
ABSTRACT
Introduction
Inborn errors of metabolism (IEMs) often result from single-gene mutations and collectively cause liver dysfunction in neonates leading to chronic liver and systemic disease. Current treatments for many IEMs are limited to maintenance therapies that may still require orthotopic liver transplantation. Gene therapies offer a potentially superior approach by correcting or replacing defective genes with functional isoforms; however, they face unique challenges from complexities presented by individual diseases and their diverse etiology, presentation, and pathophysiology. Furthermore, immune responses, off-target gene disruption, and tumorigenesis are major concerns that need to be addressed before clinical application of gene therapy.
Areas covered
The current treatments for IEMs are reviewed as well as the advances in, and barriers to, gene therapy for IEMs. Attention is then given to ex vivo and in vivo gene therapy approaches for hereditary tyrosinemia type 1 (HT1). Of all IEMs, HT1 is particularly amenable to gene therapy because of a selective growth advantage conferred to corrected cells, thereby lowering the initial transduction threshold for phenotypic relevance.
Expert opinion
It is proposed that not only is HT1 a safe indication for gene therapy, its unique characteristics position it to be an ideal IEM to develop for clinical investigation.
1. Introduction
Inborn errors of metabolism (IEMs) are a group of genetically inherited diseases that are often caused by single gene mutations [Citation1]. While individually rare, collectively 0.1% of all live births are associated with impaired liver function due to at least one of these IEMs [Citation2]. The majority of these diseases are due to mutations in genes that encode enzymes involved in specific cellular metabolic pathways. Most of the disorders involve accumulation of potentially toxic metabolite(s) in the target organ (often liver) or other consequences of impairment of metabolic pathways by inhibition of normal enzymatic function.
Despite the enormity of number and the wide range of systems that are affected by these diseases, the majority of them arise due to liver dysfunction at birth. Importantly, the effects often extend beyond the liver and may impair a vast range of organ functionality in affected patients. While each of these IEMs are associated with their own unique symptoms, many ultimately result in liver enlargement, dysfunction, and failure [Citation3] Decades of research have characterized the molecular mechanisms of most IEMs and have facilitated advanced approaches to developing therapies for these rare diseases. Of these, the unique characteristics of hereditary tyrosinemia type I (HT1) offer key advantages as a preliminary indication, which will be discussed further below. For perspective, representative significant metabolic pathways, as well as their associated gene and enzyme derivatives that are impaired in certain IEMs of the liver, are presented in . The advanced therapies needed to address these diseases are likely to be nearly as diverse as the diseases themselves. Therefore, a variety of approaches are discussed below.
Table 1. List of known inborn errors of metabolism with key features of each disease.
1.1. Current therapeutic approaches
1.1.1. General inborn errors of metabolism
Today’s treatments for IEMs of the liver include a regimen involving lifestyle alterations, small molecule drugs, cofactor supplementation, vitamins, and/or enzyme-replacement therapies. However, patient compliance with restricted diets presents a significant challenge to long-term efficacy of these treatments [Citation4–Citation7]. Over months and years these diseases often result in cellular damage that can be irreversible, and many diseases will continue to progress despite partial efficacy maintenance therapy, ultimately leading to complete organ failure. Furthermore, therapeutic potential of vitamin or cofactor supplement therapy depends on the specific genetic mutation inherited by the patient, as many of these diseases can result from a multitude of allelic variants that cause aberrant-to-absent enzymatic function. Some therapeutic approaches and their efficacy available to date for these diseases are presented in . Evidently, with the current therapies available only very few of these diseases can be treated to a satisfactory level, leaving most others with serious lifelong debilitating illness.
Table 2. Current treatment strategies for inborn errors of metabolism.
While research identifying the molecular mechanisms of IEMs has led to interim maintenance therapies for some diseases, proper cures has remained elusive for decades, leaving orthotopic liver transplant (OLT) as the only curative option. While OLT is indeed potentially curative for some IEMs, the feasibility is attenuated by availability of organ donors. There is a severe shortage of donor organs in the US and world-wide (see https://optn.transplant.hrsa.gov/data/view-data-reports/national-data/#); and those patients that do receive a transplant face lifelong consequences of immunosuppression and the potential for organ rejection. Currently, all available treatments for IEMs share a common attribute, that is – they do not provide an actual cure. This fact further underscores the need for improved therapies beyond current pharmacologic options and liver transplantation.
1.2. Current treatment strategies for hereditary tyrosinemia type 1
HT1 is an autosomal recessive IEM that affects approximately 1:100,000 live births [Citation8] with certain regions around the world experiencing rates as high as 1:16,000. Patients with HTI lack the intracellular enzyme fumarylacetoacetate hydrolase (FAH) responsible for the downstream metabolism of tyrosine [Citation9,Citation10]. This prevents proper metabolism of this essential amino acid and results in the buildup of maleylacetoacetic acid and fumarylacetoacetic acid, two hepatotoxic metabolic intermediates of tyrosine metabolism that can severely injure hepatocytes (). These metabolites are primarily responsible for the progression of disease, leading to inflammation, fibrosis, cirrhosis, liver failure, and often hepatocellular carcinoma (HCC) [Citation9,Citation11].
Figure 1. Tyrosine metabolism, with genetic cause of HT1 and pharmacologic intervention of NTBC noted.
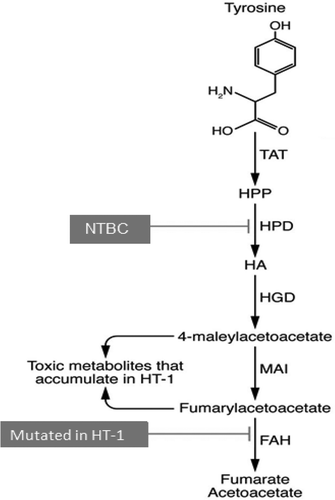
Among the few treatable IEMs, an effective maintenance therapy is available for HT1 in many developed countries. These patients are assigned to a regimen of dietary modification and oral administration of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC). Initially developed as a pesticide [Citation12], NTBC reversibly inhibits 4-hydroxyphenylpyruvate dioxygenase (HPD), usually reducing or delaying development of HT1-associated liver fibrosis, cirrhosis, and HCC. This enzyme is upstream of the causative FAH enzyme, thus significantly reducing formation of the downstream toxic intermediates of tyrosine metabolism responsible for the more severe phenotypes of the disease. NTBC treatment provides significant reduction or delay of HT1 symptoms and also decreases the tendency to develop HCC in the first decade of life [Citation13]. When administered prior to 1 month of age, NTBC treatment has resulted in no detectable liver disease for more than 5 years in a study of patients in Quebec [Citation13]. However, the compromise of this treatment is replacing HT1 for HT3, as NTBC’s inhibition of HPD mimics this less-severe tyrosinemia. Like HT3 patients, HT1 patients on NTBC can suffer from progressive and significant cognitive and ocular impairment [Citation14,Citation15], and an increasing body of literature is showing that HT1 patients on stable NTBC regimens can still develop HCC and require liver transplantation later in life [Citation16]. Further, the cost of NTBC is high, equaling approximately 300 USD US for one 10 mg capsule [Citation17], resulting in a cost of 1200 USD/day for effective treatment of a 20 kg child dosed at a typical 2 mg/kg [Citation18] (). In 2019, a generic and bioequivalent version of NTBC called NITYR was introduced to the market. Furthermore, programs exist as of the preparation of this review that offer a 0 USD co-pay to eligible commercially insured patients up to 15,000 USD annually [Citation19], which would further defray the costs of this expensive treatment regimen for eligible patients.
Figure 2. Cost per day of NTBC from 3 mo to 20 yr.

While gene therapy has the potential for a one-time curative therapy, recent evidence suggests that the natural protein tolerance in patients with HT1 improves with age, thereby potentially decreasing the difficulty with dietary adherence among patients who have not already undergone liver transplantation [Citation20]. Further, the costs of gene therapy are nonetheless substantial. Although dependent on the final manufacturing and clinical costs, the total cost of gene therapy for a young (small) HT1 patient is likely 500 USD k–1 M USD, with at least 300 USD k needed just for production of the vector based on current costs for large amounts of lentiviral vector particles. However, over time the quality of life benefits and savings relative to NTBC maintenance () ultimately support this cost.
By way of comparison, hemophilia A currently has an available gene therapy option, and this is actually more cost effective when compared with the disease’s standard prophylactic treatment with factor VIII. Over 10 years, total gene therapy costs were 1.0 USD M per person versus prophylaxis cost of 1.7 USD M per person. Quality-adjusted life-years (QALYs) were also superior for gene therapy (8.33) versus standard therapy (6.62) [Citation21]. Further investigation into other existing cell and gene therapies for spinal muscular atrophy, oncology, inherited retinal disease, and other genetic diseases in addition to hemophilia has revealed significant increase of QALY gains of cell and gene therapies versus conventional drugs and biologics for all disorders [Citation22].
1.3. Development of gene therapy
1.3.1. Gene therapy in general inborn errors of metabolism
While recent advances in gene therapy and delivery methods have shown promise for replacing defective genes with corrected functional isoforms, or even editing the actual mutations, multiple significant challenges remain [Citation23]. Current research is thus calibrating not only delivery and activation of corrected genes, but also ways to overcome these challenges such as avoiding unwanted immunological responses, disruption of key genes in target cells during delivery, chances of infection, tumorigenesis and long-term disease-free survival. Furthermore, it is also crucial to maximize transduction efficiencies or engineer selective advantages for the gene-corrected hepatocyte so that it can repopulate the damaged liver to replace native diseased hepatocytes without causing significant injury or stimulating an immune response.
Gene therapy offers a chance for a true cure for IEMs, though techniques for gene therapy must be designed and implemented in a fashion that is catered to the individual attributes of each disease or disease subtypes (). While individually unique, based on certain commonalities in the molecular pathways, which play crucial role in determining potential challenges to gene delivery methods (i.e., location of enzyme action and disease phenotype), all IEMs of the liver can be categorized into three main groups for gene therapy. Overlapping features in each category may allow efficient approaches to target each set of disorders collectively. On this basis, IEMs have been clustered based on (a) presence of a positive selective advantage for corrected cells, (b) liver-centric vs. systemic disease progression (i.e. site of action of either the affected enzyme or distribution of aberrant substrates/metabolites), and (c) immunologic consequences of correction (i.e. immune responses to the novel gene product) [Citation24]. Based on the age of the patient, site of action of the enzyme that is associated with each IEM, and the number of cells needed to be corrected, many IEMs can be targeted by a similar gene therapy approach depending on the criteria indicated above. Proper understanding of the mode of action and the molecular consequences of the corrected enzyme is important for development of effective gene therapy cures of these diseases.
Phenylketonuria (PKU) is representative of a particular type of IEM of the liver. Although lack of PAH enzyme in the liver leads to accumulation of serum phenylalanine, the toxic effect primarily influences other sites outside the liver, while the liver remains largely unaffected by the disease [Citation25]. Consequently, in contrast to correction or replacement of a mutant FAH gene in HT1, PAH gene modification/replacement does not allow any positive selection for the cured hepatocytes. This feature puts PKU and most other IEMs in a distinct category. For these IEMs a modified approach for gene therapy will be needed, as inadequate numbers of hepatocytes are corrected with current approaches. To overcome this limitation, advancements will need to be made in three possible areas: (1) the ability to reintroduce the gene therapy without immunologic consequence, (2) improved initial transduction and targeting of cells, and/or (3) deliver a supplemental selective advantage to the corrected hepatocytes so the corrected cells have a survival advantage as seen in HT1. To address the specific challenge in PKU, there is current research into taking advantage of the hepatotoxicity of acetaminophen in endogenous cells, while corrected cells would also be edited to avoid the toxic metabolic formation of N-acetyl-p-benzoquinone imine (NAPQI) from this dosing. However, this work is still in development, and presentation has been limited to abstract form. The ultimate goal of this or other similar strategies would be to introduce a selective advantage for the corrected cells over the uncorrected cells, in this case allowing healthy hepatocytes to replace native-diseased hepatocytes that are harboring only mutant PAH alleles. Similarly, other IEMs of this category have to be targeted for not only correction of the mutant gene but also will require creation of a selective advantage for the cured hepatocyte sufficient to create a phenotypically relevant biomass of corrected cells. An alternative is to use a bioreactor to grow selectively corrected hepatocytes from the patient and transplant these corrected cells over time to achieve a sufficient mass of corrected hepatocytes back to the patient.
Alpha-1 antitrypsin deficiency (A1AD) represents perhaps the most challenging subtype of IEMs of the liver. A1AD is a genetic disorder caused by a mutation in the SERPINA1 gene, which leads to a buildup of the α1-antitrypsin protein in hepatocytes and unblocked neutrophil elastase activity in the lung and liver [Citation26]. α1-antitrypsin (aka alpha-1-antiproteinase) is an enzyme that is secreted by hepatocytes for systemic availability, where it has a primary role in maintaining proper lung functionality. In A1AD the enzyme is not properly secreted by the liver and builds up inside hepatocytes causing severe liver injury. Correcting hepatocytes suffering from A1D1 may lead to a survival advantage for the engineered cells, but any attempted gene therapy in these hepatocytes for a corrected protein may elicit an immune response to the novel gene product. This occurs because the protein is not present during negative T-cell selection in utero and does not remain intracellular, allowing for a possible immune response to the corrected secreted enzyme. While no standard method is available for gene therapy for these sets of diseases, these hepatocytes are likely to require a minimum of three-fold modification; correction of the mutant gene, invention of selective advantage for the hepatocyte and also a tolerization to, or inducible immune suppression for, the novel antigen of the newly introduced corrected enzyme.
1.4. Gene therapy in HT1
HT1 represents the best example of a disorder with a selective advantage for a corrected hepatocyte. For HT1, the selective advantage comes from the presence of toxic metabolites in the diseased cells that cause their demise, creating the stimulus and matrix for expansion of even small numbers of corrected cells [Citation27]. Cycling treated subjects off of NTBC, the drug that causes reversible inhibition of HPD, results in death of the native uncorrected cells. In animal models, this has allowed the corrected cells to rapidly expand in their stead and repopulate the injured liver [Citation28–Citation31]. With the selective pressure of the disease and the regenerative capability of the liver, initial correction of as low as 0.1–1.0% of native hepatocytes can lead to complete repopulation of the liver via expansion of only corrected cells [Citation28], although it is reasonable to postulate that higher numbers of initially corrected cells would result in more efficient cure. Thus, IEMs of this category require targeting of a single allele or delivery of a working copy of the gene somewhere in the genome. Therefore, HT1 provides a favorable context for gene therapy due to this low threshold for initial transduction or correction, which has resulted in cures for animal models [Citation28,Citation31,Citation32], and high predictive value that this mechanism would also support a cure in human patients.
1.4.1. Vectors utilized for HT1 gene therapy
To date, the predominant vectors utilized for HT1 gene therapy are adeno-associated viral vectors (AAV) and lentiviral vectors (LV). AAV vectors primarily exist as episomes in the host cell and therefore are not transferred to both daughter cells after cell division. Several rounds of division may be overcome with high copy numbers of vector per cell, but AAV gene delivery has been shown in numerous studies to lose efficacy over a period of days to weeks in neonatal animal models preventing their effective use in treating HT1. Very low frequency genomic integration is possible with these vectors, and they have been shown to be carcinogenic when using certain liver specific promotors [Citation33]. Because AAV gene therapy loses efficacy over a relatively short period in the neonatal patient, additional treatments with the AAV vectors are required to maintain any therapeutic benefit. This repeat administration, however, can lead to a severe, potentially fatal immune response [Citation34,Citation35].
To date, LV gene therapy has not been shown to be carcinogenic and displays benign genomic integration profiles [Citation36–Citation38]. LV is stably maintained in the host genome and therefore readily passed to both daughter cells during cell division. With low levels of initial transduction being efficacious in HT1, there is no need for multiple administrations. A primary concern with LV gene therapy is the potential for integration into sites responsible for tumor regulation. Such integration could alter expression of oncogenes, potentially resulting in tumorgenicity [Citation39].
1.4.2. Ex vivo gene therapy approaches for HT1
In ex vivo approaches, diseased cells are harvested from the donor, followed by gene therapy applied to the cells in vitro before autologous transplantation back into the subject [Citation40]. Gene correction is generally done with the help of viral or nonviral vectors. Liver focused IEMs are treated by correction of the hepatocytes that are carrying the mutant gene.
HT1 has been experimentally targeted by ex vivo gene delivery approaches where hepatocytes are collected from a syngeneic donor (mouse) [Citation41] or via laparoscopic partial resection of the liver in the actual test animal (pig) [Citation29,Citation42]. These hepatocytes are cultured and transduced with LV vectors carrying a wildtype FAH to rescue normal function and cure the disease [Citation40]. This method has demonstrated significant efficacy in mouse and pig models of HT1 without increasing the potential for HCC in pigs [Citation29,Citation37]. Ex vivo AAV-based CRISPR/Cas9-mediated gene editing has successfully been achieved in HT1 mouse [Citation30,Citation32]. Additionally, a pig treated ex vivo was maintained for 3 years after dosing, and showed no adverse effects or tumorigenicity [Citation29]. An alternative approach designed to disrupt the HPD gene (the target of NTBC therapy) by using similar gene editing methods showed efficacy in treating test animals, but this results in subjects with a second homozygous or compound heterozygous genetic mutation, which results in sustained HT1 and HT3 phenotypes [Citation43]. Notably, in this approach the genomic copy of the mutant FAH gene remains unchanged, and these subjects would require more than correction of the mutant FAH if/when a gene therapy is available due to this additional upstream mutation in HPD ().
In some instances, gene targeting methods are used where, in contrast to gene delivery, the mutant genomic allele itself is subjected for gene editing. In this method, a vector is used to deliver a nuclease (often Cas9) and a homology-directed guide (such as a guide RNA), while a subsequent vector is used to deliver up to a 1.2 kb homology template (i.e. AAV-HT) [Citation30]. This method significantly improved the frequency of hepatocyte correction in FAH mutant mice [Citation29]. However, production of functional Cas9 using viral vectors relies on the transcription and translation of the ectopic vector and often has limitations. Although tissue tropism can be controlled to some extent with vector manipulations, proper pseudotyping, and tissue-specific promoters, there is significant potential risk associated with the systemic administration of nucleases, such as Cas9 due to the possibility of off-target genomic cutting in target and non-target tissues. Use of synthetic ribonucleoproteins (RNPs) or exosomes that use purified Cas9 protein and purified cr-RNA and tracer-RNA to deliver a final product to the cells has turned out to be efficacious; this method uses standard transfection or electroporation of the target cells prior to reintroduction to the host subject.
1.4.3. Ex vivo gene therapy approaches for other IEMs
Application of similar gene therapy approaches for many other related IEMs shows promise in animal models. AAV-based gene transfer methods in murine models have produced successful results for glycogen storage diseases (GSD) [Citation44,Citation45], Phenylketonuria (PKU) [Citation45] and urea cycle disorders (Citrullinemia) [Citation46]. Similar AAV- based gene delivery methods have resulted in positive outcomes for many other rare diseases including- Niemann-Pick C [Citation47,Citation48], primary oxalosis [Citation49,Citation50], methylmalonic acidemia [Citation51–Citation53], amyloidosis [Citation54,Citation55], and congenital glycosylation disorders [Citation56]. However, AAV approaches for various types of porphyria have produced limited success [Citation57]. While both AAV and RNAi methods have been attempted for acute intermittent porphyria (AIP) [Citation58], proper gene therapy approaches are still to be identified for other types of porphyria. The major limitation with AAV gene therapy is loss of transgene expression. This is a significant limitation in neonatal and pediatric populations that will undergo significant cell division as they grow.
1.4.4. In vivo gene therapy approaches for HT1
In the in vivo approach, subjects are intravenously administered LV or AAV vectors that contain the transgene under an appropriate promoter for the indication- constitutive, tissue-specific, or inducible. In vivo gene therapy precludes the necessity for surgery, though there are important limitations to in vivo gene delivery in adults. These limitations include muted efficiency due to immune responses to the viral vector delivery systems, poor tissue tropism with many nonviral delivery vehicles, and low frequency of cell cycling in mature tissue that contributes to low rates of homology directed repair (HDR) in gene editing. AAV vectors pose the limitation of preexisting neutralizing antibodies after exposure to naturally occurring virus [Citation34,Citation59] and HIV-derived LV vector use requires immunosuppression due to complement activation than can lead to a cytotoxic response [Citation60,Citation61]. Gene therapy applied prior to the development of a fully functional immune system may not only sidestep concerns of sensitization and immune reaction to the viral vector itself, but it may induce stable tolerance to the transgene introduced [Citation62]. In addition, any gene therapy method known to date poses some threat of off-target deleterious effect on the genome. Thus, optimizing condition at which HDR can efficiently occur to repair double strand breaks of the DNA and rescue genomic normalcy is important. While, other methods of DNA repair methods such as NHEJ or excision repair pathways are used by cells under certain conditions, HDR is the most error-proof method that needs to be emphasized during genome editing.
Both mouse [Citation31] and now larger animal models of HT1 (Nicolas, in preparation) that were administered LV-containing functional FAH in this fashion demonstrated reconstitution of hepatocyte metabolic function and repopulation of the liver by the transduced cells following weaning from NTBC [Citation31,Citation40]. A recent study demonstrated that a therapeutic dose of a LV construct had no negative impact on toxicology, clinical pathology, or histology findings in wild type mice [Citation31]. In vivo administration of a LV vector expressing FAH directly is currently underway and has demonstrated metabolic cure of the disease. This approach is innovative for two major reasons; (1) current gene therapy approaches for this and similar diseases are primarily focused on using AAV vectors for gene delivery because of high tissue tropism and (2) direct administration to the portal vein via percutaneous injection is difficult due to accessibility to the procedure. Thus far, portal vein delivery of LV-huFAH into the Fah-/- pig model has produced encouraging results, with no tumorigenic events observed in any dose group. Interestingly, animals given in vivo LV-huFAH show NTBC-independent body weight gains earlier after treatment than those receiving ex vivo LV-huFAH [Citation28], requiring only four cycles of the maintenance drug prior to weaning from it (Nicolas, in preparation).
Alternatively, gene editing by in utero injection of LV or AAV vectors is also showing successful correction of the mutant FAH allele in HT1 mouse and pig models (under current study), further demonstrating the powerful selective advantage for even a small percentage of corrected hepatocytes in this disease context. In utero gene editing using a CRISPR-based editor system in an HT1 mouse model has edited the Hpd gene upstream of Fah, successfully turning the disease into the less severe HT3 [Citation43]. These experiments deliver proof of principle of the feasibility of performing prenatal gene therapy in animal models of human fetal physiology. Targeting the immune privileged fetal environment can limit the issue of unintended immune responses and can simultaneously prevent early organ damage from disease. This effect is important in the case of HT1, where enabling curative treatment to take place before oxidative insult occurs could offer an opportunity for improvement of both hepatic and neurologic outcomes. There has been recent success with porcine in utero gene therapy for HT1 (manuscript in progress) in addition to in vivo neonatal success.
1.5. In vivo gene therapy approaches for other IEMs
Encouraged by these results in HT1 models, similar in vivo gene editing strategies have been engineered to treat PKU. In this method gene correction is driven by a combination of lentiviral and AAV- vectors that are designed to correct a mutant PAH gene in hepatocytes of various animal models. These approaches are allele-specific by nature, and there are at least over 1000 known alleles that cause PKU [Citation63], theoretically each requiring specific modification to any gene targeting platform employed. However, while direct gene therapy is possible for some of the IEMs such as HT1, it is more challenging for diseases that confer no selective advantage for corrected hepatocytes or may introduce potential for immune responses due to expression of novel proteins. Thus, development of therapy by in vivo or ex vivo techniques for many IEMs requires additional modifications to promote initial correction efficiency to phenotypically-relevant levels or create an inducible selection pressure for corrected cells.
A number of in vivo gene therapy methods have been reported for other IEMs of the liver. For study of the relatively more challenging Wilson disease, rabbit models were generated by using CRISPR/Cas9 driven homology-directed precision point mutations in the ATP7B gene [Citation64]. Lentiviral transfer of corrected ATP7B gene at an early gestational phase have produced successful outcomes in a murine model [Citation65]. In addition, AAV driven delivery of ATP7B cDNA has also resulted in reduction of serum transaminases and urinary copper secretion, indicative of efficacy in the WD model [Citation66]. Aside from WD, in utero AAV-based CRISPR/Cas9 driven gene transfer as well as nanoparticle-based gene delivery has been successful in treating murine models of Hemophilia B [Citation67–Citation69].
1.6. Challenges of gene therapy
While gene therapy approaches have been promising to date in murine and porcine models of HT1 and a few other IEMs, more work is needed prior to safe application in clinical trials. Some of the major challenges are disease specific; however, the commonalities for most of these IEMs come from methods of gene delivery and activation. Based on the gene delivery method, cultured cells from the animal or cells in the organ itself become exposed to an integrating virus (lenti) or AAV that contains a wildtype copy of the gene for replacement or carrying a CRISPR/Cas9 cassette that is capable of therapeutic gene editing in vivo. Either of these methods can expose the rest of the genome to additional (off target) mutations, and thus create potential for further genotoxic consequences, such as- disruption of fundamental cellular function, cellular senescence, cell death, or tumorigenesis.
In addition, while only some of these metabolic genes are linked to tumors to date, and it is unknown whether any specific gene mutation that is causal to metabolic disease is also linked with tumorigenicity, the major concern still comes from off target genomic editing. This raises the potential for future tumor development due to interruption of any tumorigenic signaling or suppression and thus, remains a major debate topic against applying gene therapy.
Use of nanoparticles has emerged as an alternate approach for gene delivery into human hepatocytes both in culture and in mouse xenograft models. In this method copies of DNA or mRNA are delivered to the diseased organ with polyethylenimine or poly-lactic-co-glycolic acid- or lipid-based nanoparticles [Citation70], or in combination with viral vectors, as has been done to cure the HT1 mouse [Citation71]. Although these vectors have some advantages and preliminary encouraging results, this method presents unique challenges that are associated with the delivery system, rapid degradation and clearance from the circulation (insufficient half-life), nonspecific uptake, reduced receipt by target cells, and general toxicity. In addition, numerous factors such as understanding of the structure-function relationships of each IEM, anatomical barriers, nucleic acid stability in the presence of liver enzymes, and delivery routes present major clinical challenges.
Gene therapy often raises additional challenges, such as unintended immune responses due to expression of viral vectors that are used in the delivery method as well the newly produced proteins that are replacing the mutant enzyme in the target hepatocytes [Citation34]. This may cause inflammation and, in severe cases, organ failure. While temporary immune suppression has been attempted to help introduction of the corrected gene into the hepatocytes it can enhance chances of infection from outer sources as well as the viruses itself. While direct ex vivo gene therapy is possible for some IEMs such as HT1, it is currently significantly hindered for diseases that confer no selective advantage for corrected hepatocytes or introduce potential for immune responses due to expression of novel proteins. In addition, low efficiency correction of the targeted cells can be another rate limiting factor for most delivery approaches. Given the expensive lifetime treatment required for HT1 with conventional therapy, it is likely that patients will be receptive to gene therapy options for this disease when a safe and cost-effective option is available (see above). Regardless, potential gene therapy patients must be made aware about all real and hypothetical risks of standard versus gene therapy.
1.7. Benefits of safe gene therapy
Gene therapy has great potential to provide effective cures with a single procedure for diseases that currently cannot be treated. However, these approaches are neither simple nor inexpensive, and the long-term effects of the associated genomic modifications are practically impossible to completely de-risk preclinically. Rare and genetically inherited IEMs that otherwise do not have proper cure can be treated with ex vivo gene therapy that uses autologous hepatocyte harvest and the subsequent transplantation of the corrected cells in the liver. The invasiveness of this procedure may still be justified by providing patients relief from disease and the lifetime expenses of dietary modifications, maintenance drugs and surveillance programs, especially when it can extend/save lives or obviate more invasive procedures like OLT.
Current gene therapy methods require careful application and further refinement for safe and effective deployment to human medicine. The public stigma around genomic tinkering has only been sensitized by recent events, such as the creation of the ‘CRISPR twins’ [Citation72] that propagate the perceived potential for species-wide disaster. Development of safe in vivo delivery should consider calibrating the safest time for the integration of the corrected gene in the hepatocyte genome. For example, to reduce the probability of nonspecific gene disruption, gene integration can be conducted in certain phases of the cell cycle when cellular proliferative and replicative activities are optimum, thus allowing efficient repair of DNA double strand breaks. Knowledge from ongoing research to calibrate timing and developmental state would be incorporated during application of these therapies. Temporal attenuation of certain error-prone DNA repair pathways and selective enhancement of homology directed repair (HDR) can also be evaluated to reduce the possibility of undesired genomic mutations. Thorough analysis of genomic integration and gene expression profiles remains fundamental to the understanding of the biology and can offer crucial refinement to these approaches. Furthermore, follow up functional studies that are guided by gene expression analysis have potential for finding inducible switches that can be used to enhance efficiency and safety of these ectopic gene modifications. Rigorous functional characterization of cells in vitro that has undergone gene therapy is crucial to ensure safety of each approach. Recent research indicates that gene editing procedures can be seamless and well tolerated with transient inhibition of p53 to avoid acute DNA damage responses [Citation73].
Gene therapy has shown promise in humans for several conditions to date. The actual application of gene therapy varies widely depending on the medical condition that is being treated. A successful clinical trial for Hemophilia A, for instance, involved a single intravenous dose of AAV serotype 5 vector encoding the domain of the deleted human factor VIII, which is missing in affected patients. In the high-dose cohort, factor VIII activity level gradually increased until reaching a plateau at or above physiologic levels between weeks 20–24, which was maintained at 1 year [Citation74]. A phase 1/2 clinical trial is currently underway with LV gene therapy for subjects with Type 1 Gaucher Disease involving a single infusion with long-term follow-up (see clinicaltrials.gov). While further studies are needed, with informed fine-tuning of the current approaches and homology guided in vivo gene integration methods, gene therapy can provide legitimate hope for curing many complex and rare diseases including liver IEMs, starting with HT1 due to its accommodating attribute of corrected-cell expansion.
2. Conclusion
Current therapies for inborn errors of metabolism range from ineffective to sustaining, but none are curative, and most only delay significant disease manifestation while imparting onerous lifestyle inhibitions and costs on patients that challenge the resolute compliance required for efficacy. Gene therapy is a bright future for the treatment of many diseases, including inborn errors of metabolism. However, these diseases have unique contexts, and there is no single approach to address them all. Favored modalities will have to consider the anatomical sites affected by the disease (within the liver or remote/systemic), the potential for immunologic activation by novel surface or secreted proteins, and the inherent or artificial nature of the selection applied to corrected cells. Furthermore, there may be multiple causative alleles within a single disease indication that require unique modifications to any proposed gene-targeted therapy to be applicable to an entire patient population. Refinements to current methods and entirely new platforms are constantly being developed and investigated to address these issues. Until then, patients will have to continue to endure lifestyle compromises and uncertain futures, with the possibility of needing liver transplants, reduced quality of life, and shortened life spans. These needs will continue to push innovation in gene therapy until science produces true cures for these patients.
3. Expert opinion
The need for gene therapy for IEMs is undeniable, but these indications are actually quite ideally suited to gene therapy development. Vector-born addition of a single functional transgene or successful correction of a single mutant allele (often in the context of compound heterozygosity) in the target hepatocyte is sufficient to cure most of these recessive diseases. Therefore, the goal can be simplified to be the safe and specific delivery of functional correction in a sufficient number of target cells to impact the phenotype. Of these four keys to the success of gene therapy, hereditary tyrosinemia eliminates one of these major obstacles by conferring a selective growth advantage to corrected cells, lowering the initial transduction threshold for phenotypic relevance. While it is estimated that curing PKU would require a minimum of 10% of hepatocytes to begin to produce PAH [Citation75], initial transduction targets in HT1 could be substantially less while ultimately generating complete correction of the liver. Residual unconverted pockets of cells (if any) would ultimately be eliminated by the toxicity of aberrant metabolism while lacking the overall fibrotic environment needed to develop HCC in human patients.
Therefore, we propose that not only is HT1 a safe indication for gene therapy, it should be the first indication for clinical development since it eliminates a key obstacle. Although low transduction efficiency is a very important topic for investigation and refinement, it is currently overcome in part by increasing the initial exposures. This is not necessary in HT1 due to the expansion of corrected cells, meaning less vector is needed to treat these patients. This has positive impacts in both reduced manufacturing cost and reduced exposure, where the latter means less foreign material available to elicit an immune response or cause off-target toxicity.
Regardless, a predominant sentiment in the field has been that HT1 is not a viable target for gene therapy because any small percentage of uncorrected cells could eventually seed HCC formation, and converting 100% of hepatocytes was not realistic. Indeed, in the initial description of curing the HT1 mouse with a retroviral vector-based gene therapy. Grompe et al. describe effective cure of the disease phenoptype [Citation27] but later describe that many treated mice still develop HCC in the long-term despite high levels of correction [Citation76]. However, we have not observed this in any of the HT1 mice or pigs cured with lentiviral vectors in our laboratory, and there is growing additional support for HT1 as an indication for gene therapy. Mouse models predicted [Citation77], and human clinical data are bearing out, that NTBC is not curative for these patients. Indeed, for many the disease still progresses requiring OLT, often due to HCC, despite clinically effective NTBC administration. Ultimately, patients still die from this disease [Citation78,Citation79].
NTBC has certainly been a life-giving option for HT1 patients [Citation13], but we should not rest at the development of NTBC and dismiss HT1 for the purposes of gene therapy. Instead, we must continue to innovate on behalf of this patient population in need. With separate bodies of research demonstrating (1) the continued progression of disease in HT1 patients maintained on NTBC and (2) the safety of lentiviral vectors in gene therapy, we find ourselves at an intersection where the two must meet, and the most translatable model available predicts a highly favorable outcome [Citation80].
Article highlights
Inborn errors or metabolism are ideal candidates for gene therapy due to their recessive monogeneic nature.
IEMs can be classified based on key characteristics that provide insight into the best-suited therapeutic approaches.
Use of AAV or LV should be based on indication, target population, and severity of the disease being treated.
Gene therapy is the undeniable future for treatment of IEMs, although demonstration of safety and cost mitigation barriers remain.
Hereditary tyrosinemia type I is a perfect candidate for gene therapy based on an inherent growth advantage for corrected cells that overcomes current inefficiencies in gene therapy.
Declaration of interest
JBL is an inventor on a patent regarding gene therapy for metabolic diseases. RAK and JBL are founding board members of Cytotheryx, Inc., with affiliated ownership. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Hansen K, Horslen S. Metabolic liver disease in children.Liver Transpl. 2008 [May];14(5):713–733.
- Jalan AB. Treatment of inborn errors of metabolism. Mol Cytogenet. 2014;7(Suppl 1 Proceedings of the International Conference on Human):I42.
- Hegarty R, Hadzic N, Gissen P, et al. Inherited metabolic disorders presenting as acute liver failure in newborns and young children: King’s college hospital experience. Eur J Pediatr. 2015 [Oct];174(10):1387–1392.
- Zeybek AC, Kiykim E, Soyucen E, et al. Hereditary tyrosinemia type 1 in Turkey: twenty year single-center experience. Pediatr Int. 2015 [Apr];57(2):281–289.
- Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type I: long-term outcome in French patients. J Inherit Metab Dis. 2008 [Feb];31(1):81–87.
- Al Hafid N, Christodoulou J. Phenylketonuria: a review of current and future treatments.Transl Pediatr. 2015 [Oct];4(4):304–317.
- Belanger-Quintana A, Burlina A, Harding CO, et al. Up to date knowledge on different treatment strategies for phenylketonuria. Mol Genet Metab. 2011;104(Suppl):S19–25.
- Tanguay R. Hereditary tyrosinemia, pathogenesis, screening, and management. In: Tanguay R, editor. Advances in Experimental Medicine and Biology:. Vol. 1. Cham, Switzerland: Springer; 2017:5.
- Lindblad B, Lindstedt S, Steen G. On the enzymic defects in hereditary tyrosinemia.Proc Natl Acad Sci USA. 1977 [Oct];74(10):4641–4645.
- Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1.Semin Liver Dis. 2001 [Nov];21(4):563–571.
- Endo F, Sun MS. Tyrosinaemia type I and apoptosis of hepatocytes and renal tubular cells.J Inherit Metab Dis. 2002 [May];25(3):227–234.
- Lock EA, Ellis MK, Gaskin P, et al. From toxicological problem to therapeutic use: the discovery of the mode of action of 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC), its toxicology and development as a drug. J Inherit Metab Dis. 1998 [Aug];21(5):498–506.
- Larochelle J, Alvarez F, Bussieres JF, et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Quebec. Mol Genet Metab. 2012;107(1–2):49–54. Sep.
- Thimm E, Richter-Werkle R, Kamp G, et al. Neurocognitive outcome in patients with hypertyrosinemia type I after long-term treatment with NTBC. J Inherit Metab Dis. 2012 [Mar];35(2):263–268.
- Bendadi F, de Koning TJ, Visser G, et al. Impaired cognitive functioning in patients with tyrosinemia type I receiving nitisinone. J Pediatr. 2014 [Feb];164(2):398–401.
- van Spronsen FJ, Bijleveld CM, van Maldegem BT, et al. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifluoro- methylbenzoyl)-1, 3-cyclohexanedione treatment. J Pediatr Gastroenterol Nutr. 2005;40(1):90–93. Jan.
- [cited 2020 Jun 25]. Available from: www.drugbank.ca.
- van Ginkel WG, Rodenburg IL, Harding CO, et al. Long-term outcomes and practical considerations in the pharmacological management of Tyrosinemia Type 1. Paediatr Drugs. 2019 [Dec];21(6):413–426.
- [cited 2020 Jun 25]. Available from:www.nityr.us.
- Hjalmarson O, Holme E, Lindstedt S. NTBC therapy in Tyrosinemia Type I: long time results 114.Pediatr Res. 1996 [1996 September 01];40(3):534.
- Machin N, Ragni MV, Smith KJ. Gene therapy in hemophilia A: a cost-effectiveness analysis.Blood Adv. 2018 [Jul 24];2(14):1792–1798.
- Chambers J, Silver MC, Lin PJ, et al. Cell and Gene therapies are associated with substantially larger quality-adjusted life year gains than conventional drugs and biologics. Value Health. 2019 [May];22:S263–S263.
- Zabaleta N, Hommel M, Salas D, et al. Genetic-based approaches to inherited metabolic liver diseases. Hum Gene Ther. 2019 [Oct];30(10):1190–1203.
- VanLith C, Kaiser RA, nicolas CT, et al. Ex vivo Gene therapy: a “cultured” surgical approach to curing inherited liver disease [Mini review]. OAJS. 2019 [2019 Mar 21];10(3):4.
- Grisch-Chan HM, Schwank G, Harding CO, et al. State-of-the-Art 2019 on Gene therapy for Phenylketonuria. Hum Gene Ther. 2019;30(10):1274-1283.
- Strnad P, McElvaney NG, Lomas DA. Alpha1-Antitrypsin deficiency.N Engl J Med. 2020 [Apr 9];382(15):1443–1455.
- Overturf K, Al-Dhalimy M, Tanguay R, et al. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996;12(3):266–273. Mar.
- Hickey RD, Mao SA, Glorioso J, et al. Curative ex vivo liver-directed gene therapy in a pig model of hereditary tyrosinemia type 1. Sci Transl Med. 2016;8(349):349ra99. Jul 27.
- Hickey RD, Nicolas CT, Allen KL, et al. Autologous Gene and cell therapy provides safe and long-term curative therapy in a large Pig model of hereditary Tyrosinemia Type 1. Cell Transplant. 2019 Jan;28(1):79-88.
- VanLith C, Guthman R, Nicolas CT, et al. Curative Ex Vivo Hepatocyte-directed Gene editing in a Mouse model of hereditary Tyrosinemia Type 1. Hum Gene Ther. 2018 [Nov];29(11):1315–1326.
- Kaiser RA, Nicolas CT, Allen KL, et al. Hepatotoxicity and Toxicology of In Vivo Lentiviral vector administration in healthy and liver-injury Mouse models. Hum Genet Ther Clin Dev. 2019;30(2):57–66. Apr 11.
- VanLith CJ, Guthman RM, Nicolas CT, et al. Ex Vivo Hepatocyte reprograming promotes homology-directed DNA repair to correct metabolic disease in Mice after transplantation. Hepatol Commun. 2019 [Apr];3(4):558–573.
- Chandler RJ, LaFave MC, Varshney GK, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015 [Feb];125(2):870–880.
- Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy.Blood. 2013 [Jul 4];122(1):23–36.
- Colella P, Ronzitti G, Mingozzi F Emerging Issues in AAV-mediated In Vivo Gene therapy. Mol Ther Methods Clin Dev 2017 Dec 1;8:87–104.
- Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013 [Aug 23];341(6148):1233158.
- Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013 [Aug 23];341(6148):1233151.
- Rittelmeyer I, Rothe M, Brugman MH, et al. Hepatic lentiviral gene transfer is associated with clonal selection, but not with tumor formation in serially transplanted rodents. Hepatology. 2013 [Jul];58(1):397–408.
- Visigalli I, Delai S, Ferro F, et al. Preclinical testing of the safety and tolerability of Lentiviral vector-mediated above-normal Alpha-L-Iduronidase expression in Murine and human Hematopoietic cells using toxicology and biodistribution good laboratory practice studies. Hum Gene Ther. 2016 [Oct];27(10):813–829.
- Kaiser RA, Mao SA, Glorioso J, et al. Lentiviral vector-mediated gene therapy of hepatocytes Ex Vivo for autologous transplantation in Swine. J Vis Exp. 2018;141. Nov 4.
- Overturf K, Al-Dhalimy M, Manning K, et al. Ex vivo hepatic gene therapy of a mouse model of hereditary Tyrosinemia Type I. Hum Gene Ther. 1998 [Feb 10];9(3):295–304.
- Nicolas CT, Hickey RD, Allen KL, et al. Hepatocyte spheroids as an alternative to single cells for transplantation after ex vivo gene therapy in mice and pig models. Surgery. 2018 [Sep];164(3):473–481.
- Rossidis AC, Stratigis JD, Chadwick AC, et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat Med. 2018 [Oct];24(10):1513–1518.
- Lee YM, Jun HS, Pan CJ, et al. Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology. 2012 [Nov];56(5):1719–1729.
- Ding Z, Harding CO, Rebuffat A, et al. Correction of murine PKU following AAV-mediated intramuscular expression of a complete phenylalanine hydroxylating system. Mol Ther. 2008 [Apr];16(4):673–681.
- Chandler RJ, Tarasenko TN, Cusmano-Ozog K, et al. Liver-directed adeno-associated virus serotype 8 gene transfer rescues a lethal murine model of citrullinemia type 1. Gene Ther. 2013 [Dec];20(12):1188–1191.
- Xie C, Gong XM, Luo J, et al. AAV9-NPC1 significantly ameliorates Purkinje cell death and behavioral abnormalities in mouse NPC disease. J Lipid Res. 2017 [Mar];58(3):512–518.
- Chandler RJ, Williams IM, Gibson AL, et al. Systemic AAV9 gene therapy improves the lifespan of mice with Niemann-Pick disease, type C1. Hum Mol Genet. 2017 [Jan 1];26(1):52–64.
- Salido E, Rodriguez-Pena M, Santana A, et al. Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Mol Ther. 2011 [May];19(5):870–875.
- Zabaleta N, Barberia M, Martin-Higueras C, et al. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat Commun. 2018 [Dec 21];9(1):5454.
- Senac JS, Chandler RJ, Sysol JR, et al. Gene therapy in a murine model of methylmalonic acidemia using rAAV9-mediated gene delivery. Gene Ther. 2012 [Apr];19(4):385–391.
- Harrington EA, Sloan JL, Manoli I, et al. Neutralizing Antibodies Against Adeno-Associated Viral Capsids in Patients with mut Methylmalonic Acidemia. Hum Gene Ther. 2016 [May];27(5):345–353.
- An D, Schneller JL, Frassetto A, et al. Systemic messenger RNA therapy as a treatment for Methylmalonic Acidemia. Cell Rep. 2017 [Dec 19];21(12):3548–3558.
- Kiyota T, Yamamoto M, Schroder B, et al. AAV1/2-mediated CNS gene delivery of dominant-negative CCL2 mutant suppresses gliosis, beta-amyloidosis, and learning impairment of APP/PS1 mice. Mol Ther. 2009 [May];17(5):803–809.
- Kiyota T, Zhang G, Morrison CM, et al. AAV2/1 CD74 gene transfer reduces beta-amyloidosis and improves learning and memory in a Mouse model of Alzheimer’s disease. Mol Ther. 2015 [Nov];23(11):1712–1721.
- Kanagawa M, Yu CC, Ito C, et al. Impaired viability of muscle precursor cells in muscular dystrophy with glycosylation defects and amelioration of its severe phenotype by limited gene expression. Hum Mol Genet. 2013 [Aug 1];22(15):3003–3015.
- Yasuda M, Bishop DF, Fowkes M, et al. AAV8-mediated gene therapy prevents induced biochemical attacks of acute intermittent porphyria and improves neuromotor function. Mol Ther. 2010 [Jan];18(1):17–22.
- Jiang L, Berraondo P, Jerico D, et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat Med. 2018 [Dec];24(12):1899–1909.
- Mingozzi F, High KA. Immune responses to AAV in clinical trials.Curr Gene Ther. 2011 [Aug];11(4):321–330.
- Cantore A, Ranzani M, Bartholomae CC, et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med. 2015 Mar 4;7(277):277ra28.
- Bessis N, GarciaCozar FJ, Boissier MC. Immune responses to gene therapy vectors: influence on vector function and effector mechanisms.Gene Ther. 2004 [Oct];11(Suppl 1):S10–7.
- Tran ND, Porada CD, Almeida-Porada G, et al. Induction of stable prenatal tolerance to beta-galactosidase by in utero gene transfer into preimmune sheep fetuses. Blood. 2001 [Jun 1];97(11):3417–3423.
- Himmelreich N, Shen N, Okun JG, et al. Relationship between genotype, phenylalanine hydroxylase expression and in vitro activity and metabolic phenotype in phenylketonuria. Mol Genet Metab. 2018 [Sep];125(1–2):86–95.
- Jiang W, Liu L, Chang Q, et al. Production of Wilson disease model Rabbits with homology-directed precision point mutations in the ATP7B gene using the CRISPR/Cas9 system. Sci Rep. 2018 [Jan 22];8(1):1332.
- Roybal JL, Endo M, Radu A, et al. Early gestational gene transfer with targeted ATP7B expression in the liver improves phenotype in a murine model of Wilson’s disease. Gene Ther. 2012 [Nov];19(11):1085–1094.
- Murillo O, Luqui DM, Gazquez C, et al. Long-term metabolic correction of Wilson’s disease in a murine model by gene therapy. J Hepatol. 2016 [Feb];64(2):419–426.
- Wang L, Takabe K, Bidlingmaier SM, et al. Sustained correction of bleeding disorder in hemophilia B mice by gene therapy. Proc Natl Acad Sci U S A. 1999 [Mar 30];96(7):3906–3910.
- Grimm D, Zhou S, Nakai H, et al. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood. 2003 [Oct 1];102(7):2412–2419.
- Yamada T, Iwasaki Y, Tada H, et al. Nanoparticles for the delivery of genes and drugs to human hepatocytes. Nat Biotechnol. 2003 [Aug];21(8):885–890.
- Chen J, Guo Z, Tian H, et al. Production and clinical development of nanoparticles for gene delivery. Mol Ther Methods Clin Dev. 2016;3:16023.
- Yin H, Song CQ, Dorkin JR, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016 [Mar];34(3):328–333.
- Santillan-Doherty P, Grether-Gonzalez P, Medina-Arellano MJ, et al. Considerations on genetic engineering: regarding the birth of twins subjected to gene edition. Gac Med Mex. 2020;156(1):53–59.
- Schiroli G, Conti A, Ferrari S, et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell. 2019 [Apr 4];24(4):551–565 e8.
- Rangarajan S, Walsh L, Lester W, et al. AAV5-Factor VIII gene transfer in severe Hemophilia A. N Engl J Med. 2017 [Dec 28];377(26):2519–2530.
- Hamman K, Clark H, Montini E, et al. Low therapeutic threshold for hepatocyte replacement in murine phenylketonuria. Mol Ther. 2005 [Aug];12(2):337–344.
- Grompe M, Overturf K, al-Dhalimy M, et al. Therapeutic trials in the murine model of hereditary tyrosinaemia type I: a progress report. J Inherit Metab Dis. 1998 [Aug];21(5):518–531.
- Al-Dhalimy M, Overturf K, Finegold M, et al. Long-term therapy with NTBC and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol Genet Metab. 2002 [Jan];75(1):38–45.
- Gokay S, Ustkoyuncu PS, Kardas F, et al. The outcome of seven patients with hereditary tyrosinemia type 1. J Pediatr Endocrinol Metab. 2016 [Oct 1];29(10):1151–1157.
- Gerets HH, Tilmant K, Gerin B, et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol Toxicol. 2012 [Apr];28(2):69–87.
- Hickey RD, Nicolas CT, Allen K, et al. Autologous gene and cell therapy provides safe and long-term curative therapy in a large Pig model of hereditary Tyrosinemia Type 1. Cell Transplant. 2019 [Jan];28(1):79–88.