which allows not only to screen mutations in known disease-causing genes but also to identify novel alleles involved in the disease. Despite the high efficiency of the NGS, the volume of data generated requires specialized bioinformatics resources in order to have conclusive results for the diagnosis. In cases where the clinical diagnosis is evident and there is a reasonable suspicion about the gene responsible for the disease, conventional Sanger sequencing is performed.
7. Gene therapy for X-linked RP
7.1. Overview
Gene therapy is a powerful strategy to slow down or stop the progression of a disease by delivery of the normal gene into cells. The retina is an excellent target for gene therapy since the accessibility allows relatively noninvasive procedures, the effect of the treatment can be easily monitored and the blood–retinal barrier could limit the immunological response to the treatment by reducing systemic spread. Recessive single gene retinal degenerations are the most amenable to be treated by gene therapy because the only affected gene leads to the total or partial absence of functional protein. Thus, the ideal delivery of the healthy copy of the gene would revert the phenotype without toxicity issues due to its overexpression.
The adeno-associated virus (AAV) is the best-characterized viral vector in ophthalmology and one of the main advantages is being a non-enveloped virus, thus it is less likely to induce an inflammatory response. Recombinant AAV is replication-deficient and does not integrate the transgene into the host genome [Citation68,Citation69]. The exceptional safety profile of rAAVs is also supported by the observation that rAAVs have been used for more than 20 years in ocular gene transfer studies in animal models and more than 10 year in human clinical trials with no sign of malignant transformation [Citation70]. However, the packaging capacity is only about 4.8kb of single stranded DNA. This relatively small size of the internal payload is limiting to treat some diseases where the affected gene is too large to fit into the AAV vector, such as ABCA4 cDNA responsible for Stargardt disease (6.8 kb). X-linked RP caused by mutations in RPGR is an ideal disease to be treated with AAV gene therapy because the size of the coding sequences fits well into AAV vectors (3.5 kb for RPGRORF15). Also, the relatively high prevalence and the severity of RPGR-associated retinal degeneration implies a favorable risk/benefit ratio for a gene therapy clinical trial.
7.2. The challenge to develop a therapeutic vector for RPGR gene therapy
As commented above, X-linked RP caused by mutations in RPGR is an ideal disease to be treated with AAV gene therapy. However, the development of the therapeutic vector for gene therapy constitutes a challenge because the purine rich, repetitive sequence of RPGRORF15 makes it less stable and more prone to spontaneous mutations during the cloning of recombinant viral vectors. Several studies have followed different strategies to replace RPGR in mouse and dog models with the disease. In 2012, Beltran and colleagues reported a successful therapeutic approach in the XLRPA canine model [Citation71]. In this study, the recombinant AAV2/5 vector was used to deliver human RPGR under the control of either a human photoreceptor-specific IRBP (interstitial retinol-binding protein) or GRK1 (rhodopsin kinase) promoter in the subretinal space. The vector cDNA was made from a full-length human RPGRORF15 clone (variant C; NM_001034853), based on the published sequence from Alan Wright and colleagues [Citation39]. However, despite the preservation of the retinal structure and function in these two models after the treatment, the AAV2/5.RPGR vector was found to have multiple mutations within the purine rich exon 15 region. These changes, caused by the instability of the ORF15 sequence, could be the reason for the toxic effects observed in treated mice when titers considered safe in the mouse retina (1013 vector genome (vg)/ml) were used [Citation72]. In another study, the safety and dose-efficacy of the AAV8.GRK1.RPGRORF15 vector were evaluated in the Rpgr-KO mouse [Citation73]. The strategy followed in this study to synthetize the vector was based on a stepwise cloning: the purine-rich region was prepared first and then ligated to a synthetic DNA fragment encoding the upstream exons. Although the vector stability was improved with this strategy, two out of five AAV vector preparations contained minor deletions. Additionally, the analysis of human RPGR expression in Rpgr-KO mouse retinas injected with the vector revealed (1) a set of proteins with lower molecular weight from the predicted 200 kilodalton (kDa) full-length protein, indicating that truncated or alternatively spliced forms of the protein were being expressed from the vector, and (2) the vector-expressed RPGRORF15 was mislocalized to the inner segments and the synaptic terminals in vector-treated mice. Although partial rescue of retinal structure was achieved in injected eyes, a toxic effect was seen in eyes injected with 3 × 109 vg, which showed much lower ERG amplitudes. With the same purpose, Li and colleagues tested two human AAV8.GRK1.RPGRORF15 vectors in the Rpgr-KO mouse with the particularity that these vectors contained in-frame deletions in the purine-rich region [Citation74]. As a result, the mutated vectors generated a short form of RPGR with 314 codons missing (more than 80% of the amino acids in ORF15) and a long form with a deletion of 126 codons (one-third of this region). While the long form showed significant rescue of photoreceptors cells and function, the short form was unable to localize to the connecting cilium and did not show any functional improvement in the disease phenotype. Two nearly identical constructs to these short and long forms, with very similar deletions in the same region, were further analyzed in terms of glutamylation. While the full-length RPGRORF15 is strongly glutamylated, the long form shows a significant reduction in glutamylation to about 28% of the full-length levels and no detectable glutamylation is observed in the short form [Citation48]. It has been suggested that glutamylation is important to preserve RPGRORF15 function, probably acting as signals to recruit key regulatory factors to RPGRORF15. Thus, cloning RPGR for gene therapy to obtain the full-length and fully glutamylated protein is a challenge due to the sequence instability. This challenge may be overcome by codon optimization.
7.3. RPGR gene therapy vector design by codon optimization
In the early 1970s, Walter Fiers and colleagues discovered a strikingly non-random pattern of codon usage in a bacteriophage genome and first hypothesized that non-random codon and tRNA frequency might be involved in regulating rates of gene expression [Citation75]. Indeed, highly expressed genes preferentially use so-called major codons, i.e. codons with the highest abundance of matching tRNA molecules to make translation more efficient, a concept called ‘codon bias.’ In case of Leucine for example, six codon sequences (UUA, UUG, CUU, CUC, CUA, or CUG) would lead to the correct amino acid being inserted. But if the most abundant tRNA molecule would carry the anti-codon for UUA, this codon would result in more efficient translation than any of the other nucleotide combinations. This observation was used to generate codon adaptation indices (CAI), which aim to reflect the use of high abundance codons in the investigated sequence [Citation76].
In the context of gene therapy with rAAV as vector system with its limited packaging capacity, codon optimization offers the potential to increase transgene expression without additional regulatory elements such as WPRE in cis. Moreover, changing the nucleotide sequence without altering the translated amino acid sequence (silent substitutions) of the transgene e. g. allows to change CG content, remove unwanted repeat sequences, cryptic splice sites and/or restriction sites that may interfere with cloning.
Cloning the full-length human RPGRORF15 without random mutations being introduced is difficult – as is direct sequencing of the adenine/guanine rich regions since polymerases have a tendency to stop at guanine repeats. The underlying problem of the genetic instability is most likely explained by properties of the mutational hotspot, the ORF15 region. It contains far fewer T and C nucleotides than would be predicted in the genome, for instance, the 750 base pair sequence between positions 2410 and 3160 contains no C nucleotides at all in the wild type sequence. This leads to many repeating sequences that may recombine incorrectly during cloning and vector production.
We and others recently described a codon optimized version of RPGR with superior sequence stability [Citation77,Citation78]. Since most codons start with G at position 1 in this region, in our study, the addition of C nucleotides to position 3 of the preceding codon during codon optimization has been done with consideration of limiting the number of CpG dinucleotides to no more than would be predicted in the eukaryotic exon [Citation77]. This is because too many of these sequences may be identified and become prone to methylation of the C nucleotide. For instance, in the 750 base pair sequence between positions 2410 and 3160, a total of 45 C nucleotides were added through the codon optimization process, resulting in 45 CpG dinucleotides (6.00%). This compares favorably to the predicted wild type frequency of these dinucleotides (6.25%). Furthermore, the C codon optimization was applied where possible to the fourfold degenerate codon ‘GGN,’ encoding glycine. This is because any subsequent methylation of the C nucleotide of the CpG dinucleotide within the transgene and subsequent deamination to thymine (T), even if it did occur, would not change the RPGR protein sequence because GGC and GGT both encode glycine. Insertion of T nucleotides within the GA rich region of RPGR has also been limited to avoid creating anomalous polyA signals (e.g. AATAAA) and possible splice donor sites (GT). The latter is a consideration as this region contains many splice acceptor (AG) sequences with repeating G pyrimidine bases and potential A nucleotide branch points in the 5ʹ direction. The codon optimization pattern was extensively modeled in silico to determine the optimal modification to reduce the GA repeats and also to reduce the risk of anomalous splicing and creation of premature polyA signals. The codon optimized gene is therefore more stable than the wild-type cDNA sequence, which may generate alternatively spliced variants and truncated proteins when reintroduced into the transcriptional machinery through gene therapy. Indeed, the greatest benefit of codon optimizing RPGRORF15 lies in its increased sequence fidelity or genetic stability.
7.4. Preclinical testing of the codon optimized vector
Once the superior sequence stability of the codon optimized sequence of RPGRORF15 was confirmed in vitro, the safety and efficacy of AAV.coRPGRORF15 as gene therapeutic agent for XLRP was tested [Citation77]. A single treatment of wild-type mice with AAV.coRPGRORF15 did not seem to induce any toxic effects and showed a good safety profile. In vivo retinal imaging suggests that there was no negative impact of AAV.coRPGRORF15 treatment either on retinal structure or function. The therapeutic effect of AAV.coRPGRORF15 was demonstrated in two well-characterized mouse models of XLRP due to Rpgr mutations (Rpgr-KO and Rd9 mice). A trial conducted in a total of c. 80 animals provided evidence of efficacy as indicated by significant rescue of electrophysiological measurements in both models. The treatment did not become evident at two months of age, most likely due to the slow disease progression in both animal models. However, AAV.coRPGRORF15 treatment was associated with significant ERG rescue in both animal models at month 4 and 6. This rescue was more evident in the dark adapted intensity series, which reflects the sum potential of rod photoreceptors in the lower intensity range (single flashes up to ca. 0.01 cd*s/m2) [Citation79].
Taken together, treatment with the codon optimized version seems to be safe and effective. Successful transduction of photoreceptors with AAV.coRPGRORF15 in wild type mice did not lead to toxic effects, which might have been associated with the expression of RPGRORF15 on a background of physiological levels of native Rpgr. Furthermore, treatment of animal models of XLRP showed a statistically significant rescue of ERG responses in the treated, but not in the untreated eyes. These data were the first evidence that a codon optimized coding sequence of RPGRORF15 can be used to safely treat eyes, which lack expression of this important ciliary protein.
7.5. Gene therapy clinical trials for X-linked RP
Currently, there are three clinical trials to treat XLRP caused by mutations in RPGR. The three phase 1/2 trials will evaluate the safety and efficacy of AAV vectors containing the human RPGR in subjects with XLRP caused by mutations in RPGRORF15. These nonrandomized, open-label studies will test different doses in an escalation manner. The main and most important difference between the three studies is the therapeutic vector used. In March of 2017, the first clinical trial, sponsored by Nightstar Therapeutics, was initiated (NCT03116113). In this study, known as XIRIUS, a recombinant AAV2/8 vector containing the codon-optimized human RPGRORF15 under the control of the GRK1 promoter [Citation77] was subretinally injected in male subjects with genetically confirmed RPGR mutation.
The second clinical trial, started in July of 2017 and sponsored by MeiraGTx UK Ltd (NCT03252847), the safety and efficacy of the subretinal administration of the therapeutic vector will be evaluated in adults and children with XLRP for 18 months. The RPGR gene therapy vector, AAV2/5.hRKp.RPGR is based on the AAV2/5 serotype but the RPGR sequence is mutated in the ORF15 region and carries a random deletion of 126 codons – about one-third of the human ORF15 coding sequence is missing [Citation74]. This region appears to be particularly important for post-translational modification of RPGRORF15 and a very similar deletion has been shown to reduce glutamylation by 80% compared to wildtype levels [Citation48].
Finally, the trial sponsored by Applied Genetic Technologies Corp (AGTC) (NCT03316560) is reported to be initiated in 2018. The vector used in this study expresses the human RPGRORF15 driven by the GRK1 promoter and packaged in AAV2 capsids with single tyrosine to phenylalanine (YF) mutations, described to enhance the efficiency of transduction. This trial arises from a proof-of-concept gene therapy study in dogs in which a newly developed codon-optimized human RPGR cDNA vector was able to transduce both rods and cones preserving the outer nuclear layer structure [Citation78].
8. Conclusion
X-linked RP is a severe form of retinal dysfunction without an approved effective treatment to date. With the gene therapy as one of the most powerful strategies to treat monogenic retinal diseases, several studies have been carried out in the last years to design and develop a gene therapy vector for XLRP. However, most of the studies carried out have highlighted the difficulties to produce RPGR gene therapy vectors without generating new mutations in the therapeutic vector. Codon optimization has been successfully used as approach to bypass those problems, by increasing the genetic stability. The promising preclinical tests using AAV8.GRK1.coRPGR allowed to start a phase I/II clinical trial in subjects with XLRP caused by mutations in RPGRORF15.
9. Expert opinion
Gene therapy is a potential therapeutic strategy for monogenetic diseases, in which the mutation causing the disease results in the loss of function of the gene product. Thus, the gene augmentation by delivery of a normal copy of the gene can correct the loss of function, restoring the normal phenotype. Whilst Leber congenital amaurosis (LCA) is about to become the first gene therapy treatment approved by the United States Food and Drug Administration (FDA), and choroideremia currently achieving remarkable results in Phase I/II clinical trials, there are many other inherited retinal degenerations that remain without an effective treatment.
X-linked RP is ideally suited for gene therapy for different reasons:
Clinically significant prevalence: up to 20% of all RP cases are caused by mutations in one gene, RPGR.
Severe phenotype and rapid progression of the disease facilitates assessing clinical trial endpoints within a relatively short period of time.
Accurate diagnosis: the X-linked family history study provides an important clue about the disease that can be then easily diagnosed by clinical features and at molecular level by sequencing.
As a general rule for all gene therapy programs, several aspects have to be considered in order to achieve the safest and the most effective method to replace the mutated gene in the specific retinal cell type: the vector used as delivery system, the promoter that will drive the gene expression and the surgical technique to administrate the vector in the eye. The adeno-associated viral (AAV) vector has been proven to be the ideal delivery system for retinal gene therapy so far, because of its ability to transduce efficiently nondividing cells in vivo and the fact that to date no safety concerns exist about its use. Capsid proteins determine the tissue tropism and the infection efficiency, with AAV8 being one of the most efficient for photoreceptor transduction in vivo. Investigations have been carried out to modify AAV capsids by exchanging surface protein residues that prevents the vector ubiquitination and the consequent degradation, thus enhancing the transduction of specific cell types. Further investigations would be needed to assure its safety, since neutralizing antibody response may be triggered against these modified capsid proteins. On the other hand, the use of cell-specific promoters, such as the photoreceptor-specific rhodopsin kinase (GRK1) promoter, contributes to limit the expression to target cells. In regard to the surgical technique for retinal gene therapy, the two main routes of administration are through subretinal or intravitreal injection. While the penetration of the AAV vector to the outer layers of the retina are inefficient with intravitreal injections, subretinal injection allows to target retinal pigment epithelial (RPE) cells and photoreceptors in a safe procedure with limited trauma.
Specifically for XLRP, the great challenge of the research to develop a therapeutic strategy has been the construction of a viral vector encoding stable RPGRORF15. The instability of the ORF15 region can easily lead to the production of mutated vectors that would trigger the expression of truncated forms of the protein, thus resulting in an inefficient therapy or, even worse, a toxic therapy. Although several preclinical studies have been carried out with mutated versions of RPGRORF15 with positive results with some deletions and negative in others, it is important to consider the differences in the sequence between species and also, the fact that the mechanisms responsible for gene expression and protein processing will vary between species. With this issue as the limiting step in the development of a gene therapy treatment for XLRP, an improvement in the vector design has become necessary to ensure the safety and the efficacy of the clinical trials. In this context, the most important advantage of the codon optimization in difficult sequences such as RPGRORF15 lies in the potential to improve sequence fidelity. Changing nucleotides without changing the resulting amino acid sequence carries the potential to make the sequence more stable and less prone to spontaneous mutations during the production of vectors for gene therapy. Preclinical data on the testing of AAV.coRPGRORF15 gene therapy vector have been promising, and the results of the clinical trial are awaited full of expectation.
Article highlights
Gene therapy is a powerful therapeutic strategy for recessive single gene retinal degenerations.
X-linked retinitis pigmentosa (XLRP) is mainly caused by mutations in the RPGR gene. Because of its severity and its high incidence, the search for an effective treatment for this disease has drawn a great deal of interest among the scientific community in the last years.
The development of RPGR gene therapy has meant a challenge due to the instability of RPGRORF15 sequence that made it difficult to produce a therapeutic vector without generating new mutations.
Codon optimisation has been successfully used as an approach to improve sequence fidelity during the development of the vector. This approach allowed the progression towards the first gene therapy clinical trial for XLRP.
This box summarizes the key aspects contained in this review.
This box summarizes key points contained in the article.
Declaration of Interest
RE MacLaren is the academic founder and director of Nightstar Therapeutics, a gene therapy company established by the University of Oxford and originally funded by the Wellcome Trust through Syncona Partners. RE MacLaren and M. Dominik Fischer are names inventors on a patent filed on behalf of the University of Oxford, relating to the expression cassette and codon optimisation of RPGR coding sequence in general. They are also consultants to Nightstar Therapeutics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Ferrari S, Di Iorio E, Barbaro V, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12(4):238–249.
- Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40.
- Williams DS. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vision Res. 2008;48(3):433–441.
- Beales PL, Elcioglu N, Woolf AS, et al. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–446.
- Anasagasti A, Irigoyen C, Barandika O, et al. Current mutation discovery approaches in Retinitis Pigmentosa. Vision Res. 2012;75:117–129.
- Daiger SP Retinal information network 1996–2017. [cited 2017 Oct 9]. Available from: https://sph.uth.edu/RetNet/home.htm
- Wu H, Luo J, Yu H, et al. Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron. 2014;81(1):103–119.
- Raghupathy RK, Gautier P, Soares DC, et al. Evolutionary characterization of the retinitis pigmentosa GTPase regulator gene. Invest Ophthalmol Vis Sci. 2015;56(11):6255–6264.
- Webb TR, Parfitt DA, Gardner JC, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. 2012;21(16):3647–3654.
- Flaxel CJ, Jay M, Thiselton DL, et al. Difference between RP2 and RP3 phenotypes in X linked retinitis pigmentosa. Br J Ophthalmol. 1999;83(10):1144–1148.
- Campochiaro PA, Mir TA. The mechanism of cone cell death in retinitis pigmentosa. Prog Retin Eye Res. 2018;62:24-37.
- Jaissle GB, May CA, Van De Pavert SA, et al. Bone spicule pigment formation in retinitis pigmentosa: insights from a mouse model. Graefes Arch Clin Exp Ophthalmol. 2010;248(8):1063–1070.
- Jolly JK, Wagner SK, Moules J, et al. A novel method for quantitative serial autofluorescence analysis in retinitis pigmentosa using image characteristics. Transl Vis Sci Technol. 2016;5(6):10.
- Holder GE. Pattern electroretinography (PERG) and an integrated approach to visual pathway diagnosis. Prog Retin Eye Res. 2001;20(4):531–561.
- Robson AG, El-Amir A, Bailey C, et al. Pattern ERG correlates of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci. 2003;44(8):3544–3550.
- Robson AG, Michaelides M, Saihan Z, et al. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence; a review and update. Doc Ophthalmol. 2008;116(2):79–89.
- Robson AG, Tufail A, Fitzke F, et al. Serial imaging and structure-function correlates of high-density rings of fundus autofluorescence in retinitis pigmentosa. Retina. 2011;31(8):1670–1679.
- Hood DC, Lazow MA, Locke KG, et al. The transition zone between healthy and diseased retina in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52(1):101–108.
- Birch DG, Locke KG, Wen Y, et al. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with X-linked retinitis pigmentosa. JAMA Ophthalmol. 2013;131(9):1143–1150.
- Hoffman DR, Locke KG, Wheaton DH, et al. A randomized, placebo-controlled clinical trial of docosahexaenoic acid supplementation for X-linked retinitis pigmentosa. Am J Ophthalmol. 2004;137(4):704–718.
- Comander J, Weigel-DiFranco C, Sandberg MA, et al. Visual function in carriers of X-linked retinitis pigmentosa. Ophthalmology. 2015;122(9):1899–1906.
- Andreasson S, Ponjavic V, Abrahamson M, et al. Phenotypes in three Swedish families with X-linked retinitis pigmentosa caused by different mutations in the RPGR gene. Am J Ophthalmol. 1997;124(1):95–102.
- Cideciyan AV, Jacobson SG. Image analysis of the tapetal-like reflex in carriers of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1994;35(11):3812–3824.
- Fishman GA. Weinberg AB and McMahon TT, X-linked recessive retinitis pigmentosa. Clinical characteristics of carriers. Arch Ophthalmol. 1986;104(9):1329–1335.
- Grover S, Fishman GA, Anderson RJ, et al. A longitudinal study of visual function in carriers of X-linked recessive retinitis pigmentosa. Ophthalmology. 2000;107(2):386–396.
- Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002;70(4):1049–1053.
- Hartong DT. Berson EL and Dryja TP, Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809.
- Ebenezer ND, Michaelides M, Jenkins SA, et al. Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci. 2005;46(6):1891–1898.
- Iannaccone A, Breuer DK, Wang XF, et al. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J Med Genet. 2003;40(11):e118.
- Zito I, Downes SM, Patel RJ, et al. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J Med Genet. 2003;40(8):609–615.
- Hunter DG, Fishman GA, Mehta RS, et al. Abnormal sperm and photoreceptor axonemes in Usher’s syndrome. Arch Ophthalmol. 1986;104(3):385–389.
- Hurd TW, Fan S, Margolis BL. Localization of retinitis pigmentosa 2 to cilia is regulated by Importin 2. J Cell Sci. 2011;124(5):718–726.
- Sharon D, Bruns GA, McGee TL, et al. X-linked retinitis pigmentosa: mutation spectrum of the RPGR and RP2 genes and correlation with visual function. Invest Ophthalmol Vis Sci. 2000;41(9):2712–2721.
- Chang S, Vaccarella L, Olatunji S, et al. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Curr Genomics. 2011;12(4):267–275.
- Jayasundera T, Branham KE, Othman M, et al. RP2 phenotype and pathogenetic correlations in X-linked retinitis pigmentosa. Arch Ophthalmol. 2010;128(7):915–923.
- Mookherjee S, Hiriyanna S, Kaneshiro K, et al. Long-term rescue of cone photoreceptor degeneration in retinitis pigmentosa 2 (RP2)-knockout mice by gene replacement therapy. Hum Mol Genet. 2015;24(22):6446–6458.
- Meindl A, Dry K, Herrmann K, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet. 1996;13(1):35–42.
- He S, Parapuram SK, Hurd TW, et al. Retinitis pigmentosa GTPase regulator (RPGR) protein isoforms in mammalian retina: insights into X-linked retinitis pigmentosa and associated ciliopathies. Vision Res. 2008;48(3):366–376.
- Vervoort R, Lennon A, Bird AC, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25(4):462–466.
- Zhao J, Bacolla A, Wang G, et al. Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci. 2010;67(1):43–62.
- Sharon D, Sandberg MA, Rabe VW, et al. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73(5):1131–1146.
- Ferreira PA. Insights into X-linked retinitis pigmentosa type 3, allied diseases and underlying pathomechanisms. Hum Mol Genet. 2005;14(Spec No. 2):R259–67.
- Hadjebi O, Casas-Terradellas E, Garcia-Gonzalo FR, et al. The RCC1 superfamily: from genes, to function, to disease. Biochim Biophys Acta. 2008;1783(8):1467–1479.
- Megaw RD. Soares DC and Wright AF, RPGR: its role in photoreceptor physiology, human disease, and future therapies. Exp Eye Res. 2015;138:32–41.
- Shu X, Fry AM, Tulloch B, et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum Mol Genet. 2005;14(9):1183–1197.
- Natarajan K, Gadadhar S, Souphron J, et al. Molecular interactions between tubulin tails and glutamylases reveal determinants of glutamylation patterns. EMBO Rep. 2017;18(6):1013–1026.
- Sergouniotis Panagiotis I, Chakarova C, Murphy C, et al. Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am J Hum Genet. 2014;94(5):760–769.
- Sun X, Park JH, Gumerson J, et al. Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc Natl Acad Sci U S A. 2016;113(21):E2925–34.
- Linari M, Ueffing M, Manson F, et al. The retinitis pigmentosa GTPase regulator, RPGR, interacts with the delta subunit of rod cyclic GMP phosphodiesterase. Proc Natl Acad Sci U S A. 1999;96(4):1315–1320.
- Won J, Gifford E, Smith RS, et al. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum Mol Genet. 2009;18(22):4329–4339.
- Khanna H, Hurd TW, Lillo C, et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J Biol Chem. 2005;280(39):33580–33587.
- Murga-Zamalloa CA, Atkins SJ, Peranen J, et al. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: implications for cilia dysfunction and photoreceptor degeneration. Hum Mol Genet. 2010;19(18):3591–3598.
- Wright RN. Hong D-H and Perkins B, RpgrORF15Connects to the usher protein network through direct interactions with multiple whirlin isoforms. Invest Opthalmol Vis Sci. 2012;53(3):1519.
- Patnaik SR, Raghupathy RK, Zhang X, et al. The Role of RPGR and Its Interacting Proteins in Ciliopathies. J Ophthalmol. 2015;2015:414781.
- Rao KN, Anand M, Khanna H. The carboxyl terminal mutational hotspot of the ciliary disease protein RPGRORF15 (retinitis pigmentosa GTPase regulator) is glutamylated in vivo. Biol Open. 2016.
- Wright AF, Shu X. Focus on Molecules: RPGR. Exp Eye Res. 2007;85(1):1–2.
- Zeiss CJ. Acland GM and Aguirre GD, Retinal pathology of canine X-linked progressive retinal atrophy, the locus homologue of RP3. Invest Ophthalmol Vis Sci. 1999;40(13):3292–3304.
- Zhang Q, Acland GM, Wu WX, et al. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum Mol Genet. 2002;11(9):993–1003.
- Kropatsch R, Akkad DA, Frank M, et al. A large deletion in RPGR causes XLPRA in Weimaraner dogs. Canine Genet Epidemiol. 2016;3:7.
- Hong DH, Yue G, Adamian M, et al. Retinitis pigmentosa GTPase regulator (RPGR)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2000;276(15):12091–12099.
- Huang WC, Wright AF, Roman AJ, et al. RPGR-associated retinal degeneration in human X-linked RP and a murine model. Invest Ophthalmol Vis Sci. 2012;53(9):5594–5608.
- Thompson DA, Khan NW, Othman MI, et al. Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15. PLoS One. 2012;7(5):e35865.
- Mitamura Y, Mitamura-Aizawa S, Nagasawa T, et al. Diagnostic imaging in patients with retinitis pigmentosa. J Med Invest. 2012;59(1–2):1–11.
- Genead MA, Fishman GA and Lindeman M. Structural and functional characteristics in carriers of X-linked retinitis pigmentosa with a tapetal-like reflex. Retina. 2010;30(10):1726–1733.
- Wegscheider E, Preising MN and Lorenz B. Fundus autofluorescence in carriers of X-linked recessive retinitis pigmentosa associated with mutations in RPGR, and correlation with electrophysiological and psychophysical data. Graefes Arch Clin Exp Ophthalmol. 2004;242(6):501–511.
- Popovic P, Jarc-Vidmar M and Hawlina M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2005;243(10):1018–1027.
- Churchill JD, Bowne SJ, Sullivan LS, et al. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(2):1411–1416.
- Bell P, Wang L, Lebherz C, et al. No evidence for tumorigenesis of AAV vectors in a large-scale study in mice. Mol Ther. 2005;12(2):299–306.
- Li H, Malani N, Hamilton SR, et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood. 2011;117(12):3311–3319.
- Balaggan KS, Duran Y, Georgiadis A, et al. Absence of ocular malignant transformation after sub-retinal delivery of rAAV2/2 or integrating lentiviral vectors in p53-deficient mice. Gene Ther. 2012;19(2):182–188.
- Beltran WA, Cideciyan AV, Lewin AS, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci U S A. 2012;109(6):2132–2137.
- Deng W-T, Dyka FM, Dinculescu A, et al. Stability and safety of an AAV vector for treatingRPGR-ORF15X-linked retinitis pigmentosa. Human Gene Therapy. 2015;26(9):593–602.
- Wu Z, Hiriyanna S, Qian H, et al. A long-term efficacy study of gene replacement therapy for RPGR-associated retinal degeneration. Hum Mol Genet. 2015;24(14):3956–3970.
- Pawlyk BS, Bulgakov OV, Sun X, et al. Photoreceptor rescue by an abbreviated human RPGR gene in a murine model of X-linked retinitis pigmentosa. Gene Therapy. 2015;23(2):196–204.
- Fiers W, Grosjean H. On codon usage. Nature. 1979;277(5694):328.
- Sharp PM, Li WH. The codon adaptation index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15(3):1281–1295.
- Fischer MD, McClements ME, Martinez-Fernandez De La Camara C, et al. Codon-optimized rpgr improves stability and efficacy of AAV8 gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol Ther. 2017;25(8):1854–1865.
- Beltran WA, Cideciyan AV, Boye SE, et al. Optimization of retinal gene therapy for X-linked retinitis pigmentosa due to RPGR mutations. Mol Ther. 2017;25(8):1866–1880.
- Tanimoto N, Muehlfriedel RL, Fischer MD, et al. Vision tests in the mouse: functional phenotyping with electroretinography. Front Biosci (Landmark Ed). 2009;14:2730–2737.