
ABSTRACT
Introduction: Netherton syndrome (NS) is a rare and severe ichthyosis characterized by superficial scaling, skin inflammation, a specific hair shaft defect, severe atopic manifestations and multisystemic complications. It is an orphan disease with currently no satisfactory treatment. NS is caused by loss-of-function mutations in SPINK5 encoding the serine protease inhibitor LEKTI. NS patients present with ichthyosiform erythroderma or ichthyosis linearis circumflexa and show considerable clinical variability.
Areas covered: Uncontrolled serine protease activity leads to a profound skin barrier defect and the release of pro-inflammatory and pro-allergic mediators by keratinocytes and immune cells. Improved understanding of NS pathogenesis has led to the successful use of repurposed biologics such as intravenous immunoglobulins and anti-IL-17A blockers. Between April 1, 2020 and November 18, 2020, authors searched for NS-relevant information in the following databases: MEDLINE, DrugBank, ClinicalTrials.gov, and patent datasets accessed through lens.org.
Expert opinion: Specific KLK5 and/or KLK7 inhibitors represent the most promising disease-modifying treatments. They are currently being developed by several companies. Comprehension of the determinants of NS variability, flares and modification over time will be the foundation for precision medicine. While improved knowledge of the inflammatory and allergic pathways involved is still needed, clinical trials using repurposed biologics have already begun.
1. Introduction
Inborn errors of keratinization comprise a wide group of heterogeneous genetic diseases referred as ichthyoses [Citation1]. Among them, Netherton syndrome (NS) is a rare and particular condition characterized by superficial desquamation associated with constant and severe atopic manifestations and multisystemic complications. This article reviews our current knowledge of this orphan disease and addresses its clinical aspects, pathophysiologic mechanisms, as well as current and emerging treatments.
1.1. Clinical features of Netherton syndrome
Netherton syndrome (NS), also known as Comel-Netherton syndrome, was first described by Comel in 1949 [Citation2] and Netherton in 1958 [Citation3]. The eponym of Netherton’s disease was given by Wilkinson in 1964 [Citation4].
NS is a rare and severe autosomal recessive condition characterized by the diagnostic triad of ichthyosiform (scaly) erythroderma (IE), a specific hair shaft defect known as trichorrhexis invaginata (TI) and atopic manifestations . NS prevalence is estimated to be 1 in 200 000, but its frequency may be underestimated because of early neonatal mortality [Citation5,Citation6].
Figure 1. Clinical features of Netherton syndrome. Images of children (A-B) and adults (C-D) with scaly erythroderma or ichthyosis linearis circumflexa. Panels to the right are close-ups of affected skin areas for each patient
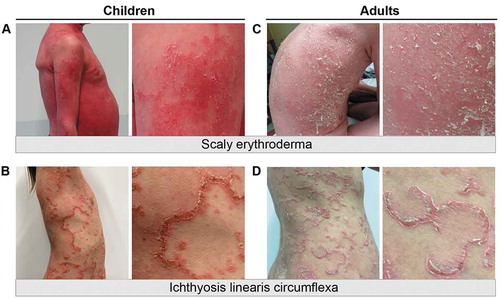
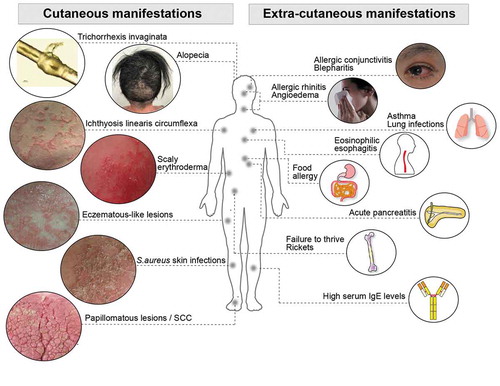
Figure 2. Netherton syndrome is a multisystem disease. A scheme summarizing the common cutaneous and extra-cutaneous manifestations observed in Netherton syndrome patients. Other extracutaneous manifestations such as hypothyroidism, thymic atrophy and acute bilateral renal vein thrombosis have been reported in rare cases. IgE, immunoglobulin E
Newborns affected with NS classically present with congenital IE (generalized redness and scaling of the skin), with variable intensity and extension. Hair, eyebrows and eyelashes can be absent at birth and grow slowly or are present and subsequently become abnormal. The neonatal period is a critical period with high morbidity and life-threatening complications including hypernatraemic dehydration due to a severe skin barrier defect, hypothermia, recurrent skin, respiratory tract and systemic infections and gastrointestinal symptoms including abdominal pain, vomiting and diarrhea. Failure to thrive is frequent and has multiple causes such as increased catabolic rate, chronic inflammation, malabsorption, food allergies and recurrent infections. As a result, growth retardation and short stature are almost constant features. Rare associated findings have been reported including acute pancreatitis, acute bilateral renal vein thrombosis and persistent pulmonary hypertension .
The skin manifestations often remain severe during infancy and childhood, but tend to improve in teenagers and adults. IE can be patchy or generalized in infants and is often more pronounced on the face, where redness and peeling of the skin is intense (, ) [Citation7]. IE can evolve into characteristic and specific ichthyosis linearis circumflexa (ILC) which consists in erythematous serpiginous and migratory lesions surrounded by a double-edged scale (, ). Although very suggestive of NS, ILC is not constant and IE can persist through adulthood with no ILC, defining two different endotypes . The skin lesions have a fluctuating course with periods of inflammatory and scaly flares, with intense itching, separated by periods of remission. Pruritus is a constant feature especially during flares and is a significant factor of disease worsening via irritability, sleep disturbance, scratching and skin infections. Hair abnormalities are highly suggestive of NS but are delayed (usually after one year of age) and remarkably variable. Hair is often sparse, thin, short, and fragile with a spiky appearance and typically slow to grow. The same applies to eyebrow, eyelashes and body hair .
Under light microscopy, hairs show a pathognomonic abnormality known as trichorrhexis invaginata (TI), or bamboo hair, resulting from the invagination of the distal part of the hair shaft into the cup formed by the proximal hair shaft . Although highly specific for NS, TI is not always present and does not involve all hairs. Alopecia can be diffuse or localized with areas of broken hair surrounded by long unbroken, but abnormal hairs. In rare cases, patients have no alopecia but long hair of normal appearance, with or, infrequently, without TI.
1.1.1. Atopic manifestations
Atopy is a constant and early feature of patients with NS. Skin lesions include eczematous-like and oozing lesions, not limited to the classical topography of AD . Other atopic manifestations are frequent and include allergic asthma, rhinitis and conjunctivitis and angioedema. Food and airborne allergies are very frequent. Blood hyper-eosinophilia is frequent. Eosinophilic esophagitis has been reported and contributes to the difficulty of eating and to failure to thrive. Serum immunoglobulin E (IgE) levels are usually high to very high, with increased specific IgE levels to multiple food and airborne allergens [Citation8].
1.1.2. Complications
NS patients develop recurrent skin infections predominantly with Staphylococcus aureus. Secondary infections with S.aureus can trigger flares (personal observation) [Citation9]. Blepharitis due to Staphylococcus aureus are also frequently observed. Fungal and viral infections (except for HPV in papillomatous lesions) are unusual. Other skin complications include lichenification in flexor creases with thickening of the skin as in atopic dermatitis. NS patients can also develop papillomatous skin lesions in particular in the groin, perineal and genito-anal regions, which can be HPV positive and can evolve into a giant condyloma of Buschke-Löwenstein tumor [Citation10–12]. Squamous cell carcinomas of the skin have been reported in a few cases of adult NS patients [Citation13].
1.1.3. Growth retardation
Failure to thrive can be attributed to a combination of factors including increased catabolic rate, impaired skin barrier resulting in transepidermal water loss and caloric loss due to heat evaporation, chronic skin infections and inflammation, enteropathy causing chronic diarrhea and malabsorption in infancy and childhood [Citation14]. Moreover, a previous study reports that SPINK5 and KLKs are expressed in the pituitary gland, and that KLKs can degrade human growth hormone, thus providing a possible molecular mechanism for growth retardation observed in NS patients [Citation15]. NS patients with growth failure require appropriate nutritional support and a hypoallergic diet since early age. Despite these measures, NS patients most often show growth retardation with short stature.
1.1.4. Immunological abnormalities
Different immunological abnormalities have been reported in NS patients with variable consistency. Blood eosinophilia is often observed with various abnormalities in T and B lymphocyte counts and/or subpopulations. Immunophenotyping of a limited number of NS patients showed no evidence of immune deficiency, but some T and B cell imbalances in NS children and a Th17/Th22 bias, with no signs of Th2 skewing [Citation16–18]. NK cell function was reported to be reduced in two studies [Citation16,Citation19]. Plasmablasts were found to be decreased in one study [Citation16]. Total IgG, IgA and IgM are most often normal or sometimes raised. Total serum IgE levels are almost always significantly enhanced with increased specific IgE against food and airborne allergens. LEKTI being expressed in the Hassall’s corpuscles of the thymus, it is possible that T cell differentiation is altered. However, except for increased serum IgE levels, immune abnormalities in NS are inconstant. It is therefore also possible that these abnormalities are secondary to chronic skin infections and inflammation. Further phenotyping and functional studies of lymphocyte subclasses in a sufficient number of NS patients by age category are required.
1.1.5. Diagnosis
The diagnosis of NS is often difficult in early infancy because the clinical presentation of erythroderma with failure to thrive is common with other conditions, such as immune deficiency syndromes. The absence of hair or the delayed onset of hair abnormalities precludes the search for TI, and atopic manifestations and ILC have not developed yet. In children and in adults, specific hair abnormalities are usually present, and their association with atopic manifestations, IE or ILC is highly suggestive of NS. As hair shaft defect is not always present even in severe NS patients, the clinical frequency of NS could be higher. Therefore, diagnosis is routinely confirmed by histology and molecular analyses.
Histological examination of a skin biopsy shows marked epidermal hyperplasia with elongated and enlarged rete ridges, stratum corneum detachment, lack of or reduced granular layer and variable cell infiltrates in the papillary dermis . Confirmation of the diagnosis relies on the demonstration by immunostaining of complete absence, or more rarely marked reduction, of LEKTI expression in the granular layer of the epidermis and in the inner root sheath of hair follicles [Citation20]. The definitive proof of NS comes from the identification of bi-allelic loss-of-function mutations in SPINK5, the majority of which lead to premature termination codons [Citation21]. At present, more than 85 distinct disease-causing SPINK5 mutations have been reported in the Human Gene Mutation Database [Citation22], including some founder mutations in specific populations as well as hot-spot mutations favored by stretches of repeated nucleotides.
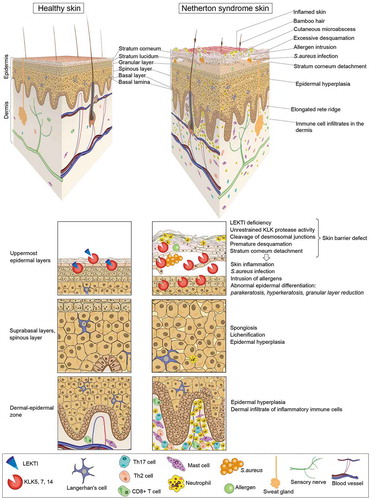
Figure 3. Cellular and histopathological hallmarks of NS patient skin. Schematic representation of NS patient skin in comparison to healthy skin. The skin of NS patients is often red (a sign of ongoing skin inflammation). Hairs are sparse and, when present, are short, fragile or broken. A typical hair defect in NS patients is the ‘bamboo’ hair in which the distal part of the hair invaginates into its proximal part (trichorrhexis invaginata). In the absence of LEKTI, kallikrein-related peptidases (KLKs) secreted by the cells of the upper granular layer uncontrollably degrade corneodesmosin, corneodesmosomal cadherins, filaggrin and lipid processing enzymes, thus leading to premature desquamation and stratum corneum detachment. In parallel, unrestrained activity of epidermal serine proteases activates a cascade of pro-inflammatory signals independent of the skin barrier defect and environmental factors. The barrier defect together with these intrinsic inflammation signals result in major pathologic changes of NS patient skin such as abnormal keratinocyte differentiation, skin infections and chronic skin inflammation. Signs of abnormal differentiation are parakeratosis (retention of nuclei in the stratum corneum), altered lamellar body secretion in the stratum corneum resulting in abnormal or absent lamellar lipid layers and partial or complete lack of granular layer with loss of keratohyalin granules. These features are associated with epidermal hyperplasia, elongation of rete ridges, and spongiosis (expanded intercellular spaces between spinous layer keratinocytes due to intercellular oedema). Defective epidermal barrier facilitates bacterial infections and allergen penetration through the skin. This results in skin inflammation mediated by protease activity and cytokine signaling cascades in the epidermis and recruitment of immune cell infiltrates mainly consisting in mast cells, neutrophils, Th17 and Th2 cells
1.1.6. Differential diagnoses
Differential diagnoses include peeling skin syndrome, Omenn syndrome and other primary immune deficiency syndromes, hyper IgE syndromes, severe atopic dermatitis and severe skin dermatitis, multiple allergies and metabolic wasting (SAM) syndrome.
1.2. Pathophysiology of Netherton syndrome
Netherton syndrome is caused by loss-of-function mutations in the gene SPINK5, which encodes lymphoepithelial Kazal-type-related protease inhibitor (LEKTI) [Citation23]. LEKTI is a reversible serine protease inhibitor that is strongly expressed in the most differentiated viable layers of stratified epithelia such skin, esophagus, suprabasal epithelial layers of gingival, vaginal and uterine ectocervix mucosa, tonsillar epithelium as well as in the thymic Hassall’s corpuscules [Citation20]. In the epidermis, LEKTI is expressed by the cells of the upper granular layer and its active proteolytic fragments are secreted at the interface between the granular and cornified layers [Citation20,Citation24]. LEKTI is also expressed in the inner root sheath of hair follicles [Citation20,Citation25]. LEKTI expression and function are conserved in mammals [Citation26]. Spink5-deficient mice mimic the disease and therefore have been instrumental in understanding the molecular mechanisms and cellular alterations in Netherton syndrome [Citation27–33].
LEKTI is a Kazal-type related inhibitor structured into 15 inhibitory domains [Citation34,Citation35]. Alternative splicing generates three LEKTI isoforms that are proteolytically processed by Furin to result in several single or multi-domain bioactive protein fragments [Citation20]. Each of these fragments exhibits different serine protease inhibitory activity [Citation36–38].
Extracellular serine proteases such kallikrein-related peptidases (KLKs) and LEKTI co-localize and interact in the upper granular and cornified layers of the epidermis [Citation24]. In the epidermis, LEKTI is a direct inhibitor of the serine proteases KLK5, KLK6, KLK7, KLK13, KLK14 and Cathepsin G [Citation37–42] and an indirect inhibitor of Elastase 2 (ELA2) [Citation43] . LEKTI can also inhibit the cysteine protease Caspase 14 in the epidermis [Citation44].
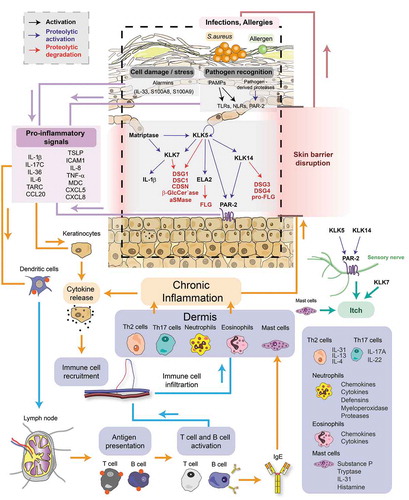
Figure 4. Signaling pathways underlying the pathophysiology of Netherton syndrome. LEKTI deficiency leads to unrestrained activity of KLKs in the upper granular and cornified layers of the epidermis. Matriptase and KLK5 initiate a proteolytic cascade through which other KLKs such as KLK7 and KLK14, and ELA2 become activated. Uncontrolled serine protease activity results in (1) skin barrier defect through the cleavage of corneodesmosin (CDSN) and the corneodesmosomal cadherins Desmoglein 1 (DSG1) and Desmocollin 1 (DSC1), increased pro-Filaggrin (pro-FLG) processing and Filaggrin (FLG) degradation, and cleavage of the lipid-processing enzymes β-glucocerebrosidase (β-GlcCer’ase) and acidic sphingomyelinase (aSMase) (2) hair defects through the degradation of Desmoglein 3 (DSG3) and Desmoglein 4 (DSG4) (3) inflammation through proteolytic activation of keratinocyte expressed proteinase-activated receptor 2 (PAR-2), increased processing of antimicrobial peptides LL-37 and proteolytic activation of pro-Interleukin 1β (pro-IL-1β) and (4) itch through proteolytic activation of PAR-2 expressed on sensory nerves. PAR-2 activation leads to the synthesis of pro-inflammation molecules in keratinocytes. Barrier defects expose the skin to microbes and allergens. Sensing of danger signals by keratinocytes through alarmins and/or pathogen recognition receptors also triggers the production of pro-inflammation factors in keratinocytes. Downstream signaling induces the differentiation and recruitment of Th2 and Th17 cells and activation and recruitment of mast cells, neutrophils and eosinophils to the skin. These immune cells secrete an arsenal of inflammation mediators such as cytokines, chemokines or proteases that act on the epidermis by stimulating further production of pro-inflammatory cytokines by keratinocytes, blocking epidermal differentiation, causing tissue damage and inducing itch. Hence, a vicious circle of skin inflammation and barrier disruption is established, ultimately resulting into a chronic skin disease. PAMPs, pathogen-associated molecular patterns; TLRs, Toll-like receptors; NLRs, NOD-like receptors; S100A8, S100 calcium binding protein A8; S100A9, S100 calcium binding protein A9; TSLP, thymic stromal lymphopoietin; MDC, macrophage-derived chemokine; TARC, thymus- and activation-regulated chemokine; ICAM1, intercellular adhesion molecule 1; TNF-α, tumor necrosis factor alpha; IL-4, interleukin 4; IL-6, interleukin 6; IL-8, interleukin 8; IL-13, interleukin 13; IL-17A, interleukin 17A; IL-17 C, interleukin 17 C; IL-22, interleukin 22; IL-31, interleukin 31; IL-36, interleukin 36; IgE, immunoglobulin E; Th2 cell, T helper 2 cell; Th17 cell, T helper 17 cell
At the interface between the granular and cornified layers, KLKs are tightly inhibited by LEKTI. This inhibitory interaction depends on the extracellular pH that forms a gradient from 6.3–7.3 in the upper granular layer to 4.3–5.8 in the uppermost cornified layers. At acidic pH in the upper cornified layers, LEKTI dissociates from its target enzymes and the inhibition of KLKs is relieved, thereby permitting cleavage of corneodesmosome junctions and subsequent desquamation. Thus, the balance between LEKTI-mediated inhibition of KLK proteases and KLK proteolytic activity is one of the mechanisms that ensures a gradual desquamation process, hence a normal renewal of the epidermis [Citation38]. In Netherton syndrome, LEKTI deficiency leads to unrestrained proteolytic activity in the epidermis, resulting in premature stratum corneum detachment and skin barrier defect [Citation45] .
Because KLKs participate in various processes such as desquamation, antimicrobial defense and lipid permeability [Citation46], their unrestrained activity in the context of LEKTI deficiency can have several impacts on the skin that can be classified into two main categories: (1) structural and (2) related to innate immunity and skin inflammation. Transgenic mouse models engineered to express human or mouse KLKs in the epidermis have been instrumental in delineating the contribution of each of these proteases to the skin barrier defects and skin inflammation in NS [Citation29,Citation43,Citation47–52].
At the structural level, KLKs hyperactivity causes premature desquamation by cleavage of corneodesmosin and the corneodesmosomal cadherins Desmoglein 1 (DSG1) and Desmocollin 1 (DSC1) [Citation37,Citation53–55], proteolytic processing of pro-Filaggrin [Citation56] and degradation of Filaggrin [Citation27,Citation28]. Furthermore, cleavage of DSG3 and DSG4 by KLK14 contributes to hair abnormalities and alopecia [Citation48]. KLKs regulate stratum corneum lipid permeability barrier, hence transepidermal water loss, by proteolytic degradation of the lipid-processing enzymes β-Glucocerebrosidase and Acid sphingomyelinase [Citation57,Citation58]. Furthermore, by activating PAR-2 receptor, KLKs downregulate lamellar body secretion by corneocytes [Citation59]. This correlates with observations of altered intercellular lipid composition and altered expression of lipid-processing enzymes as well as abnormal lipid organization in the stratum corneum of NS patients [Citation60,Citation61]. Epidermal Elastase 2 is also involved in the induction of lipid abnormalities as transgenic ELA2 mice lack lipid lamellae in the extracellular space of the cornified layer [Citation43]. Altered lamellar body secretion in Netherton syndrome leads to absent or irregular lipid inter-corneocyte lamellae [Citation62] .
KLK hyperactivity in the epidermis can trigger pro-inflammatory signaling independent of external stimuli. KLK5 and KLK14 proteolytically activate PAR-2 expressed in keratinocytes of the granular layer, which, through activation of NFkB, results in the production and secretion of the pro-inflammatory cytokines IL-8, intercellular adhesion molecule 1 (ICAM1), tumor necrosis factor alpha (TNF-α), and thymic stromal lymphopoietin (TSLP) [Citation31,Citation32,Citation54,Citation63,Citation64]. TSLP activates Langerhan’s cells leading to Th2 immune response, whereas IL-8, TNF-α and ICAM1 stimulate immune cell activation and recruitment to the skin. Through a PAR-2 independent mechanism, KLK activity triggers MDC and TARC expression in keratinocytes, which attract Th2 cells to the skin. Cytokines released by Th2 cells (IL-4, IL-5 and IL-13) induce B cell class switch to produce IgE antibodies that activate eosinophils and mast cells. KLK7 cleaves pro-IL-1β into active IL-1β in the epidermis, thus inducing pro-inflammatory responses [Citation65] .
Upon skin barrier disruption, penetration of microbes and allergens triggers the release of antimicrobial peptides. KLK5 and KLK7 activity increases processing of the cathelicidin hCAP18 and inactivation of LL-37 antimicrobial peptide, further facilitating bacterial invasion [Citation66]. Infection and keratinocyte damage/stress signals activate pattern recognition receptor signaling and induce release of alarmins, thereby triggering the expression of pro-inflammatory cytokines such as IL-6, IL-17C or IL-36 cytokines in keratinocytes. Downstream chemokine release leads to neutrophil recruitment and activation of dendritic cells, triggering the IL-23/Th17 immune signaling axis [Citation18,Citation48,Citation67]. Immune cell infiltrates in NS skin consist mainly of neutrophils, mast cells, eosinophils, Th2 and Th17 cells. Once in the skin, immune cells release signals that amplify the pro-inflammatory state of keratinocytes .
Apart from KLK activity, Th2-derived cytokines IL-4 and IL-13 are also known to suppress the production of antimicrobial peptides and thus promote bacterial skin infections [Citation68]. Moreover, Th2 cytokines downregulate ceramide synthesis and the expression of skin barrier proteins such as filaggrin, loricrin and involucrin, thus contributing to skin barrier disruption [Citation69–71].
A recent study shows that the microbiome of NS patient affected and non-affected skin differs considerably from healthy skin, with S.aureus and S.epidermidis being the predominant bacteria in NS patient skin [Citation9]. Although the causes of this dysbiosis remain unknown, virulence factors and proteases derived from S.aureus and the commensal S.epidermidis additionally disrupt the proteolytic balance in NS skin and promote inflammation.
By acting on sensory nerves in the skin, KLK activation and Th2 cytokine signaling produce itch. KLK5 and KLK14 activity induces PAR-2 mediated itch [Citation31,Citation52], whereas KLK7 and Th2 cytokines induces itch via PAR-2 independent mechanisms [Citation51,Citation72].
Altogether, the molecular signaling triggered by KLKs unrestrained proteolytic activity in NS is ultimately translated into morphological/structural abnormalities of the epidermis such as excessive desquamation, stratum corneum lipid abnormalities, hyperplasia, edema, dysregulated keratinization, abnormal epidermal differentiation, and hair abnormalities. Barrier disruption exposes the skin to infections and allergens that induce additional pro-inflammatory signals and recruitment of immune cells to the skin. The inflammatory cell infiltrates in the skin amplify the immune response and further aggravate skin barrier disruption by triggering production of pro-inflammatory signals and downregulating the expression of structural proteins in keratinocytes, ultimately leading to tissue damage. Thus, a vicious cycle of barrier disruption and chronic skin inflammation is formed . The skin phenotype of NS, similar to any other dermatosis, is shaped by a balance between pathogenic and life-enabling compensatory tissue responses. Some of the compensatory mechanisms that restore the barrier include accelerated lamellar body secretion and upregulation of DSG3/DSC3 upon DSG1/DSC1 degradation [Citation57]. This ‘dynamic’ balance of tissue responses underlies the natural history of the disease, marked by episodes of flares and remissions and evolving to a stable/improved phenotype in adults .
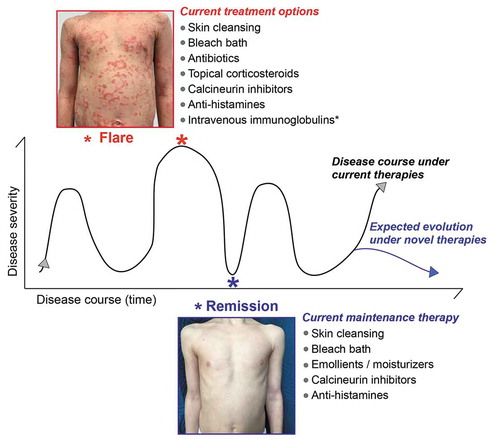
Figure 5. Common therapeutic approaches to breaking the vicious cycle of epidermal barrier defects and inflammation in Netherton syndrome. A schematic illustrating disease progression with time especially observed in NS patients with ILC. Netherton syndrome patients can experience disease exacerbation (flares), during which skin lesions appear or aggravate, separated by temporary remissions, during which skin lesions improve or disappear transiently. The duration of flares and remissions can vary from patient to patient. The time between each peak of flare is also variable and can range from 1 week to several months. Likewise, flare intensity for a given patient can fluctuate. The images show abdomen skin of the same NS patient during flare and remission. Although in this particular case remission is complete, other NS patients often show incomplete remission. The current, common therapeutic regimes for each disease state are indicated. These current therapies do not prevent the relapse of flares nor make them less severe, but they can help reduce their frequency. Future, novel therapies for Netherton syndrome are expected to prevent flares and/or reduce their intensity and frequency
The skin inflammation landscape of Netherton syndrome shares similarities with that of other inflammatory skin diseases such as atopic dermatitis and psoriasis. Type 2 inflammation is the predominant response in atopic dermatitis comparable to that observed in NS, whereas the IL-36 and IL-23/Th17 immune signaling axes are common to psoriasis and NS [Citation67,Citation73,Citation74]. Comparative molecular analyses of affected skin from several ichthyoses revealed that the molecular signature of NS resembles more that of psoriasis than that of atopic dermatitis [Citation18].
Collectively, studies on the mechanisms of Netherton syndrome to date have demonstrated that the main actors/initiators of skin barrier defects in NS are KLKs. Chronic skin inflammation, being a consequence of skin barrier disruption caused by unrestrained KLK activity, plays a major role in disease perpetuation. The function and contribution of KLKs to NS pathophysiology has been studied extensively, allowing to define precise therapeutic targets. Although the inflammation landscape of NS has received much attention in recent years, precise immune signaling molecular therapeutic targets remain to be determined.
2. Therapeutic strategies for NS
Netherton syndrome pathogenesis involves two main mechanisms: (1) a severe skin barrier defect due to excessive/premature detachment of the stratum corneum and (2) skin inflammation, infections and allergies which can evolve into systemic manifestations. Therefore, the therapeutic strategies for NS can be classified into two broad categories: (1) therapies aiming to restore the skin barrier and (2) therapies aiming to suppress/modulate the immune response, allergy and skin infections and thus decrease skin/systemic features. Because skin inflammation/infection/allergy and skin barrier defect are mutually causal, employing one therapeutic strategy is not a solution. Rather, a combination of therapies from both of these strategies could result in the most effective treatment.
In the following sections, we describe the treatment options currently available for NS, targeted therapies for NS under development as well as potential new therapeutic targets which can be applied to NS treatment. We further discuss the advantages and disadvantages of each therapeutic approach.
2.1. Current treatment options and disease management
Most of the current approved therapies for NS are also used for the management of other dermatological conditions such as psoriasis and/or atopic dermatitis. At present, there is no curative nor specific therapy targeting NS pathophysiology and NS treatment is mostly palliative.
2.1.1. Cleansing of the skin
A gentle/soft non-detergent liquid cleansing oil, preferably with an acidic pH (5) to counteract overactive serine proteases, is recommended for daily bath and/or shower. In the absence of infected and inflamed skin flare, antiseptic solutions are not necessary. They should be used during acute flares to treat and/or prevent infection of the lesions.
2.1.2. Emollients, moisturizers, keratolytics
Because NS patient skin is most often dry, scaly and peeling, emollients and moisturizers are essential adjuvants in the treatment of NS. Emollients are preparations that soften the skin, whereas moisturizers contain humectants which hydrate the stratum corneum [Citation75]. Both aim at improving the integrity of the skin barrier and should be started since birth when neonates and infants are at high risk of hypernatremic dehydration. In childhood and adulthood, their regular application improves the skin aspect and SC elasticity, promotes skin barrier recovery and prevents water loss. Their use as a permanent maintenance therapy or during the relapse-free phases of the disease in combination with other therapies is recommended. They also contribute to reduce pruritus.
Emollients and moisturizers can be delivered in a variety of topical formulations such as creams, ointments, gels, emulsions, lotions, or balms. They include for instance Dexeryl (glycerol 15%, vaseline 8%, paraffin 2%), Ictyane (vaseline, glycerine), Lipikar, Trixera, AtopiControl, Eucerin, Xemose, Atoderm, Cerave, Cold Cream … Creams and lotions are often more acceptable than greasy preparations. Some of them could be irritating to some patients and it is therefore often needed to try various preparations to find the most suitable one. Neutral oinment-based emollients such as a mixture (50–50) of white soft paraffin and liquid paraffin are recommended for newborns and infants.
Keratolytics such as salicylic acid, urea or alpha-hydroxy acids are often irritative and not well tolerated by NS patients.
The efficacy of these topical agents is variable depending on skin lesion severity and infection status [Citation76]. Adverse reactions to ingredients of topical creams can be observed and are facilitated by the skin barrier defect. For example, excessive use of emollients containing liquid paraffin can lead to penetration of paraffin into the skin, resulting in lymphadenopathy [Citation77].
Because emollients, although hydrating, can be harmful to the barrier, their choice has to be optimized [Citation78].
2.1.3. Anti-pruritics and anti-histamines
Oral antihistamines with sedative and anti-cholinergic effects such as hydroxyzine dichlorohydrate are used to alleviate itching, facilitate sleeping when pruritus is nocturnal and treat allergic rhinitis. Antihistamines with anti-allergic but non-sedative action such as Desloratadine are sometimes preferred. Antihistamines are often effective on allergic manifestations of NS, but their efficacy on pruritus is inconstant. Mechanistic studies using mouse models of inflammatory skin diseases have shown that topical application of antihistamines (H1/2 receptor antagonists) decreased inflammation and enhanced barrier function [Citation79]. Topical antihistamines are not currently used for treatment of NS patients, but, in view of these in vivo studies, they represent a promising therapeutic approach.
2.1.4. Antibiotics
The skin of NS patients is prone to frequent bacterial infections and displays an altered microbiome [Citation9]. Limited infections are treated with topical antibiotics for a short period of time (1 to 2 weeks) to avoid the selection of resistant bacterial strains. When skin infection is extensive, especially if it is associated with fever and enlarged lymph nodes, oral antibiotics targeting Staphylococcus aureus and Streptococcus, such as tetracyclines (above the age of 8 years), macrolides (pristinamycine or josacine) or large spectrum penicillin (amoxicillin and clavulanate) are required.
2.1.5. Antimicrobial bleach bath
By analogy with patients with eczema for whom bleach baths (sodium hypochlorite diluted in water) are recommended, NS patients, in particular children, also benefit from this care [Citation80]. This procedure has antimicrobial effects and, when performed twice or three times a week, reduces bad smells and the frequency of skin infections.
2.1.6. Topical corticosteroids
Topical application of synthetic corticosteroids is the main treatment option for many inflammatory and hyperproliferative dermatological disorders including Netherton syndrome [Citation81,Citation82]. Corticosteroids exert anti-inflammatory and immunosuppressive effects as well as anti-proliferative effect on keratinocytes [Citation83–85].
Although topical corticosteroids can be prescribed for inflamed, localized and non-infected lesions in NS patients, their use should be as limited as possible. Only corticosteroids of moderate potency are acceptable in children with NS (Locoid, Tridesonit cream, …) during a short period of time, while more potent corticoids (Diprosone (Betametazone 0.05% cream)) are often used in adults.
Treatment of NS patients with topical corticosteroids often improves skin lesions rapidly, however the duration and area of application of topical corticosteroids should be limited due to adverse effects. Because Netherton syndrome patients have a severe skin barrier defect, excessive percutaneous absorption of topical corticosteroids can impair renal reabsorption and result in aminoaciduria [Citation86]. Prolonged and/or excessive use of topical corticosteroids by NS patients has also been associated with the development of Cushing syndrome, severe skin atrophy, acute adrenal insufficiency or even fatality [Citation87,Citation88]. Excessive use of topical steroids can also aggravate the defective skin barrier by inducing additional loss of stratum corneum. Other adverse effects include growth retardation, hypertension, as well as weakness and lethargy upon discontinuation of topical treatment.
2.1.7. Retinoids
Naturally occurring and chemically synthetized derivatives of vitamin A exert effects on keratinocyte proliferation and differentiation, and also on skin inflammation. Their mode of action is mediated by interactions with the nuclear retinoic acid receptors (RAR) and retinoid-X receptors (RXR) [Citation89].
Systemic retinoid therapy in NS patients has shown varying degree of efficacy and tolerance [Citation90]. Previous case studies using either acitretin or isotretinoin, reported success [Citation91,Citation92], partial improvement or skin aggravation [Citation93–96].
The major side effects/risks associated with continuous retinoid therapy are bone toxicity and teratogenicity [Citation97,Citation98]. Groves et al. described the possible long-term (10 years) effect of etretinate therapy on statural growth in two NS patients [Citation99]. Systemic retinoids strictly contraindicate pregnancy during and after a period of time which depends on the retinoid used. Their effect is delayed (3 weeks) and their teratogenicity is prolonged after the medication is discontinued.
2.1.8. Calcipotriol
Calcipotriol is a synthetic analog of vitamin D3 that is commonly used as a topical therapy for plaque psoriasis [Citation100]. An ointment (Daivobet) containing both calcipotriol and the corticosteroid betamethasone has proven more efficacious in plaque psoriasis patients than the individual ingredients alone [Citation101]. Calcipotriol exerts its effects in the skin by binding to the vitamin D receptor (VDR) expressed on keratinocytes and immune cells, which in turn regulates gene expression, thus inhibiting proliferation and enhancing differentiation of keratinocytes [Citation102]. Mechanistic studies have demonstrated that calcipotriol can induce the expression of antimicrobial peptides in the skin of psoriasis patients [Citation103]. A recent study revealed that calcipotriol exerts its anti-inflammatory effects in the skin by binding to VDR on keratinocytes and suppressing IL-23/IL-17 signaling [Citation104].
Two case studies reported partial clinical improvement with no side effects in two NS patients treated with topical calcipotriol [Citation105,Citation106]. More patients should be treated to evaluate efficacy and safety of this medication.
2.1.9. Calcineurin inhibitors
Topical calcineurin inhibitors (tacrolimus and pimecrolimus) are one of the therapies used for inflammatory skin diseases such as psoriasis, atopic dermatitis and Netherton syndrome. Tacrolimus and pimecrolimus bind with high affinity to the immunophilin FKBP-12, thus inhibiting the calcium-dependent phosphatase calcineurin, which prevents translocation of NFAT into the nucleus. This leads to inhibition of T cell activation by blocking the transcription of early cytokines such as IL-2, IFNγ, IL-4 and IL-10 [Citation107,Citation108]. Tacrolimus and pimecrolimus also abolish the release of inflammatory cytokines and mediators from mast cells [Citation109]. Pimecrolimus has lower percutaneous absorption than tacrolimus [Citation110–112].
Topical tacrolimus shows efficacy in NS patients, with daily applications reducing erythema. However, to diminish side effects from systemic absorption, only a limited body surface area can be treated and long-term exposure to tacrolimus should be avoided [Citation113].
Allen et al. describe dramatic skin improvement (reduction of crusting, scaling and erythema), decreased pruritus and slow regrowth of hair in 2 out of 3 children with Netherton syndrome and erythroderma treated with 0.1% tacrolimus ointment. However, all 3 patients experienced significant percutaneous absorption of the drug [Citation114]. Another study reports the efficacy of topical tacrolimus without significant percutaneous absorption [Citation115].
A case study reports marked clinical improvement of one 10-year-old boy after three weeks of treatment with topical tacrolimus. Subsequent treatment with topical pimecrolimus resulted in 75% reduction of skin severity score within 4 weeks. Continuation of therapy for 3 months maintained this reduction of skin severity score and reduced flares of erythroderma [Citation112].
A study done to evaluate the safety profile of topical pimecrolimus in 3 children with NS demonstrated marked improvements in nearly all of the parameters evaluated. Patients treated with pimecrolimus responded rapidly, within the first month of treatment, and improvement persisted throughout the study period [Citation116]. Further case studies demonstrate the efficacy of tacrolimus and pimecrolimus in NS patients [Citation117,Citation118].
Collectively, these reports and our clinical experience identify topical calcineurin inhibitors as an efficacious therapy for NS patients. However, their dose and frequency of use should be strictly monitored to avoid toxicity due to systemic absorption, additional barrier damage or possible persistence of lesions [Citation119,Citation120].
2.1.10. Phototherapy
Narrowband UVB (NB-UVB) phototherapy and psoralen-UVA (PUVA) photochemotherapy are methods often used for treatment of skin diseases such as psoriasis and eczema [Citation121]. NB-UVB therapy consists in the repeated controlled delivery of the 311 nm centered narrowband region of the UVB spectrum. The mechanism of action of NB-UVB is still not completely understood, but studies point to alteration of cytokine expression, induction of apoptosis of both keratinocytes and immune cells, promotion of immunosuppression or cell cycle arrest [Citation122]. PUVA involves the intake of psoralen (a plant-derived organic compound that intercalates with DNA) and the subsequent exposure to long wave ultraviolet A irradiation, which acts on the DNA-intercalated psoralen to induce DNA strand crosslinking, thus inhibiting DNA synthesis and cell division.
Several case reports describe clinical improvement of skin lesions in NS patients after treatment with NB-UVB [Citation96,Citation123,Citation124], PUVA [Citation125] or UVA1 [Citation126] phototherapy.
The adverse effects of NB-UVB phototherapy have been linked to dose and frequency. While erythemal doses damage the barrier, suberythemal doses enhance barrier function and production of antimicrobial peptides [Citation127,Citation128]. The use of phototherapy is limited due to its adverse effects (mainly erythema) and the risk of skin cancer [Citation129]. The epidermal barrier dysfunction in NS increases the skin penetration of UV irradiation, thus potentially affecting immune cells as well. NS patients with erythroderma are particularly sensitive to UV irradiation due to loss of skin pigmentation. Combination of immunosuppressive therapies and phototherapy in inflammatory skin diseases has been associated with increased incidence of skin cancer [Citation13,Citation130,Citation131]. Sun exposure is variably tolerated by NS patients (personal communication). Sweating tends to aggravate skin lesions in NS, which could be due to the fact that sweat contains active KLKs [Citation132,Citation133]. Finally, a combination of phototherapy/UV exposure with other topical treatments could interfere with active substances in the topical treatment agent [Citation134].
2.2. Therapies for NS under development
2.2.1. Inhibition of kallikrein-related peptidase activity
2.2.1.1. Tissue kallikrein expression and physiological function
Tissue kallikreins (KLKs) are a family of 15 serine proteases comprising human tissue kallikrein (KLK1) and kallikrein-related peptidases (KLK2-KLK15) [Citation135]. These are single-domain serine proteases secreted as inactive zymogens (pro-KLKs). Under normal physiological conditions, KLKs are expressed in a variety of tissues with certain members displaying tissue-restricted patterns, whereas others having broad tissue expression patterns [Citation136–138]. Reflecting their broad spectrum of tissue expression, KLKs are involved in a variety of biological processes and have been associated with several diseases such as cancer, inflammation and neurodegeneration [Citation139]. Therefore, they have emerged as therapeutic targets for several diseases including Netherton syndrome [Citation140–142].
Normal human epidermis and its appendages express KLK1, KLK4 – KLK11, KLK13 and KLK14 [Citation132,Citation136,Citation138,Citation143,Citation144]. Among them, KLK5 shows the highest expression level at both protein and mRNA levels and KLK5, KLK7, KLK8 and KLK14 are the major active serine proteases in normal human epidermis. These proteases are produced by keratinocytes from the upper granular layers where they are co-secreted with other proteins transported by lamellar bodies to the stratum corneum [Citation145–147].
KLKs contribute to epidermal homeostasis and skin barrier function by acting in several processes: (1) desquamation by cleavage of corneodesmosin and the corneodesmosomal cadherins DSG1 and DSC1 [Citation37,Citation53–55] pro-Filaggrin processing [Citation56] and degradation of Filaggrin [Citation27,Citation28]; (2) regulation of stratum corneum lipid permeability barrier by proteolytic degradation of lipid processing enzymes [Citation57,Citation58,Citation61], (3) innate immune signaling in the skin by PAR-2 activation and downstream induction of TSLP secretion [Citation31,Citation54,Citation63] proteolytic processing of C3 [Citation148], IL-1β [Citation65] and antimicrobial peptides [Citation66].
2.2.1.2. Regulation of KLK activity in the skin
The activity of KLKs in the skin is regulated by several mechanisms whose fine balance ensures a normal skin barrier function. KLK activation in the stratum corneum occurs through a proteolytic cascade initiated by pro-KLK5 auto-cleavage [Citation149]. Activated KLK5 can then cleave and activate pro-KLK7, pro-KLK8 and pro-KLK14 [Citation150,Citation151]. In a positive feedback loop, active KLK14 can proteolytically activate pro-KLK5. Matriptase, a transmembrane serine protease expressed in the upper granular layer of the epidermis, can also activate pro-KLK5 and pro-KLK7 in mice [Citation152]. In human skin, matriptase expression and activity are restricted to the basal and spinous layers of the epidermis. Therefore, it seems unlikely that matriptase can activate human KLKs [Citation153,Citation154]. Another epidermal protease, PRSS3 (serine protease 3, also known as mesotrypsin) is able to activate pro-KLK5 and pro-KLK7 [Citation155].
Other mechanisms of KLK activity regulation involve enzyme inhibition through several factors such as metal ions, environmental pH, or endogenous protein inhibitors. Zinc metal ions are abundant in the epidermis and are known to reversibly inhibit KLK5, KLK7, KLK8 and KLK14 activity [Citation156–160].
Environmental pH modulates KLK activity. The pH in the outer layer of the epidermis forms a gradient ranging from 6.3–7.3 in the upper granular layer to 4.3–5.8 in the upper stratum corneum [Citation161,Citation162]. In vitro studies have demonstrated that several skin KLKs display optimal activity at around pH 7.5–8.0 and lower activity at acidic pH [Citation149,Citation151,Citation156,Citation163,Citation164]. The interaction between LEKTI and KLKs is pH-dependent. At acidic pH, the LEKTI-KLK complex dissociates. Thus, the pH gradient along the upper granular layer and the stratum corneum is another mechanism that ensures a gradual activation of KLKs and higher protease activity in the uppermost dead layers of the SC, where shedding of corneocytes is needed [Citation38].
Other endogenous inhibitors of epidermal KLKs are proteins such as serpins, Kazal-type inhibitors, or α2-macroglobulin [Citation165]. The Kazal-type inhibitors LEKTI-1 (encoded by SPINK5), LEKTI-2 (encoded by SPINK9) and SPINK6 are potent inhibitors of KLKs [Citation165]. They inhibit KLKs in a reversible manner by binding to the substrate binding site of the enzyme. LEKTI-1 precursor is structured into 15 inhibitory domains which are cleaved by furin before being secreted into the extracellular space [Citation20,Citation35,Citation36]. These LEKTI-1 fragments were shown to inhibit KLK5, KLK6, KLK7, KLK13 and KLK14 [Citation37,Citation40,Citation156,Citation166]. SPINK6, showing similar expression pattern as SPINK5, but absent in hair follicles, inhibits KLK5, KLK7 and KLK14, but not KLK8 [Citation167]. SPINK9, expressed only in palmoplantar epidermis, inhibits KLK5 and KLK8, but not KLK7 nor KLK14 [Citation168,Citation169].
The serpin superfamily of proteins consists of 35 protein-coding genes in human and 60 genes in mouse with diverse structure, function and tissue distribution [Citation170]. Some of the serpins can function as inhibitors of serine and/or cysteine proteases. Protease inhibition by serpins occurs through the formation of an irreversible covalent complex. Among the serpins that inhibit skin KLK activity are SERPINA1 (a serine protease inhibitor also known as α1-antitrypsin) strongly inhibiting KLK7 and KLK14 [Citation156,Citation171], SERPINA4 (a serine protease inhibitor also known as kallistatin) strongly inhibiting KLK1 and KLK7 [Citation171,Citation172], SERPINA5 (also known as PCI, proteinase C inhibitor) inhibiting KLK5, KLK7, KLK8, KLK13 and KLK14 [Citation171], SERPINB6 (also known as PI-6) inhibiting KLK8 in keratinocytes [Citation173] and SERPINA12 (also known as Vaspin) inhibiting KLK7 and KLK14 [Citation174,Citation175]. WAP four-disulfide core (WFDC) domain proteins are small serine proteinase inhibitors with prominent biological role in innate immunity and inflammation [Citation176,Citation177]. WFDC4, WFDC12 and WFDC14 are expressed in the skin. WFDC4 (also known as secretory leukocyte protease inhibitor, SLPI) strongly inhibits KLK7 [Citation178]. A recent study describing the expression and function of WFDC12 (also known as whey acidic protein 2, WAP2) in human skin, reports WFDC12 as an upregulated protein in affected and non-affected skin of NS patients and as a weak inhibitor of KLK7 [Citation179]. WFDC14 (also known as peptidase inhibitor 3 or Elafin) is a weak inhibitor of KLK7 as well [Citation178,Citation180].
2.2.1.3. KLK activity in skin of NS patients
Increased level of KLK activity in the epidermis is one of the molecular hallmarks of Netherton syndrome [Citation181,Citation182]. Higher KLK7 activity has been described in SC of healthy infants as compared to SC of healthy adults [Citation183]. Analyses of skin sections by in situ zymography using substrates for trypsin- and chymotrypsin-like proteases demonstrated increased protease activity in the skin of NS patients [Citation54]. Furthermore, analyses of KLK5 protease activity in tape-strip samples from a cohort of NS patients indicates increased protease activity in affected skin (Liddle et al., manuscript in revision).
2.2.1.4. In vivo and in vitro tools for studying the role KLKs in Netherton syndrome
The expression pattern, regulation and function of KLKs are conserved in mice [Citation184–186]. The skin KLKs involved in the pathogenesis of Netherton syndrome have homologues in mice. Breeding of Spink5-deficient mice to Klk5-deficient mice [Citation187], Klk5/Klk7 double-knockout mice [Citation188], Klk6-deficient mice [Citation189] or Matriptase-deficient mice [Citation152] has confirmed the essential role of these serine proteases in NS pathogenesis and has established them as possible therapeutic targets for NS. While Klk5 deficiency alone is not sufficient to completely rescue post-natal lethality of Spink5-/- mice (Klk5-/-/Spink5-/- mice have slightly longer survival compared to Spink5-/- ones), it significantly reduces global, Klk7 and Klk14 protease activities in the skin, prevents desmosome cleavage, acanthosis, abnormal epidermal differentiation and blocks Il17a- and Par-2-mediated skin inflammation [Citation187,Citation188]. Inactivation of Klk7 alone does not rescue lethality, nor does it revert the phenotype of Spink5-/- mice, whereas double knock-out of Klk5 and Klk7 in Spink5-/- background leads to complete rescue of lethality, restoration of epidermal barrier and differentiation and attenuation of cutaneous and systemic inflammation [Citation188]. Nevertheless, the hair defect in Klk5-/-/Klk7-/-/Spink5-/- mice is not rescued, suggesting the unrestrained activity of another protease targeted by Lekti, possibly Klk14. Future studies will show whether triple knock-out of Klk5, Klk7, and Klk14 can fully reverse the NS phenotype of Spink5-deficient mice. Deficiency of Klk6 is also insufficient to rescue post-natal lethality and the severe epidermal barrier defect of Spink5-/- mice, although it suppresses the expression of inflammation mediators and the infiltration of mast cells [Citation189]. In Matriptase-/-/Spink5-/- mice, post-natal lethality is not rescued either, but the skin barrier function is improved, global protease activity and the expression of pro-inflammatory cytokines such as Il6 and Il1b in the skin are strongly reduced [Citation152].
Given the high level of expression of KLK5 and KLK7 in the skin and the effect of their combined deletion on the survival of Spink5-/- mice and reversal of NS phenotype features, KLK5 and KLK7 have emerged as the major NS-specific therapeutic targets.
Transgenic mice overexpressing KLK5 [Citation52], Klk6 [Citation47,Citation190], KLK7 [Citation49–51], or KLK14 [Citation48,Citation191] are viable and have helped to study the role of each of these proteases in the development of skin barrier defect, skin inflammation and hair defects resembling NS. Over-expression of hKLK5 in the epidermis of mice leads to the development of a skin phenotype resembling NS with features of cutaneous and systemic inflammation [Citation52]. Skin-specific overexpression of mouse Klk6 driven by the bovine keratin 5 promoter results in severe psoriasiform dermatitis and arthritis-like joint disease mediated by Par-1 activation [Citation47]. However, ubiquitous overexpression of Klk6 under the control of the human ubiquitin C promoter does not result in spontaneous development of skin barrier defect [Citation190]. The different phenotypes of these two transgenic mouse models could be due to a different promoter-dependent transgene expression level. Transgenic hKLK7 mice develop a less severe skin phenotype as compared to transgenic hKLK5 mice, mainly characterized by partial hair loss, fine scaling, epidermal thickening and itchy dermatitis [Citation49–51]. The most prominent phenotypic feature of transgenic hKLK14 is hair loss due to an intrinsic hair shaft defect. Overexpression of hKLK14 in the granular layer also leads to a skin barrier defect and cutaneous inflammation with Il-36 signature [Citation48]. Altogether, these viable KLK transgenic mouse models provide a tool for in vivo efficacy testing of KLK-targeting therapies. However, because LEKTI can inhibit several proteases, Spink5 conditional knock-out mice represent a more comprehensive model for testing a wide range of NS therapy candidates.
Additionally, in vitro models with human cultured keratinocytes and skin-humanized mouse models of NS provide alternative tools for testing drug candidates for NS [Citation192–194].
2.2.1.5. Pharmacological inhibition of KLKs for the treatment of Netherton syndrome
Several strategies have been employed to block the activity of KLKs including small organic molecule antagonists, small peptide- and protein-based inhibitors, recombinant endogenous inhibitors or monoclonal antibodies. In the following sections, we outline some of the most promising KLK inhibitors known to date and comment on their future potential as therapies of NS.
2.2.1.5.1. Small molecule inhibitors of KLKs
Several KLK-specific small molecule inhibitors have been described and some of them have already entered into a drug development stage.
Walker et al identified via a structure-based design strategy, GSK951, a small borolane-based reversible, covalent inhibitor of human KLK5 [Citation195]. GSK951 was thoroughly characterized in the perspective of a topical treatment for Netherton syndrome. Pre-clinical testing in transgenic hKLK5 mice has proven its efficacy in attenuating skin inflammation and blocking insitu KLK5 activity (Liddle et al., manuscript in revision).
Sixera Pharma has identified benzoxazinone derivatives as selective inhibitors of KLK5, KLK7 and KLK14 [Citation196]. Final pre-clinical development of the lead molecule SXR1096 and preparation for clinical testing are currently ongoing [Citation197]. SXR1096 has been granted an orphan drug designation in EU and USA for the treatment of Netherton syndrome [Citation198].
BridgeBio is developing a topically formulated, small molecule inhibitor BBP-561 which targets both KLK5 and KLK7 [Citation199,Citation200]. The BBP-561 molecule is currently in a preclinical development stage.
Other small molecules inhibitors of KLKs that have been discovered and biochemically characterized include a collection of coumarin-3-carboxylate derivatives selective against one, two or more enzymes among KLK5, KLK7, KLK14 and Matriptase [Citation201], a naturally occurring compound brazilin inhibiting KLK5-8, KLK13 and KLK14 [Citation202], 3-acyltetramic acids inhibiting KLK5 and KLK7 [Citation203], derivatives of 1,3,6-trisubstituted 1,4-diazepane-7-one inhibiting KLK7 [Citation204,Citation205], and isomannide derivatives inhibiting KLK7 [Citation206]. The development and/or pre-clinical testing of these compounds has not been published.
A case study reported treatment of a newborn NS patient with an ointment containing 40% zinc oxide and sodium bicarbonate () [Citation207]. Daily application of this ointment improved hypernatremia, hypertension, and alkalosis within 24 hours of treatment and resulted in improved growth and partial reduction of exfoliation after one week of treatment. The authors concluded that these improvements were due to skin serine protease inhibition by the alkaline and zinc-containing ointment, despite lacking molecular evidence for KLK inhibition. Zn2+ ions are known inhibitors of tissue kallikreins, which could have been the active ingredient of this cream. However, there is no rationale for using sodium bicarbonate in NS. In healthy skin, basic pH facilitates binding of LEKTI to KLKs, resulting in their inhibition. In the skin of NS patients, LEKTI is absent, therefore increasing the skin pH would not facilitate binding of LEKTI to KLKs, but rather will be detrimental, as KLKs have optimal activity at pH 8.0. Moreover, as acidic pH is an important player in permeability barrier homoeostasis, stratum corneum integrity and inhibition of pro-inflammatory cytokine signaling, it is unlikely that the bicarbonate ingredient is beneficial [Citation208]. In fact, alkaline creams/soaps are not recommended as a skin care products for NS patients.
Table 2. Clinical case studies of therapies tested in Netherton syndrome patients. List of published case studies. Partial improvement refers to improvement of some clinical features within a patient. Incomplete improvement refers to clinical response in some patients only in case studies including more than one patient
Bleach baths are also alkaline, thus despite being beneficial in terms of antimicrobial action, they can stimulate KLK activity. Therefore, it is urgent and necessary to develop KLK-specific inhibitors, which can be used in combination with anti-bacterial/anti-inflammatory therapies.
A challenge with the design of small molecule inhibitors of KLKs is achieving specificity. KLKs have highly conserved active site and the surface of interaction between the enzyme’s active site and the drug is not large enough to allow for enzyme-specific interaction. Thus, finding inhibitors selective for a certain KLK is often a bottleneck in the drug discovery and development process [Citation140]. On the other hand, the advantage of small molecule inhibitors is their easier synthesis compared to biologics, low immunogenicity and broader choice of drug delivery modes. To avoid possible off-target systemic effects, topical administration of small molecule KLK inhibitors is favored in the treatment of NS. The defective skin barrier in NS facilitates topical delivery of drugs, which is an advantage, but can also pose a risk by promoting systemic absorption of KLK inhibitors with potential toxic systemic effects. Therefore, factors that need to be considered for a successful topical KLK inhibition therapy include the concentration of the drug (high enough to exert activity, but low enough to avoid systemic exposure and toxicity) as well as a low pH vehicle to prevent protease activation.
2.2.1.5.2. Small peptide-based KLK inhibitors
Small peptide-based inhibitors are short peptides (around 20 amino acids) which are natural or synthetic and are often modified to improve their half-life and/or solubility [Citation209].
Cyclic depsipeptides are oligopeptides in which one or more amino acids are replaced by a hydroxy acid, resulting in the formation of at least one ester bond in the core ring structure. Many natural cyclic depsipeptides have been isolated from fungi, plants, and marine organisms [Citation210,Citation211]. Novartis isolated a cyclic depsipeptide from the bacteria strains Chondromyces crocatus or Chondromyces robustus, which selectively inhibits KLK7 and epidermal Elastase 2 [Citation212]. The lead molecule LM-030 (previously known as BPR277), now licensed to LifeMax, has being granted fast track designation, orphan drug designation and rare pediatric disease designation by the FDA for topical treatment of Netherton syndrome. LM-030 is the first KLK-targeting drug for Netherton syndrome ready to enter a pivotal trial [Citation213].
Sunflower trypsin inhibitor-1 (SFTI-1) is a 14-amino-acid-long naturally-occurring β-hairpin peptide with broad inhibitory activity towards serine proteases, which was isolated from Helianthus annus sunflower seeds [Citation214]. SFTI-1 has emerged as a scaffold for engineering KLK-specific inhibitors. Using a combinatorial SFTI inhibitor library, KLK5-, 7- and 14-specific inhibitors have been engineered [Citation215–218]. Currently, there are no reports on the development of these molecules as drug candidate for NS.
Small cyclic peptide inhibitors of KLK5 (also called peptidic macrocycles) have been recently identified [Citation219].
2.2.1.5.3. Small protein-based KLK inhibitors
Small protein-based inhibitors are endogenous proteins or protein domains built of approximately 60 amino acids that have been engineered to achieve optimal inhibition properties against KLKs.
Two Kunitz-domain natural protein inhibitors named ATPI-I and ATPI-II were isolated from the sea anemone A. tenebrosa. These proteins are potent inhibitors of KLK5, KLK7 and KLK14 [Citation220]. Such natural proteins represent a scaffold for the engineering of highly specific KLK inhibitors, which can be further developed as drugs for the treatment of Netherton syndrome.
Chemical synthesis of LEKTI domain 6 yielded a protein fragment which retains inhibitory activity against KLK5 [Citation221], thus providing a proof-of-principle strategy for the synthesis of inhibitory domains with known inhibition properties against KLKs. To date, there are no studies reporting preclinical testing and/or drug development of engineered small protein-based inhibitors.
2.2.1.5.4. Recombinant endogenous serine protease inhibitors
Recombinant endogenous inhibitors can serve as a scaffold to engineer KLK-specific inhibitors. Dermadis/MedDiscovery used a phage display pentapeptide library screening method to design inhibitors based on the serpin α1-antichymotrypsin (SERPINA1) [Citation222,Citation223]. One of the resulting lead inhibitors (MDPK67b) specific for KLK2 was tested on transgenic hKLK5 mice in a topical formulation. MDPK67b showed moderate efficacy, which was most prominent in mice with low-grade skin lesions [Citation223]. To date, there are no reports on the clinical development of this inhibitor for treatment of NS.
Topical treatment of five NS patients with recombinant α1-antitrypsin as 2% gel formulation for a duration of 3 weeks did not result in a significant improvement of skin lesions [Citation224]. The authors claim several reasons for the failure of this therapy such as low skin penetration of recombinant inhibitor formulation, short treatment duration or low drug concentration. There are no studies reporting further development of this therapy approach.
2.2.1.5.5. KLK-specific monoclonal antibodies
Therapeutic monoclonal antibodies have emerged as the predominant treatment method for myriads of diseases [Citation225]. Previous preclinical studies report good efficacy of KLK-targeting antibodies, such as KLK1-neutralizing antibody in an allergic sheep model of asthma [Citation226] and KLK6-neutralizing antibody in a mouse model of Theiler’s murine encephalomyelitis virus-induced spinal cord pathology [Citation227].
As of today, there are no reports describing KLK targeted antibody therapy for skin diseases. A risk with systemic administration of KLK-neutralizing antibodies is that the antibodies may not reach the epidermal granular/cornified layers – the site of active KLKs. Previous studies provide evidence for the efficacy of local administration of therapeutic antibodies in the context of skin diseases [Citation228]. Intradermal delivery of TNF-α neutralizing antibody in affected skin of patients with mild to moderate psoriasis vulgaris [Citation229] or in patients with necrobiosis lipoidica [Citation230] has proved safe and efficacious. Topical application of TNF-α neutralizing antibody (Infliximab) proved efficacious in the treatment of chronic leg ulcers [Citation231] and pyoderma gangrenosum [Citation232]. Improvement of skin lesions in atopic dermatitis patients was also achieved by topical application of pooled polyclonal IgG [Citation233]. Topical administration of anti-TNF-α scFv antibodies (DLX105 in 0.5% hydrogel formulation applied twice daily for a duration of 4 weeks) was tested in a phase 2a clinical trial to treat mild to moderate psoriasis. Although, topical DLX105 did not induce a clinical response, it mediated a significant decrease in the mRNA level of pro-inflammatory cytokines assessed in skin biopsies 2 weeks after treatment [Citation229].
Due to severe barrier defect, the skin of NS patients readily absorbs topically delivered medicines. With the increasing diversity of topical formulations, it is expected that more therapeutic antibodies will be tested as a topical treatment for skin diseases. Future research will show the bottlenecks to KLK targeting by monoclonal antibodies in the context of inflammatory skin diseases.
Blocking epidermal KLK activity is a therapeutic strategy for NS which has the potential to impact on both skin barrier defect and skin inflammation . However, given the severity of skin lesions and the already instilled skin inflammation, KLK inhibition may not be sufficient to fully revert the clinical features of NS. KLK activity is an upstream player in the pathogenesis of NS. However, once initiated, skin inflammation is fueled by an arsenal of downstream cytokines and immune cell-derived proteases. Moreover, interleukins were reported to amplify KLK-mediated proteolysis in inflamed skin by upregulating KLK gene expression [Citation234]. In view of chronic skin inflammation in NS patients, it is likely that KLK inhibition could be most efficient in the early stages of lesion development or even before lesions appear. It is, therefore, important to determine the optimal time-window and duration for KLK-based therapy in NS patients. Combination of KLK-targeted therapy and immunotherapies could be a winning strategy.
2.2.2. Immunotherapy
2.2.2.1. Immunotherapy of NS: a review of clinical trials
Registered or ongoing clinical trials of immunotherapies for NS include efficacy and safety testing of Humira (adalimumab, anti-TNF-α monoclonal antibody), Dupixent (dupilumab, anti-IL-4Rα antibody) and Cosentyx (Secukinumab, anti-IL-17A antibody) . The outcome of these trials has not been published or the trial has still not been initiated (in the case of dupilumab).
Table 1. Clinical trials of therapies for Netherton syndrome. List of completed and ongoing clinical trials
2.2.2.2. Immunotherapy of NS: a review of case studies
Several immunotherapies have been tested in NS patients within the context of case studies ().
2.2.2.2.1. TNF-α neutralization
In NS patient keratinocytes as well as in a mouse model of NS, unrestrained KLK5 activity leads to PAR-2 receptor activation and downstream NFkB pathway signaling activation, resulting in the induction of pro-inflammatory cytokines such as TNF-α [Citation31].
TNF-α is a proinflammatory cytokine which, when produced by epidermal keratinocytes, induces its own expression as well as the expression of IL-1α, IL-6, IL-8, IL-33, and IL-36 cytokines [Citation235–239]. TNF-α upregulation has been associated with the pathophysiology of psoriasis, atopic dermatitis and allergy [Citation67,Citation240,Citation241].
TNF-α neutralizing antibodies already approved for a wide range of inflammatory diseases [Citation242,Citation243] have been clinically tested in two case studies involving Netherton syndrome patients. Infliximab therapy of a 25-year-old NS patient resulted in marked clinical improvement. Molecular analyses of skin samples after one year of treatment confirmed the reduction of TSLP, IL-6 and IL-17A levels [Citation244]. Significant clearance of skin lesions, reduction of pruritus and improvement of hair was achieved in a second case study [Citation245]. Anti-TNF-α therapy of skin diseases is reported to have adverse effects such as skin infections, autoimmune disease or malignancies, which may be problematic in NS [Citation246,Citation247].
2.2.2.2.2. IL-17A neutralization
IL-17 family cytokines are key mediators of inflammation in several inflammatory skin diseases including psoriasis and Netherton syndrome [Citation18]. Blocking IL-17 signaling pathway has received a huge interest as a strategy for the treatment of inflammatory diseases, resulting in the development of several biologics targeting different or the same component of this pathway [Citation248]. IL-17A cytokine can be produced by Th17 cells, γδ T cells, iNKTs, innate lymphoid cells (ILCs), mast cells or neutrophils [Citation249–251]. Luchsinger et al. report the compassionate use therapy of four NS patients with the IL-17A antagonist antibody secukinumab (Cosentyx) [Citation252,Citation253]. Secukinumab systemic administration led to significant reduction of erythema, scaling, and itch and improvement of life quality. The clinical improvement was particularly remarkable in children with erythrodermic phenotypes. Blanchard and Prose also used secukinumab to treat a 16-year-old NS patient [Citation254]. Treatment resulted in rapid and sustained improvement of both skin erythema and ichthyosis linearis circumflexa. Yet, a third study reports the compassionate use of ixekizumab (Talz, a humanized anti-IL-17A antibody) for the treatment of 3 adult NS patients [Citation255]. In contrast to the previous study, clinical improvement was more prominent in young adult patients with ichthyosis linearis circumflexa endotype compared to scaly erythroderma endotype patients. Serum cytokine and skin immune cell profiling indicated infiltration of mast cells in NS-ILC patients, decrease of serum CCL3 and IL-8 levels and increase of serum CCL20, TNF-α and IFNγ levels.
Collectively, these studies demonstrate partial efficacy and good tolerability of systemic anti-IL-17A therapy in NS patients. However, other inflammation pathways could play role/interfere or sustain the inflammation. More studies are needed to stratify patients based on their cytokine profile and skin-expressed molecular markers in order to maximize response to therapy.
2.2.2.2.3. Blocking IL-12p40/IL-23p40
IL-12 and IL-23 belong to the IL-12 family of heterodimeric cytokines consisting of a unique α chain subunit (p35 for IL-12 and p19 for IL-23) and the common β chain subunit p40. IL-12 binds the IL-12Rβ1/IL-12Rβ2 heterodimeric receptor complex, whereas IL-23 binds to a heterodimeric receptor composed of IL-12Rβ1 (shared with IL-12) and a unique IL-23Rα chain. Downstream of receptor binding, IL-12 and IL-23 activate JAK-STAT signaling. STAT4 mediates IL-12 signaling, while IL-23 acts mainly through STAT3 and STAT4 [Citation256].
The main producers of IL-12 and IL-23 are dendritic cells, monocytes and macrophages. In conditions of inflammation, keratinocytes can also produce IL-23 and IL-12 [Citation257–259]. IL-12 promotes the cytotoxic function of NK cells and plays a key role in inducing Th1 cell effector responses, dominated by IFNγ secretion. Keratinocytes also express IL-12Rβ2 and respond to IL-12 signaling [Citation260].
On the other hand, IL-23 induces the development of Th17 cells and regulates the function of NK cells, NKT cells, γδ T cells and innate lymphoid cells, which, like Th17 cells, can also produce IL-17 and/or IL-22.
Several biologics targeting IL-12/IL-23 signaling have been developed or are under development [Citation261]. Ustekinumab is a fully human monoclonal IgG1 antibody targeting the p40 shared subunit of IL-12 and IL-23, which is approved for the treatment of psoriasis, psoriatic arthritis, Crohn’s diseases or ulcerative colitis. Volc et al. report an off-label treatment of a 15-year-old NS patient with ustekinumab [Citation262]. Significant clinical response was observed as early as four weeks after therapy start with almost complete clearance of erythroderma and scaling. This improvement was sustained until the last reported observation (1 year of 3-monthly doses of ustekinumab). The authors did not perform laboratory analyses of efficacy.
Studies with more NS patients are need in order to assess the efficacy and safety of IL-12/IL-23p40 inhibition. Because the Th1 axis in NS is not the main inflammation driver, IL-12/IL-23p40 inhibition may not provide superior efficacy compared to biologics targeting only Th17 or IL-23. Mechanistic studies with genetic mouse models have demonstrated that IL-12 protects from psoriasiform inflammation by counteracting the infiltration of IL-17 producing γδ T cells [Citation260]. Moreover, cases of squamous cell carcinoma development have been reported in patients with psoriasis following ustekinumab therapy, which is consistent with the fact that IL-12 plays a role in anti-tumor immunity [Citation263,Citation264].
Given these safety issues with ustekinumab, priority should be given to the testing of other biologics in NS patients.
2.2.2.2.4. Blocking IL-23p19
Selective inhibition of the IL-23α chain p19 subunit has the potential to specifically inhibit Th17 signaling [Citation265]. Three monoclonal antibodies (guselkumab, risankizumab and tildrakizumab) that target IL-23p19 and block its binding to the IL-23 receptor have been approved for the treatment of psoriasis [Citation256,Citation266–268].
Treatment of one NS patient with guselkumab (Tremfya) for a duration of three months did not result in clinical improvement (Alain Hovnanian, personal communication).
Given that IL-23/Th17 signaling is one of the main inflammation mediators in NS, it is worth testing the efficacy of IL-23p19 targeting biologics in more NS patients.
2.2.2.2.5. Blocking IL-4Rα
IL-4 and IL-13 cytokines are key mediators of type 2 inflammation response triggered by allergens or parasites. These cytokines can be produced by different immune cells such as CD4+ T cells, basophils, eosinophils, mast cells, NK T cells, or ILC2 cells. Once secreted, IL-4 and IL-13 bind to their receptors on different cell types and initiate STAT6-mediated signaling and downstream regulation of gene expression. IL-4 binds to type I receptors composed of IL-4Rα chain and IL-2Rγc chain and type II receptors made up of the common IL-4Rα chain and IL-13Rα1, while IL-13 signals through type II receptors only [Citation269]. Type I receptors are expressed on myeloid and hematopoietic cells. Type II receptors are expressed in myeloid cells and all non-hematopoietic cells. IL-4 signaling regulates Th2 differentiation and B cell IgG1 and IgE class switch, while IL-13 signaling induces smooth muscle cell contraction and mucus production in the airway epithelium [Citation269,Citation270].
Dupilumab is a recombinant human IgG4 antibody that targets IL-4 receptor subunit alpha. It thus inhibits IL-4 signaling via the type I receptor (IL-4Rα/IL-2Rγc), and both IL-4 and IL-13 signaling through the type II receptor (IL-4Rα/IL-13Rα1). As a result, dupilumab suppresses Th2-mediated inflammation. Dupilumab is approved for the treatment of patients with eczema, atopic dermatitis, severe asthma with type 2 inflammation and chronic rhinosinusitis with nasal polyps [Citation271–273].
Steuer and Cohen report a case study of an adult NS patient treated with dupilumab [Citation274]. Therapy with dupilumab resulted in a marked decrease of itch within 2 months of treatment initiation and sustained improvement of disease severity with biweekly doses.
Allergy and itch are common features of NS driven by Th2 cytokine signaling. Therefore, blocking Th2-mediated inflammation, in particular through dupilumab, should be beneficial for NS patients. Studies with more patients are warranted to assess the efficacy and safety of dupilumab in the setting of NS.
2.2.2.2.6. Neutralization of IgE
Asthma, allergic rhinitis and food allergies are extracutaneous disease manifestations often observed in NS patients . Patients with these conditions present elevated serum IgE levels. IgE binds to high-affinity FcεRI receptors expressed on mast cells and basophils and crosslinks specific allergens on their surface. This leads to the release of inflammation mediators such as histamine, leukotrienes, cytokines, enzymes or prostaglandins. IgE binding to low-affinity FcεRII receptors expressed on B cells results in allergen uptake and antigen presentation to T cells, thus leading to secondary immune responses [Citation275]. Th2 cytokines IL-4 and IL-13 induce gene transcription leading to IgE class switch. This process is tightly regulated through cytokines, B cell surface receptors or transcription factors [Citation275].
Strategies to block IgE signaling include anti-IgE antibodies, DARPins (designed ankyrin repeat proteins), or bi-specific fusion proteins that co-aggregate FcεRI and the inhibitory FcγRIIb receptor, resulting in the block of IgE-dependent cell activation [Citation276].
Omalizumab is a recombinant, humanized, monoclonal antibody against human IgE that has been approved for treatment of allergic asthma and chronic idiopathic urticaria [Citation277,Citation278]. Omalizumab acts mainly by neutralizing soluble IgE, but it can also inhibit IgE binding to the low-affinity FcεRII receptor and accelerate dissociation of IgE from the high affinity FcεRIα receptor [Citation279].
Yalcin et al. report a case study of a 20-year-old NS patient treated with omalizumab [Citation280]. Therapy with omalizumab for a duration of 4 months led to clinical improvement as evidenced histologically by re-epithelization below necrotic epidermis lesions. Laboratory analyses demonstrated significant decrease of IgE, d-dimer, AST, prolactin, IgG, CRP, IL-4, IL-5, IL-1β and IL-17A levels after treatment.
Treatment of another NS patient with omalizumab significantly improved asthma, but it had no clinical benefit on skin lesions (Alain Hovnanian, personal communication).
To achieve complete clinical response, IgE neutralization therapy may be combined with biologics blocking Th2/Th17 immune responses. Future studies including more patients is required to evaluate both efficacy and safety of these approaches.
2.2.2.2.7. Immunoglobulin replacement therapy
Patients suffering from primary immunodeficiency diseases often display reduced serum level and/or poor function of immunoglobulins (IgG isotype antibodies), which results in recurrent infections. Previous studies of the immune system phenotype in NS describe it as a primary immunodeficiency disorder based on the following findings: recurrent sinopulmonary and cutaneous infections, decreased serum IgG levels, decreased number of NK cells, specific antibody responses to protein and polysaccharide antigens, CD4+ T cell anergy, decreased number of switched memory B cells, decreased number of activated B cells and plasmablasts, and impaired cytotoxic and degranulation capacity of NK cells [Citation16,Citation17,Citation19,Citation281].
Immunoglobulin replacement therapy is a standard of care for many primary immunodeficiency disorders [Citation282]. It consists in the administration of a pool of IgG collected from healthy individuals through intravenous (IVIg) or subcutaneous (SCIg) injection into patients.
Several case studies report immunoglobulin replacement therapy of NS patients (mainly newborns and children), the majority of them describing a remarkable clinical response with no major adverse effects [Citation16,Citation19,Citation283,Citation284].
Overall, immunoglobulin replacement therapy is efficacious and well-tolerated in NS patients. Studies with more patients are needed to assess the long-term benefits, age-related efficacy and adverse effects of this treatment. Immunoglobulin replacement therapy is relatively costly and is conditioned by hospitalization or presence of medical personnel to administer the therapy, which may not be convenient for most patients.
2.2.2.3. Immunotherapy of NS: potential new targets
None of the above reports describe complete cure of NS. Of the immunotherapies used in NS, anti-IL-17A is the most efficient so far. Below, we discuss other possible targets for immunotherapy of NS and alternative immunotherapies with potential efficacy in NS patients, despite the fact that they have not been tested in NS patients to date.
2.2.2.3.1. Blocking Th17 cell function
Apart from IL-17A neutralization using secukinumab or ixekizumab, other therapeutic targets have been explored to inhibit Th17 cell function [Citation285].
The IL-17 cytokine family comprises six members: IL-17A, IL-17B, IL-17 C, IL-17D, IL-17E and IL-17F. Except for IL-17D, all IL-17 cytokines bind to heterodimeric receptor complexes which consist of a common subunit IL-17RA and a second, ligand-specific subunit IL-17RB-RE [Citation285,Citation286]. Therefore, targeting the IL-17RA receptor subunit has the potential to provide a broader inhibition of the Th17 inflammation response by blocking the binding of several cytokines of the IL-17 family.
Brodalumab is a recombinant fully humanized monoclonal antibody specific for IL-17RA that has been approved for the treatment of moderate-to-severe chronic plaque psoriasis [Citation287]. Based on published reports, brodalumab efficacy is similar to that of ixekizumab and superior to that of secukinumab [Citation288]. Given the major role of IL-17 signaling in NS pathogenesis, therapy with brodalumab could be efficacious in NS patients and is probably worth testing.
Another strategy to block Th17 signaling is by targeting IL-17C. IL-17C is a member of the IL-17 cytokine family which, unlike IL-17A, is expressed by epithelial cells upon triggers such as bacterial infection (mediated by TLR2/5 and/or NOD2 receptors), TNF-α, IL-1β or IL-17A [Citation286]. Expression of IL-17C in keratinocytes leads to (1) autocrine signaling resulting in reinforcement of the mechanical epithelial barrier through the expression of tight junction proteins and induction of host defense through the expression of antimicrobial proteins and pro-inflammatory cytokines and (2) paracrine signaling on Th17 cells through the induction of IL-17A expression in Th17 cells [Citation289,Citation290]. The expression of IL-17 C is upregulated in NS patient affected skin [Citation18].
A monoclonal antibody specific for IL-17C (MOR106) has been developed as a therapy for psoriasis and atopic dermatitis. Pre-clinical testing of MOR106 in mouse models of psoriasis and atopic dermatitis showed efficacy in attenuating severity of skin lesions and inflammation [Citation291]. However, the clinical development of MOR106 for treatment of atopic dermatitis has been discontinued due to lack of efficacy in phase 2 clinical trial [Citation292].
Given the major role of IL-17 signaling in skin inflammation in NS, upregulation of IL-17C in affected skin of NS patients as well as the currently available viable mouse models of NS, MOR106 represents an attractive therapy candidate for NS.
2.2.2.3.2. Blocking Th22 cell function
Th17/Th22 pathways are activated in inflammatory skin diseases including psoriasis, atopic dermatitis and NS. IL-22 cytokine produced by Th17 and Th22 cells induces epidermal hyperplasia, causes abnormal keratinocyte differentiation resulting in skin barrier defect. The IL-22 blocking antibody fezakinumab was well-tolerated and induced consistent improvements in clinical and molecular disease scores of moderate-to-severe AD as compared to placebo [Citation293]. Fezakinumab has been tested in phase I clinical trial (NCT00563524) for the treatment of psoriasis. The results of this study were not published and the development of fezakinumab for the treatment of psoriasis has been discontinued [Citation294,Citation295]. Given the upregulation of Th22 pathways in NS, it may be worth testing anti-IL-22 biologics in future case studies or clinical trials involving NS patients.
2.2.2.3.3. Blocking IL-1 signaling
The IL-1 subfamily cytokines IL-1α and IL-1β are pro-inflammatory cytokines which trigger both local and systemic inflammation [Citation296]. IL-1α and IL-1β bind to the same cell surface receptor IL-1R1, but have different expression, function and activation modes. IL-1α is constitutively expressed in many cell types in normal conditions. Both precursor pro-IL-1α and proteolytically processed IL-1α forms are bioactive. Pro-IL-1α can be localized to the nucleus, where it activates NF-κB and AP-1 transcriptional machineries in a membrane IL-1R independent manner [Citation297,Citation298]. Upon cell death by necrosis, it is rapidly released into the extracellular space, binds to IL-1R1 on tissue resident macrophages and induces the production of chemokines, which attract neutrophils and monocytes. Pro-IL-1α can be cleaved and thus additionally activated by calpain, neutrophil elastase, granzyme B or chymase [Citation299]. The IL-1α precursor can localize to the cell membrane of monocytes and lymphocytes, where it regulates the activity of IFNγ [Citation296].
IL-1β is expressed only upon stimulation triggers such as microbial products, TNF-α, IL-1α, IL-18 or IL-1β itself. The inactive pro-IL-1β form can be activated through proteolytic cleavage of its N-terminus by intracellular caspase 1 or caspase 8, or extracellular immune cell-derived cathepsin G, proteinase 3, chymase or neutrophil elastase [Citation299]. Additionally, in the skin it was demonstrated that KLK7 can process pro-IL-1β [Citation65]. Other KLKs may be involved in the proteolytic activation of pro-IL-1β in the skin. For example, Klk13 is known to activate IL-1β in the mouse submandibular gland [Citation300]. Pro-IL-1β can also be activated by bacterial proteases [Citation301]. Downstream targets of IL-1β signaling include cytokines such as IL-1α, IL-1β, IL-23A, IL-32 and IL-36G and chemokines such as CCL20, CXCL1, CXCL2, CXLC5, CXCL6 and CXCL8, whose action results in leukocyte chemotaxis, neutrophil activation, and mucosal immunity [Citation302,Citation303].
IL-1 signaling plays role in the pathogenesis of a wide range of autoinflammatory diseases including inflammatory skin diseases caused by a dysfunctional skin barrier [Citation59]. Keratinocytes constitutively express IL-1α and IL-1β [Citation304,Citation305] and expression can be further upregulated by cutaneous barrier disruption [Citation306–308].
Serum IL-1β levels of NS patients from our cohort are low except for one patient, and we could not detect IL-1β protein expression by immunostaining of skin sections (Hovnanian, personal communication). However Renner et al report elevated serum levels of IL-1β in 5 NS patients [Citation19] and in mouse models of NS Il1b mRNA expression in skin is strongly increased [Citation31]. Given the high interindividual variability, cytokine profiling of skin and serum within bigger cohorts of NS patients should elucidate the expression pattern/level of IL-1β and its variation with age and disease endotype. Low serum levels of IL-1β have also been reported in other inflammatory disorders, but nevertheless blocking IL-1 signaling has achieved remarkable clinical responses [Citation309].
Different types of biologics targeting IL-1 signaling have been approved and are being developed, such as recombinant IL-1R1 receptor antagonist, soluble IL-1 receptors, neutralizing IL-1α/IL-1β monoclonal antibodies, IL-1R1 blocking antibodies, therapeutic vaccine targeting IL-1β or small molecule inhibitor of caspase 1 [Citation310].
Anakinra is a recombinant version of the naturally occurring IL-1 receptor antagonist (IL-1Ra), which prevents the binding of IL-1α as well as IL-1β to their common receptor IL-1R1. Since its approval for the treatment of rheumatoid arthritis in 2001, it has also shown efficacy in many other inflammatory diseases [Citation309].
In vivo studies using Cdsn-/- mice – a model of skin barrier dysfunction resembling Spink5-/- mice – demonstrate that blocking IL-1 signaling with anakinra improves skin inflammation phenotype through the inhibition of TSLP/IL-4 and IL-23/IL-17/IL-22 cytokine axes and suppression of immune cell infiltrates [Citation311].
Anakinra has shown efficacy in an open-label clinical trial for treatment of hidradenitis suppurativa [Citation312]. A case study reports transient and partial clinical improvement of two patients with palmoplantar pustular psoriasis treated with anakinra [Citation313], whereas several other studies report successful treatment of generalized pustular psoriasis with anakinra [Citation314–316]. Anakinra has been tested in phase 2 clinical trial for treatment of pustular psoriasis and other inflammatory pustular skin diseases (NCT01794117), but results have not been published.
Given the excellent safety profile of anakinra and its demonstrated efficacy in inflammatory skin diseases such as psoriasis, testing its efficacy in NS patients should provide low risk-benefit ratio. Blocking IL-1 signaling in general is worth testing in NS patients, as IL-1 signaling is one of the most up-stream events triggering the chronic skin and systemic inflammation in NS.
2.2.2.3.4. Blocking IL-13
Another strategy for suppressing the Th2-mediated allergic inflammation in NS is by selectively targeting IL-13. Several drugs are currently being developed to block IL-13, among them being two biologics – lebrikizumab and tralokinumab [Citation270].
Lebrikizumab is a humanized monoclonal antibody which blocks IL-13 binding to the IL-4Rα receptor subunit, thus preventing heterodimerization of the IL-13Rα1/IL-4Rα receptor subunits and downstream signaling [Citation317]. Lebrikizumab has been tested in clinical trials for the treatment of asthma, allergic asthma, atopic dermatitis, idiopathic pulmonary fibrosis, and chronic obstructive pulmonary disease. Results from phase 2 and phase 3 trials indicate that lebrikizumab did not consistently reduce asthma exacerbations [Citation318]. However, in adult patients with moderate-to-severe atopic dermatitis, lebrikizumab showed rapid, dose-dependent efficacy and a favorable safety profile [Citation319].
Tralokinumab is a human IgG4 monoclonal antibody which binds to IL-13 and prevents its interaction with the IL-13Rα1 receptor subunit as well as with the IL-13Rα2 decoy receptor [Citation320]. Results from phase 2 clinical trial testing of tralokinumab in patients with severe uncontrolled asthma demonstrate acceptable safety and tolerability profile, but no significant reduction in asthma exacerbation rates [Citation321]. By contrast, in phase 2b clinical trial with patients suffering from moderate-to-severe atopic dermatitis, tralokinumab showed significant efficacy and a favorable safety profile [Citation322]. Positive top-line results from phase 3 trials of tralokinumab in adult patients with moderate-to-severe atopic dermatitis have been published recently [Citation323].
2.2.2.3.5. Blocking CRTH2/Prostaglandin D2 Receptor 2
Prostaglandin D2 Receptor 2 (PTGDR2/CRTH2) is a G-protein-coupled receptor that is expressed by Th2 cells and mediates the pro-inflammatory chemotaxis of eosinophils, basophils, and Th2 lymphocytes during allergic inflammation. Given the participation of Th2 cells and eosinophils in skin and esophagus inflammation in NS as well as the development of asthma in NS patients, targeting CRTH2 in NS may prove beneficial. Fevipiprant (Novartis) and timapiprant (Atopix Therapeutics/CHIESI Farmaceutici) are two small molecule antagonists of CRTH2 which have been developed for treatment of asthma and atopic dermatitis [Citation324,Citation325]. Treatment of severe eosinophilic asthma patients with timapiprant resulted in reduction of airway eosinophilic inflammation [Citation326]. However, neither timapiprant (NCT02002208), nor fevipiprant (NCT01785602) showed significant efficacy in phase 2 clinical trials for treatment of moderate to severe AD.
2.2.2.3.6. Blocking IL-33
IL-33 is an alarmin released by damaged barrier cells (endothelial and epithelial cells) that plays role in both innate and adaptive immune responses [Citation327,Citation328]. Konishi et al report expression of IL-33 in the epidermis of two NS patients [Citation329]. Increased expression of Il33 has also been described in mouse models of NS [Citation188]. AnaptysBio has developed a humanized anti-IL-33 IgG1 antibody ANB020 for the treatment of atopic diseases [Citation330]. ANB020 is currently in a phase 2 clinical trial for efficacy assessment in patients with chronic rhinosinusitis with nasal polyps (NCT03614923) and atopic dermatitis (NCT03533751) and has been evaluated in phase 2 clinical trials for activity in patients with eosinophilic asthma (NCT03469934) and patients with peanut allergy (NCT02920021). Netherton syndrome could be another disease indication for this biologic.
2.2.2.3.7. Blocking IL-31
Pruritus (itch) is one of the symptoms of Netherton syndrome that leads to scratching behavior, thus further exacerbating skin lesions and aggravating quality of life.
The Th2 cytokine IL-31 is one of the known inducers of itch [Citation331]. The IL-31 cytokine is produced mainly by activated Th2 cells. IL-31 signals through a heterodimeric receptor composed of IL-31 receptor A and oncostatin M receptor. IL-31 binding to receptor activates JAK-STAT, the RAS/ERK or the PI3K/AKT signal transduction pathways. IL-31 release has two main effects: (1) binding to IL-31A/OSMR receptor on keratinocytes upregulates the expression of IL-1α, CCL17, CCL22, S100A7, and β-defensin 2 and decreases the expression of Filaggrin and Corneodesmosin, thereby causing skin barrier disruption and inflammation, (2) IL-31 binding to IL-31A/OSMR receptors on sensory nerves and induces itch sensation via TRPV1 and TRPA1 ion channel activation [Citation331,Citation332]. Therefore, IL-31 represents yet another component of the Th2 signaling pathway that can be used as a therapeutic target to suppress itch and skin inflammation.
Nemolizumab is a humanized monoclonal antibody that targets the IL-31 receptor A subunit, thus preventing binding of IL-31 and downstream signaling. It has been tested in phase 2 clinical trials for treatment of patients with moderate-to-severe atopic dermatitis. Nemolizumab significantly improved pruritus and cutaneous signs of inflammation in patients with moderate-to-severe atopic dermatitis [Citation333–335].
Nemolizumab could be a promising therapy option for NS patients.
2.2.2.3.8. Blocking the action of neutrophils
Neutrophils are among the most prominent immune cell infiltrates in NS affected skin. Excessive activation of neutrophils leads to release of proteases which not only cause tissue damage, but also act as pro-inflammation factors, thus contributing to the pathogenesis of NS.
Since the destructive role of excessive neutrophil activation is common to many inflammatory diseases, neutrophils have emerged as therapeutic targets. Strategies to inhibit the function of neutrophils comprise blocking neutrophil production, activation, recruitment and chemotaxis, promoting neutrophil apoptosis, or blocking neutrophil-derived mediators such as proteases [Citation336]. Alvelestat, formerly known as AZD9668, is an oral neutrophil elastase inhibitor that has shown efficacy in a phase 2 clinical trial for treatment of bronchiectasis [Citation337]. Alvelestat is currently in a phase 2 clinical trial for treatment of alpha-1 antitrypsin deficiency [Citation338].
Other strategies described to inhibit neutrophil-derived proteases (elastase, cathepsin G and proteinase 3) include peptide-based pseudosubstrates [Citation339] and recombinant SLPI/elafin/trappin-2 chimeric proteins [Citation340].
None of these therapy approaches have been tested in the settings of NS. Because neutrophils are one of the main immune cell players in NS, targeting neutrophil function in NS could prove beneficial.
2.2.2.3.9. PAR-2 inhibition
PAR-2 is a G-protein-coupled cell surface receptor expressed by epithelial cells, endothelial cells, fibroblasts, immune cells (eosinophils, dendritic cells, neutrophils, mast cells) and neurons [Citation341,Citation342]. In the skin, PAR-2 expression is most prominent in the granular layer and it is also expressed in the inner root sheath of hair follicles and in myoepithelial cells of sweat glands [Citation342]. To effect downstream signaling, PAR-2 requires activation, which is mediated by proteolytic cleavage of its extracellular N-terminus. Cleavage exposes a new motif at the N-terminus, which serves as a tethered ligand, activating the receptor by interacting with its extracellular loops. This leads to recruitment of G proteins and other signaling molecules to the intracellular domains of the receptor.
Both exogenous (allergen- or pathogen-derived) and endogenous (keratinocyte- or immune cell-derived) proteases are known to activate PAR-2 in the skin [Citation343]. PAR-2 can be activated by trypsin [Citation344], mast cell tryptase [Citation345], elastase [Citation346], KLK5, 6 and 14 [Citation347]. Elastase‑mediated activation of PAR-2 signaling takes place through a non-canonical pathway in which cleavage occurs at a site distinct from the one that exposes the tethered ligand sequence, resulting in p44/42 MAPK pathway activation [Citation346]. Extracellular proteases can also modulate PARs signaling by inactivating the receptor through proteolytic cleavage downstream of the receptor activating site, resulting in tethered ligand truncation.
Initiation of PAR-2 signaling triggers different downstream pathways such as phospholipase C/Ca2+ signaling, MAPK signaling, NFkB signaling or ERK signaling [Citation348]. These signaling cascades then lead to distinct tissue-specific biological effects [Citation349]. In the skin, PAR-2 activation triggers production of IL-6, GM-CSF [Citation350], IL-8 [Citation351,Citation352], ICAM1 [Citation353], and IL-13 [Citation354,Citation355], which stimulates growth and inhibits differentiation of keratinocytes, induces maturation of Langerhan’s cells and activation of macrophages, thus mediating inflammation. Activation of PAR-2 also induces expression of the cytokine TSLP which triggers Th2/allergic inflammation responses [Citation31]. Furthermore, PAR-2 activation triggers the secretion of neuropeptides that mediate inflammatory edema by regulating vasodilatation and plasma extravasation, ultimately resulting in itch [Citation342]. Recent studies show that PAR-2 activation mediates itch via TRPV3 signaling in keratinocytes [Citation356]. PAR-2 signaling is also involved in skin pigmentation by mediating the transfer of melanosomes from melanocytes to keratinocytes [Citation357].
Because PAR-mediated signaling influences processes such inflammation, pain and itch, PARs have emerged as attractive therapeutic targets for several inflammatory diseases [Citation358]. PAR-2 expression is elevated in affected skin of patients with atopic dermatitis as compared to nonaffected skin or skin of healthy controls and PAR-2 mediated inflammatory cytokine release is involved in psoriasis [Citation351,Citation352]. In NS patients, the expression of TSLP (downstream target of PAR-2 activation) is upregulated [Citation31].
Some studies report variable/lower expression of PAR-2 in affected skin of psoriasis or AD patients compared to healthy controls [Citation351,Citation352,Citation359].
Likewise, in Cdsn-/- mouse model of epidermal barrier dysfunction, Tslp expression is upregulated in young mice (two weeks of age) and downregulated in adult mice (20 weeks of age) as compared to WT controls [Citation311]. In an ex vivo dermatitis model, PAR-2 expression was increased in response to transient upregulation of KLK5, while persistent up-regulation of KLK5 did not induce increase of PAR-2 levels [Citation359]. It is possible that these decreased levels of PAR-2 expression in chronic affected skin are a result of desensitization effects. Repeated activation of PARs by proteases leads to desensitization of the Ca2+ response in cells, whereby PARs are internalized for lysosomal degradation in the cytoplasm [Citation360]. This implies that different stages of the inflammatory skin disease could respond differently to PAR-2 inhibition.
Genetic deletion of Par2 completely rescues inflammation and ichthyosis in transgenic mice with epidermal-specific overexpression of CAP1/Prss8 [Citation361]. However, genetic deletion of Par2 does not inhibit inflammation in Spink5-/- mouse adult skin, although early production of Tslp is blocked in Spink5-/- embryos at E19.5 [Citation32]. This suggests that anti-PAR-2 therapy in NS should be combined with other therapies. Several strategies have been developed to block activation of PARs including small molecule tethered ligand antagonists, monoclonal antibodies targeting the tethered ligand, or the cell‑penetrating intracellular antagonists termed pepducins [Citation358]. Pre-clinical testing of the pepducin PZ-235 (Oasis Pharmaceuticals) in mouse models of atopic dermatitis showed good efficacy in reducing itch, skin lesion severity and immune cell infiltrates [Citation362]. To date, PAR-2 blocking approaches have not been tested in NS models or patients.
2.2.2.3.10. Blocking TSLP
Thymic stromal lymphopoietin (TSLP) is a cytokine expressed in epithelial cells of barrier tissues (skin, gut, and lung). TSLP expression is elevated in diseases involving allergic inflammation including NS. Allergen- and microbial-derived proteases or unrestrained activity of endogenous proteases such as KLKs (in the case of NS) activate PAR-2, which then induces TSLP expression [Citation31,Citation64,Citation363]. TSLP induces Th2 response by activating DCs to produce the chemokines CCL17 and CCL22. Furthermore, TSLP induces mast cells, basophils and natural killer T cells to produce cytokines, which further recruit Th2 cells and eosinophils to sites of inflammation and suppress Th1 responses [Citation364]. TLSP signaling is upstream Th2 cell recruitment and can act on both innate and adaptive immune responses. Therefore, TSLP represents an attractive therapeutic target having the potential to block early events in the skin inflammation cascade in NS. TSLP expression is upregulated in the epidermis of NS patients, Spink5-deficient mice as well as transgenic hKLK5 mice [Citation31,Citation52,Citation187].
The TSLP blocking antibody tezepelumab (MedImmune/Amgen) has been tested in phase 2a clinical trial to evaluate its efficacy and safety in adult patients with moderate-to-severe atopic dermatitis (NCT02525094). The results of this trial demonstrate that patients achieved numerical improvements over placebo in week 12 EASI50 responses; however, these improvements were not statistically significant [Citation365].
The safety, tolerability and efficacy of tezepelumab in adult patients with asthma have been evaluated in two clinical trials [Citation366]. Gauvreau et al report that tezepelumab inhibited the late allergen-induced asthmatic response [Citation367]. In the second study, treatment of asthma patients with tezepelumab antibody resulted in lower annualized rate of asthma exacerbations compared to the placebo group. The same clinical trial reports effects of tezepelumab on interleukin-4, interleukin-5, and interleukin-13 pathways, indicating that TSLP inhibition can have broader biological effects than inhibition of individual downstream Th2 cytokines [Citation368].
Other TSLP blocking biologics include an antibody fragment that binds to TSLP and is delivered by inhalation (CSJ 117, Novartis), which has completed phase 1 trial (NCT03138811), and anti-TSLP/IL-13 bispecific antibodies [Citation369].
Because NS patients are predisposed to allergies and asthma, they could benefit from therapy with tezepelumab. However, given the complex inflammation response in NS, PAR2/TSLP signaling inhibition may not be sufficient to revert/prevent skin lesions. Keratinocyte-specific genetic inactivation of Tslp or ablation of Par-2 in Cdsn-/- mouse model of epidermal barrier dysfunction abolishes Th2 response, but at the same time aggravates IL-23 expression and the Th17 response [Citation311]. This counter-regulation mechanism between TSLP/type 2 and IL-23/type 17 immune axes suggests that a combination of biologics targeting different immune signaling pathways would be the most promising approach to treatment of NS.
2.2.2.3.11. OX40 inhibition
One of the mechanisms through which TSLP induces Th2 response is by upregulating OX40L expression in dendritic cells [Citation370]. OX40 is a costimulatory immune checkpoint receptor expressed on activated T cells including Foxp3+ Treg cells. Binding of OX40L to OX40 enhances T cell responses mediated through the T-cell receptor, such as effector T cell expansion, survival, cytokine production, adhesion and migration. Depending on the pre-existing cytokine milieu, OX40-OX40L interaction can promote T cell polarization towards Th1, Th2, Th9 or Th17. Stimulation of OX40 expressed on Foxp3+ Treg cells impairs their immune suppressor function [Citation371,Citation372].
GBR830 (Glenmark Pharmaceuticals) is a humanized monoclonal antibody against OX40 developed for the treatment of moderate-to-severe atopic dermatitis (AD). Results from phase 2a clinical trial demonstrate efficacy and safety of GBR830 [Citation373].
Given the complex immune cell landscape in NS involving different subtypes of T cells, OX40 could be a promising therapeutic target for NS.
2.2.2.3.12. Inhibition of JAK/STAT signaling
Many cytokine signaling pathways active in NS such as the ones involving IL-4, IL-6, IL-12, IL-13, IL-22, IL-23, TSLP or IFNs converge on JAK/STAT-mediated signal transduction. Therefore, JAK inhibition can have a broad impact on skin inflammation in NS. Small molecule inhibitors of JAKs already approved (e.g. ruxolitinib, tofacitinib) or currently in development are being tested in clinical trials for the treatment of inflammatory skin diseases such as atopic dermatitis and psoriasis. Both topical and systemic treatment modalities are investigated. Results from some of these studies demonstrate good efficacy and overall good safety profile for a short-term treatment [Citation374–376]. Thus, JAK inhibitors could provide yet another therapy approach for NS. Future results of clinical trials with psoriasis and AD patients should be instructive for the potential use of JAK inhibitors in NS patients. In general, the use of JAK inhibitors is attractive because, being small molecule inhibitors, their manufacturing cost is substantially lower than that of biologics.
2.2.3. Gene therapy
Different gene therapy approaches are currently being developed for NS. These include gene addition approaches aiming at restoration of LEKTI expression by viral vector-mediated gene transfer, and siRNA-mediated silencing of genes encoding KLKs or pro-inflammation mediators.
Being a monogenic inherited skin disease, Netherton syndrome is amenable to gene restoration by reintroduction of the wild-type SPINK5 allele in keratinocytes. Gene delivery can be performed ex vivo or in vivo and involves integrative and non-integrative approaches, allowing permanent or transient expression of the gene of interest, respectively [Citation377].
2.2.3.1. Ex vivo gene therapy
Proof-of-concept studies have demonstrated genetic correction of NS patient primary keratinocytes using HIV-1 based lentiviral vector [Citation192] or rAAV2 vector [Citation378] mediated expression of SPINK5.
The ex vivo lentiviral gene therapy approach has led to a phase I clinical trial [Citation379,Citation380]. The study consists in the grafting of gene-modified autologous epidermal sheets of limited size on a previously de-epidermized skin lesion. Initial results demonstrated safety and feasibility, however expression of restored LEKTI in the transplanted epidermis was only transient (less than three months), despite the use of an integrative lentiviral vector .
The ex vivo gene therapy approach may not be the most efficient strategy for NS therapy because of the need to graft extensive areas of affected skin. The gene delivery approach has to be improved to ensure sustained expression of the restored gene in the grafted skin.
2.2.3.2. In vivo viral gene therapy
A non-integrative gene therapy consisting in HSV-1 mediated delivery of SPINK5 cDNA by topical application on affected skin is currently in pre-clinical development [Citation381,Citation382]. Although topical applications will need to be repeated owing to the transient expression of LEKTI (HSV-1 vectors do not integrate into the host genome, but remain episomal), this approach is very attractive because it is non-invasive, could be applied to extensive areas of affected skin and takes advantage of the skin barrier defect, which facilitates gene delivery to the skin.
2.2.3.3. In vivo siRNA gene therapy
In vivo siRNA-mediated gene silencing has emerged as a promising strategy for treatment of skin diseases due to the possibility to deliver siRNA directly to the skin by topical application [Citation383]. Leachman et al report the first in-human siRNA therapy trial for treatment of pachyonychia congenita – an autosomal dominant skin disease caused by mutations in either KRT6A, KRT6B, KRT6C, KRT16 or KRT17 [Citation384]. Intralesional injection of siRNA targeting KRT6A or vehicle (saline) into the plantar callus of one patient for a duration of 17 weeks resulted in marked regression of the callus compared to the vehicle-treated callus on the same patient [Citation384]. The authors do not report any adverse effects, except for the extremely painful nature of the injections, which led to discontinuation of the treatment.
Different technologies have been developed to deliver siRNA-based therapeutics through the skin barrier and some of them have been tested in pre-clinical models of inflammatory skin diseases [Citation385]. Most of these studies report in vivo silencing of inflammation mediators. Noninvasive iontophoresis-mediated delivery of siRNA targeting IL-10 has been tested in vivo in atopic dermatitis-like rat model [Citation386]. The same technique was used for transdermal delivery of decoy oligonucleotides targeting NFkB in a mouse model of skin inflammation [Citation387]. It significantly reduced ear skin thickness caused by phorbol ester as well as the protein and mRNA expression levels of TNF-α in the ear skin [Citation387]. Liposome-mediated topical delivery of siRNA targeting TNF-α [Citation388], DEFB4 [Citation389], or IL-13 [Citation390] have shown efficacy in pre-clinical assays with psoriasis and/or atopic dermatitis models.
Nanoparticles represent another class of material that can be coupled to synthetic RNA oligonucleotides and that results in a superior efficiency of oligonucleotide intracellular delivery compared to conventional transfection reagents [Citation391,Citation392].
A type of nanostructures called spheric nucleic acids (SNAs) are 3-dimensional arrangements of densely packed and radially oriented oligonucleotides, which possess skin penetration properties and can pass efficiently through the cell membrane without physical or chemical enhancers [Citation393]. Zheng et al report efficient topical delivery of SNAs in mice without any associated toxicity [Citation394]. Efficient penetration and target gene knock-down were also achieved in human skin equivalents. Another study reports that topically delivered spherical nucleic acids targeting TNF-α prevent development of imiquimod-induced psoriasis-like phenotype in mice and psoriatic phenotype in 3D raft models [Citation395].
Dermelix Biotherapeutics is developing a therapy based on this topical SNAs technology for the treatment of NS [Citation396]. The target dysregulated gene(s) are not disclosed and the drug development program is currently at the pre-clinical stage.
2.2.4. Microbiome skin therapy
NS patients suffer from recurrent bacterial skin infections. Microbiome sequencing of swabs from NS patient skin revealed dysbiosis in NS patient skin, with predominant colonization by S. aureus and S. epidermidis bacterial species [Citation9]. The same study demonstrates the pathogenic role of these bacteria in the context of NS. Antibiotics improve the skin of NS patients dramatically, but transiently, suggesting that prevention of bacterial infections could be an efficacious therapy. However, antibiotics are associated with bacterial resistance and adverse effects. An alternative to antibiotics therapy would be microbiome therapy in which the introduction of engineered bacterial species, bacteriophages or small molecules onto the skin interferes with interactions between microbes and the host and thus reverts microbial dysbiosis.
2.2.4.1. Phage therapy
A case study reports therapy of a 16-year-old NS patient with topical and oral antistaphylococcal bacteriophage preparations [Citation397]. The authors describe skin lesion improvement as early as 7 days after therapy initiation and sustained clinical response for a duration of 6 months. Studies with more patients are needed to establish the long term efficacy and safety of this therapy approach.
2.2.4.2. Therapy with genetically modified bacteria
Bacteria engineered to express recombinant serine protease inhibitors Elafin or SLPI have shown efficacy in a mouse model of acute and chronic colitis [Citation398,Citation399]. Topical formulations of such bacteria engineered to express LEKTI inhibitory domains could be a promising therapy for Netherton syndrome.
Azitra has engineered the ATR-12 strain of Staphylococcus epidermidis to express LEKTI. ATR-12, currently in pre-clinical testing, is provided as a non-aqueous ointment for topical application and is designed to inhibit the overactive proteases responsible for NS [Citation400].
3. Conclusions
Since the first description in 1949, the understanding of the clinical and biological manifestations of Netherton syndrome and its underlying pathogenesis have considerably improved. It is now clear that this rare and severe genetic skin disease with life-threatening complications in the neonatal period results from defective inhibition of several epidermal proteases, at the forefront of which are KLK5, 7 and 14. Loss of proteolytic regulation results in a profound skin barrier defect leading to skin inflammation, multiple organ allergy and recurrent skin infections where Staphylococcus aureus predominates. Patients’ peripheral lymphocytes display a Th17/Th22 skewing, with debated features of immune deficiency. Murine models have been instrumental to decipher the biological cascades involved, especially constitutive and conditional Spink5 knock-out mice which closely recapitulate the disease phenotype. Patient investigations and murine model characterization have allowed to identify therapeutic targets, involving deregulated proteases and pro-inflammatory cytokines. This has led to the development of different molecules aiming at inhibiting KLK5 and/or KLK7 proteolytic activities and the use of biologics blocking pro-inflammatory cytokines showing significantly increased expression profiles. Several case studies have reported clinical improvement in a small number of NS patients, including intravenous immunoglobulins and diverse biologics such as anti-IL-17A. Clinical trials using existing biologics as well as new molecules under development targeting KLK5 and/or KLK7 hold promise for improved treatment.
4. Expert opinion
The significant progress made in the understanding of the underlying genetic defect and pathophysiological mechanisms of NS has real impact on diagnosis, current care and future treatment. Genetic testing using a custom gene panel including SPINK5 is now available in many diagnostic centers and should be proposed for every erythrodermic newborn or infant who is unwell, with failure to thrive, even in the absence of scaling or hair abnormalities. Should genetic testing not be feasible, procurement of a skin biopsy fixed in formalin is the key test to demonstrate absence or marked reduction of LEKTI staining in the granular layer of the epidermis.
Intravenous immunoglobulins, anti-TNF-α and anti-IL-17 biologics as well as other repurposed biologics such as IL-12/IL-23 and IL-4Rα neutralizing antibodies have brought clinical benefit in case studies. However, proper clinical trials including a sufficient number of patients are required to assess the real safety and effectiveness of these treatments in NS patients. Moreover, several difficulties and uncertainties remain, including:
1. The rarity of NS is a limitation for the implementation of a large number of clinical trials and justifies international multicenter studies. It also hinders NS patient natural history studies which would be insightful and helpful to evaluate treatment efficacy. Indeed, NS patients are not constantly sick . They undergo acute flare-ups whose triggering causes are not well understood. A natural history study would have the potential to evaluate disease variability over time and to search for possible correlations with patient and possibly environmental characteristics.
2. It is still unknown whether repurposed biologics targeting pro-inflammatory and/or pro-allergic biological pathways have the potential to significantly improve NS patient condition, knowing that they do not address the basic defects underlying the disease, i.e. defective inhibition of specific serine proteases. To be effective, they should target a highly relevant biological pathway playing a crucial role in NS pathogenesis, whose abrogation has the potential to revert or significantly improve the disease features.
3. More specific treatments targeting the initial causative events, i.e. unopposed KLK5 and/or KLK7 activities have the greatest therapeutic potential. Several molecules inhibiting KLK5 and/or KLK7 are currently at different stages of development. These molecules have been designed for topical or systemic delivery. Their use in future clinical trials should provide very insightful information on their effectiveness to revert the disease phenotype via different modes of delivery.
4. While KLK inhibitors which are currently under development hold great promise, their mode of delivery, topical or systemic, which would allow to optimize treatment efficacy with limited toxicity is still debated. Topical delivery is expected to allow high local concentrations of the active compound with minimal systemic effects, but skin penetration is highly variable within infants, children and adults and skin barrier is profoundly compromised in NS. In contrast, systemic delivery allows the delivery of a known and reproducible dose of the compound, has the potential to access multi cutaneous and mucosal sites, but can expose to potential toxicity, if the molecule is not highly specific for KLK5 and/or KLK7.
5. There is currently no validated standardized scoring system to evaluate disease severity in NS patients. The existing scores used for ichthyoses (IASI), atopic dermatitis (SCORAD) or psoriasis (PASI) are not appropriate, and the proposed scores for NS (NASA, IASI-I and EASI-I) scores have their limitations. Definition and validation of a standardized scoring system for NS would facilitate outcome measurement in clinical trials.
While the implementation of clinical trials will be essential to validate selected molecules, an improved understanding of the biological bases underlying several important NS features would be essential.
1. It would be insightful to understand the genetic and non-genetic determinants which account for the marked differences observed between the two main disease endotypes, namely IE and ILC, as well as between NS patients with the same endotype and sometimes within the same family, who can display a remarkable variability in disease severity despite complete absence of LEKTI. The identification of the underlying biological cascades is likely to have therapeutic implications since preliminary investigations indicate that NS-ILC and NS-IE patients display distinct immune cell skin infiltrates, which correlate with different responses to anti-IL-17A treatment [Citation255].
2. A better understanding of the mechanisms of itch and scratching would be important. Indeed, these are major aggravating factors, they severely compromise NS patient quality of life and they will serve as valuable end points in clinical trials.
3. It would be of critical importance to investigate the clinical and biological disease signatures in infants, children and in adults in order to determine possible differences over time, and to adapt treatment to these findings.
4. Elucidation of disease aggravation factors and the role of the skin and possibly the gut microbiome in disease progression would allow specific therapeutic interventions.
Advances in the understanding of these aspects would be the foundation for precision medicine, by which each patient would receive a specific treatment tailored to his/her specific gene and protein signature.
In the future, it is anticipated that deciphering the molecular signatures of the main NS endotypes and their progression over time, including the characterization of their innate and adaptive immunity status, as well as the elucidation of the bases for inter-individual variability in disease expression, will pave the way for improved medical management and effective treatments . The success of new treatments will rely on the development of highly specific and potent KLK inhibitors, on repurposed biologics and possibly in vivo gene therapy using HSV-1 based vectors. These studies will benefit from the highly relevant conditional Spink5 knock-out mouse model recently developed. The most effective and the safest molecules will allow to implement new treatments customized to the disease pathogenesis as well as to patient age and characteristics.
Article highlights
Netherton syndrome is a rare and severe genetic skin disease with very high unmet medical need.
It is characterized by a clinical triad of ichthyosiform erythroderma, a specific hair shaft abnormality (trichorrhexis invaginata, or bamboo hair), atopic manifestations and multisystemic complications.
Ichthyosiform erythroderma often evolves into ichthyosis linearis circumflexa which is highly specific, but not constant.
All NS patients have bi-allelic loss-of-function mutations in the SPINK5 gene encoding the lymphoepithelial Kazal-Type-related protease inhibitor (LEKTI).
Loss of inhibition of kallikrein-related peptidases (KLK) 5, 7 and 14 plays a crucial role in NS pathogenesis, with KLK5 being central for the initiation of the proteolytic process.
A profound skin barrier defect is a major, but not exclusive, determinant of skin inflammation and allergy, in which keratinocytes and immune cells actively participate.
Immunophenotyping of peripheral blood cells shows Th17 skewing with debated features of immune deficiency.
Case studies using intravenous immunoglobulins and diverse biologics including monoclonal antibodies targeting TNF-α, IL-17A, IL4-Rα, or IL-12/IL-23 have shown clinical benefits, but involved only a limited number of patients.
Improved understanding of NS pathogenesis has allowed to develop specific therapeutic approaches aiming at blocking KLK5 and/or KLK7, or at using biologics targeting secondary but determinant inflammatory and/or allergic events.
Clinical trials have begun and should allow to assess the potential use of these drugs to alleviate the disease burden.
This box summarizes key points contained in the article.
Declaration of interest
E Petrova receives funding from the ANR project TARGET-NS-19-CE17-0017-02 to A Hovnanian. E Petrova received funding by INSERM for a research service project sponsored by UCB Pharma. A Hovnanian is a consultant for Amagma, Boehringer-Ingelheim, BridgeBio, Genentech, Kamari and LifeMax.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors thank Jesus Maria Lopez-Gay Orts (Curie Institute, Paris, France) for help with scientific drawings.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the first ichthyosis consensus conference in sorèze 2009. J Am Acad Dermatol. 2010;63:607–641.
- Comel M. Ichthyosis linearis circumflexa. Dermatologica. 1949;98:133–136.
- Netherton EW. A unique case of trichorrhexis nodosa; bamboo hairs. AMA Arch Derm. 1958;78: 483–487. DOI:10.1001/archderm.1958.01560100059009
- Wilkinson RD, Curtis GH, Hawk WA. Netherton´s disease; Trichorrhexis invaginata (bamboo hair), congenital ichthyosiform erythroderma and the atopic diathesis. A histopathologic study. Arch Dermatol. 1964;89:46–54. DOI:10.1001/archderm.1964.01590250052010.
- Ong C, Harper J. Netherton’s syndrome. In: Harper J, Orange A, Prose N, editors. Textbook of pediatric dermatology. Turin: Blackwell; 2006. p.1359-1366.
- Traupe H. The Comel-Netherton syndrome. In: Traupe H, editor. The Ichthyoses. a guide to clinical diagnosis, genetic counseling and therapy. Berlin: Springer-Verlag; 1989. p. 168-178.
- Hausser I, Anton-Lamprecht I. Severe congenital generalized exfoliative erythroderma in newborns and infants: a possible sign of Netherton syndrome. Pediatr Dermatol. 1996;13:183–199.
- Hannula-Jouppi K, Laasanen SL, Heikkilä H, et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J Allergy Clin Immunol. 2014;134:985–988.
- Williams MR, Cau L, Wang Y, et al. Interplay of staphylococcal and host proteases promotes skin barrier disruption in netherton syndrome. Cell Rep. 2020;30:2923–2933.e7. DOI:10.1016/j.celrep.2020.02.021
- Fölster-Holst R, Swensson O, Stockfleth E, et al. Comèl-Netherton syndrome complicated by papillomatous skin lesions containing human papillomaviruses 51 and 52 and plane warts containing human papillomavirus 16. Br J Dermatol. 1999;140:1139–1143.
- Guerra L, Fortugno P, Sinistro A, et al. Betapapillomavirus in multiple non-melanoma skin cancers of Netherton syndrome: case report and published work review. J Dermatol. 2015;42:786–794.
- Ashton R, Moledina J, Sivakumar B, et al. Considerations in surgical management of a Buschke-Lowenstein tumor in Netherton syndrome: a case report. Pediatr Dermatol. 2017;34:e328–e330.
- van der Voort EAM, Prens EP. Netherton syndrome with multiple non-melanoma skin cancers. Acta Derm Venereol. 2013;93:727–728.
- Moskowitz DG, Fowler AJ, Heyman MB, et al. Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr. 2004;145:82–92.
- Komatsu N, Saijoh K, Otsuki N, et al. Proteolytic processing of human growth hormone by multiple tissue kallikreins and regulation by the serine protease inhibitor Kazal-Type5 (SPINK5) protein. Clin Chim Acta. 2007;377:228–236.
- Eränkö E, Ilander M, Tuomiranta M, et al. Immune cell phenotype and functional defects in Netherton syndrome. Orphanet J Rare Dis. 2018;13:1–10. DOI:10.1186/s13023-018-0956-6
- Hannula-Jouppi K, Laasanen SL, Ilander M, et al. Intrafamily and interfamilial phenotype variation and immature immunity in patients with netherton syndrome and finnish SPINK5 founder mutation. JAMA Dermatol. 2016;152:435–442.
- Paller AS, Renert-Yuval Y, Suprun M, et al. An IL-17–dominant immune profile is shared across the major orphan forms of ichthyosis. J Allergy Clin Immunol. 2017;139:152–165. DOI:10.1016/j.jaci.2016.07.019
- Renner ED, Hartl D, Rylaarsdam S, et al. Comèl-Netherton syndrome defined as primary immunodeficiency. J Allergy Clin Immunol. 2009;124:536–543. DOI:10.1016/j.jaci.2009.06.009
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum Mol Genet. 2003;12:2417–2430. DOI:10.1093/hmg/ddg247
- Lacroix M, Lacaze-Buzy L, Furio L, et al. Clinical expression and new SPINK5 splicing defects in Netherton syndrome: unmasking a frequent founder synonymous mutation and unconventional intronic mutations. J Invest Dermatol. 2012;132:575–582.
- Stenson PD, Ball EV, Mort M, et al. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr Protoc Bioinforma. 2012;Chapter 1(Unit1):13.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141–142. DOI:10.1038/75977
- Ishida-Yamamoto A, Deraison C, Bonnart C, et al. LEKTI is localized in lamellar granules, separated from KLK5 and KLK7, and is secreted in the extracellular spaces of the superficial stratum granulosum. J Invest Dermatol. 2005;124:360–366. DOI:10.1111/j.0022-202X.2004.23583.x
- Raghunath M, Tontsidou L, Oji V, et al. SPINK5 and Netherton syndrome: novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. J Invest Dermatol. 2004;123:474–483.
- Galliano MF, Roccasecca RM, Descargues P, et al. Characterization and expression analysis of the Spink5 gene, the mouse ortholog of the defective gene in Netherton syndrome. Genomics. 2005;85:483–492.
- Descargues P, Draison C, Bonnart C, et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat Genet. 2005;37:56–65. DOI:10.1038/ng1493
- Hewett DR, Simons AL, Mangan NE, et al. Lethal, neonatal ichthyosis with increased proteolytic processing of filaggrin in a mouse model of Netherton syndrome. Hum Mol Genet. 2005;14:335–346. DOI:10.1093/hmg/ddi030
- Keuylian Z, Hovnanian A. Mechanistic insight from murine models of Netherton syndrome. Biol Chem. 2016;397:1223–1228.
- Yang T, Liang D, Koch PJ, et al. Epidermal detachment, desmosomal dissociation, and destabilization of corneodesmosin in Spink5-/- mice. Genes Dev. 2004;18:2354–2358. DOI:10.1101/gad.1232104
- Briot A, Deraison C, Lacroix M, et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206:1135–1147. DOI:10.1084/jem.20082242
- Briot A, Lacroix M, Robin A, et al. Par2 inactivation inhibits early production of TSLP, but not cutaneous inflammation, in netherton syndrome adult mouse model. J Invest Dermatol. 2010;130:2736–2742.
- Kasparek P, Ileninova Z, Haneckova R, et al. A viable mouse model for Netherton syndrome based on mosaic inactivation of the Spink5 gene. Biol Chem. 2016;397:1287–1292.
- Mägert HJ, Ständker L, Kreutzmann P, et al. LEKTI, a novel 15-domain type of human serine proteinase inhibitor. J Biol Chem. 1999;274:21499–21502. DOI:10.1074/jbc.274.31.21499
- Tartaglia-Polcini A, Bonnart C, Micheloni A, et al. SPINK5, the defective gene in netherton syndrome, encodes multiple LEKTI isoforms derived from alternative pre-mRNA processing. J Invest Dermatol. 2006;126:315–324.
- Fortugno P, Bresciani A, Paolini C, et al. Proteolytic activation cascade of the netherton syndrome-defective protein, LEKTI, in the epidermis: implications for skin homeostasis. J Invest Dermatol. 2011;131:2223–2232. DOI:10.1038/jid.2011.174
- Borgoño CA, Michael IP, Komatsu N, et al. A potential role for multiple tissue kallikrein serine proteases in epidermal desquamation. J Biol Chem. 2007;282:3640–3652.
- Deraison C, Bonnart C, Lopez F, et al. LEKTI fragments specifically inhibit KLK5, KLK7, and KLK14 and control desquamation through a pH-dependent interaction. Mol Biol Cell. 2007;18:3607–3619. DOI:10.1091/mbc.e07-02-0124
- Bennett K, Heywood W, Di WL, et al. The identification of a new role for LEKTI in the skin: the use of protein “bait” arrays to detect defective trafficking of dermcidin in the skin of patients with Netherton syndrome. J Proteomics. 2012;75:3925–3937.
- Egelrud T, Brattsand M, Kreutzmann P, et al. hK5 and hK7, two serine proteinases abundant in human skin, are inhibited by LEKTI domain 6. Br J Dermatol. 2005;153:1200–1203.
- Mitsudo K, Jayakumar A, Henderson Y, et al. Inhibition of serine proteinases plasmin, trypsin, subtilisin A, cathepsin G, and elastase by LEKTI: A kinetic analysis. Biochemistry. 2003;42:3874–3881.
- Schechter NM, Choi EJ, Wang ZM, et al. Inhibition of human kallikreins 5 and 7 by the serine protease inhibitor lympho-epithelial Kazal-type inhibitor (LEKTI). Biol Chem. 2005;386:1173–1184.
- Bonnart C, Deraison C, Lacroix M, et al. Elastase 2 is expressed in human and mouse epidermis and impairs skin barrier function in Netherton syndrome through filaggrin and lipid misprocessing. J Clin Invest. 2010;120:871–882. DOI:10.1172/JCI41440
- Bennett K, Callard R, Heywood W, et al. New role for LEKTI in skin barrier formation: label-free quantitative proteomic identification of caspase 14 as a novel target for the protease inhibitor LEKTI. J Proteome Res. 2010;9:4289–4294.
- Komatsu N, Takata M, Otsuki N, et al. Elevated stratum corneum hydrolytic activity in Netherton syndrome suggests an inhibitory regulation of desquamation by SPINK5-derived peptides. J Invest Dermatol. 2002;118:436–443.
- Eissa A, Diamandis EP. Human tissue kallikreins as promiscuous modulators of homeostatic skin barrier functions. Biol Chem. 2008;389:669–680.
- Billi AC, Ludwig JE, Fritz Y, et al. KLK6 expression in skin induces PAR1-mediated psoriasiform dermatitis and inflammatory joint disease. J Clin Invest. 2020;130:3151–3157.
- Gouin O, Barbieux C, Leturcq F, et al. Transgenic kallikrein 14 mice display major hair shaft defects associated with desmoglein 3 and 4 degradation, abnormal epidermal differentiation, and IL-36 signature. J Invest Dermatol. 2020;140:1184–1194. DOI:10.1016/j.jid.2019.10.026
- Bäckman A, Ny A, Edlund M, et al. Epidermal overexpression of stratum corneum chymotryptic enzyme in mice: A model for chronic itchy dermatitis. J Invest Dermatol. 2002;118(3):444–449. DOI:10.1046/j.0022-202x.2001.01684.x
- Ny A, Egelrud T. Transgenic Mice over-expressing a serine protease in the skin: evidence of interferon γ-independent MHC II expression by epidermal keratinocytes. Acta Derm Venereol. 2003;83:322–327.
- Ny A, Egelrud T. Epidermal hyperproliferation and decreased skin barrier function in mice overexpressing stratum corneum chymotryptic enzyme. Acta Derm Venereol. 2004;84:18–22.
- Furio L, De Veer S, Jaillet M, et al. Transgenic kallikrein 5 mice reproduce major cutaneous and systemic hallmarks of Netherton syndrome. J Exp Med. 2014;211:499–513. DOI:10.1084/jem.20131797
- Caubet C, Jonca N, Brattsand M, et al. Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J Invest Dermatol. 2004;122:1235–1244.
- Descargues P, Deraison C, Prost C, et al. Corneodesmosomal cadherins are preferential targets of stratum corneum trypsin- and chymotrypsin-like hyperactivity in Netherton syndrome. J Invest Dermatol. 2006;126:1622–1632. DOI:10.1038/sj.jid.5700284
- Simon M, Jonca N, Guerrin M, et al. Refined characterization of corneodesmosin proteolysis during terminal differentiation of human epidermis and its relationship to desquamation. J Biol Chem. 2001;276:20292–20299.
- Sakabe JI, Yamamoto M, Hirakawa S, et al. Kallikrein-related peptidase 5 functions in proteolytic processing of profilaggrin in cultured human keratinocytes. J Biol Chem. 2013;288:17179–17189.
- Hachem JP, Wagberg F, Schmuth M, et al. Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. J Invest Dermatol. 2006;126:1609–1621.
- Hachem JP, Man MQ, Crumrine D, et al. Sustained serine proteases activity by prolonged increase in pH leads to degradation of lipid processing enzymes and profound alterations of barrier function and stratum corneum integrity. J Invest Dermatol. 2005;125:510–520.
- Elias PM, Wakefield JS. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134(781–91):e1.
- Van Smeden J, Janssens M, Boiten WA, et al. Intercellular skin barrier lipid composition and organization in netherton syndrome patients. J Invest Dermatol. 2014;134:1238–1245.
- van Smeden J, Al-Khakany H, Wang Y, et al. Skin barrier lipid enzyme activity in Netherton patients is associated with protease activity and ceramide abnormalities. J Lipid Res. 2020;61:859–869. DOI:10.1194/jlr.RA120000639
- Fartasch M, Williams ML, Elias PM. Altered lamellar body secretion and stratum corneum membrane structure in Netherton syndrome: differentiation from other infantile erythrodermas and pathogenic implications. Arch Dermatol. 1999;135:823–832.
- Oikonomopoulou K, Hansen KK, Saifeddine M, et al. Kallikrein-mediated cell signalling: targeting proteinase-activated receptors (PARs). Biol Chem. 2006;387:817–824.
- Stefansson K, Brattsand M, Roosterman D, et al. Activation of proteinase-activated receptor-2 by human kallikrein-related peptidases. J Invest Dermatol. 2008;128:18–25.
- Nylander-Lundqvist E, Egelrud T. Formation of active IL-1 beta from pro-IL-1 beta catalyzed by stratum corneum chymotryptic enzyme in vitro. Acta Derm Venereol. 1997;77:203–209.
- Yamasaki K, Schauber J, Coda A, et al. Kallikrein‐mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. Faseb J. 2006;20:2068–2080.
- Malik K, He H, Huynh TN, et al. Ichthyosis molecular fingerprinting shows profound T H 17 skewing and a unique barrier genomic signature. J Allergy Clin Immunol. 2019;143:604–618. DOI:10.1016/j.jaci.2018.03.021
- Kobayashi T, Nagao K. Host-microbial dialogues in atopic dermatitis. Int Immunol. 2019;31:449–456.
- Kim BE, Leung DYM, Boguniewicz M, et al. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126:332–337.
- Howell MD, Kim BE, Gao P, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2009;124:R7–R12.
- Hatano Y, Terashi H, Arakawa S, et al. Interleukin-4 suppresses the enhancement of ceramide synthesis and cutaneous permeability barrier functions induced by tumor necrosis factor-alpha and interferon-gamma in human epidermis. J Invest Dermatol. 2005;124:786–792.
- Steinhoff M, Bienenstock J, Schmelz M, et al. Neurophysiological, neuroimmunological, and neuroendocrine basis of pruritus. J Invest Dermatol. 2006;126:1705–1718.
- Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396:345–360.
- Greb JE, Goldminz AM, Elder JT, et al. Psoriasis. Nat Rev Dis Prim. 2016;2:16082.
- Lodén M. Role of topical emollients and moisturizers in the treatment of dry skin barrier disorders. Am J Clin Dermatol. 2003;4:771–788.
- Wehr RF, Hickman J, Krochmal L. Effective treatment of Netherton’s syndrome with 12% lactate lotion. J Am Acad Dermatol. 1988;19:140–142.
- Cockayne SE, Lee JA, Harrington CI. Oleogranulomatous response in lymph nodes associated with emollient use in Netherton’s syndrome. Br J Dermatol. 1999;141:562–564.
- Elias PM, Wakefield JS, Man M-Q. Moisturizers versus current and next-generation barrier repair therapy for the management of atopic dermatitis. Skin Pharmacol Physiol. 2019;32:1–7.
- Lin T-K, Man M-Q, Santiago J-L, et al. Topical antihistamines display potent anti-inflammatory activity linked in part to enhanced permeability barrier function. J Invest Dermatol. 2013;133:469–478.
- Teng J, Marqueling A, Benjamin L. Therapy in pediatric dermatology: management of pediatric Skin Disease. Cham: Springer; 2017.
- Bandyopadhyay D. A treatise on topical corticosteroids in dermatology. Use, misuse and abuse. Indian J Dermatol Venereol Leprol. 2018. DOI:10.4103/ijdvl.IJDVL_755_18
- Ference JD, Last AR. Choosing topical corticosteroids. Am Fam Physician. 2009;79:135–140.
- Ramamoorthy S, Cidlowski JA. Corticosteroids: mechanisms of action in health and Disease. Rheum Dis Clin North Am. 2016;42:15–31.
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723.
- Uva L, Miguel D, Pinheiro C, et al. Mechanisms of action of topical corticosteroids in psoriasis. Int J Endocrinol. 2012;2012:561018.
- Greene SL, Muller SA. Netherton’s syndrome: report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329–337.
- Halverstam CP, Vachharajani A, Mallory SB. Cushing syndrome from percutaneous absorption of 1% hydrocortisone ointment in Netherton syndrome. Pediatr Dermatol. 2007;24:42–45.
- Valette C, Ofaiche J, Severino M. Évolution fatale du syndrome de Netherthon due à une utilisation excessive de dermocorticoïdes chez un adulte. Ann Dermatol Venereol. 2020;147:36–40.
- Beckenbach L, Baron JM, Merk HF, et al. Retinoid treatment of skin diseases. Eur J Dermatology. 2015;25:384–391. DOI:10.1684/ejd.2015.2544
- Digiovanna JJ, Mauro T, Milstone LM, et al. Systemic retinoids in the management of ichthyoses and related skin types. Dermatol Ther. 2013;26:26–38.
- Lazaridou E, Apalla Z, Patsatsi A, et al. Netherton’s syndrome: successful treatment with isotretinoin. J Eur Acad Dermatol Venereol. 2009;23:210–212.
- Leung AKC, Barankin B, Leong KF. An 8-year-old child with delayed diagnosis of netherton syndrome. Case Rep Pediatr. 2018;2018:1–4.
- Haußer I, Anton-Lamprecht I, Hartschuh W, et al. Netherton’s syndrome: ultrastructure of the active lesion under retinoid therapy. Arch Dermatol Res. 1989;281:165–172.
- Nevet MJ, Indelman M, Ben-Ari J, et al. A case of Netherton syndrome with intestinal atresia, a novel SPINK5 mutation, and a fatal course. Int J Dermatol. 2017;56:1055–1057.
- Caputo R, Vanotti P, Bertani E. Netherton’s Syndrome in two adult brothers. Arch Dermatol. 1984;120:220–222.
- Maatouk I, Moutran R, Tomb R. Narrowband ultraviolet B phototherapy associated with improvement in Netherton syndrome. Clin Exp Dermatol. 2012;37:364–366.
- Happle R, van de Kerkhof PC, Traupe H. Retinoids in disorders of keratinization: their use in adults. Dermatologica. 1987;175(Suppl):107–124.
- Orfanos CE, Zouboulis CC, Almond-Roesler B, et al. Current use and future potential role of retinoids in dermatology. Drugs. 1997;53:358–388.
- Groves S, Dezfoulian B, Bonardeaux C, et al. Netherton’s syndrome in two sisters. A ten year experience of therapy with retinoids. J Eur Acad Dermatol Venereol. 1995;5:173–176.
- Scott LJ, Dunn CJ, Goa KL. Calcipotriol ointment. A review of its use in the management of psoriasis. Am J Clin Dermatol. 2001;2:95–120.
- Guenther LC. Calcipotriol/betamethasone dipropionate :daivobet/dovobet. Therapy. 2005;2:343–348.
- Trémezaygues L, Reichrath J. Vitamin D analogs in the treatment of psoriasis: where are we standing and where will we be going? Dermatoendocrinol. 2011;3:180–186.
- Peric M, Koglin S, Dombrowski Y, et al. Vitamin D analogs differentially control antimicrobial peptide/‘“ alarmin ”’ expression in psoriasis. PLoS One. 2009; 4(7):e6340.
- Germán B, Wei R, Hener P, et al. Disrupting the IL-36 and IL-23/IL-17 loop underlies the efficacy of calcipotriol and corticosteroid therapy for psoriasis. JCI Insight. 2019;4(2):e123390.
- Lucker GP, van de Kerkhof PC, van Dïjk MR, et al. Effect of topical calcipotriol on congenital ichthyoses. Br J Dermatol. 1994;131:546–550.
- Godic A, Dragos V. Successful treatment of Netherton’s syndrome with topical calcipotriol. Eur J Dermatol. 2004;14:115–117.
- Grassberger M, Steinhoff M, Schneider D, et al. Pimecrolimus - An anti-inflammatory drug targeting the skin. Exp Dermatol. 2004;13:721–730.
- Nakahara T, Morimoto H, Murakami N, et al. Mechanistic insights into topical tacrolimus for the treatment of atopic dermatitis. Pediatr Allergy Immunol off Publ Eur Soc Pediatr Allergy Immunol. 2018;29:233–238.
- Ma Z, Jiao Z. Mast cells as targets of pimecrolimus. Curr Pharm Des. 2011;17:3823–3829.
- Billich A, Aschauer H, Aszódi A, et al. Percutaneous absorption of drugs used in atopic eczema: pimecrolimus permeates less through skin than corticosteroids and tacrolimus. Int J Pharm. 2004;269:29–35.
- Meingassner JG, Aschauer H, Stuetz A, et al. Pimecrolimus permeates less than tacrolimus through normal, inflamed, or corticosteroid-pretreated skin. Exp Dermatol. 2005;14:752–757.
- Oji V, Beljan G, Beier K, et al. Topical pimecrolimus: a novel therapeutic option for Netherton syndrome. Br J Dermatol. England. 2005;1067–1068. DOI:10.1111/j.1365-2133.2005.06884.x
- Shah KN, Yan AC. Low but detectable serum levels of tacrolimus seen with the use of very dilute, extemporaneously compounded formulations of tacrolimus ointment in the treatment of patients with netherton syndrome. Arch Dermatol. 2006;142(10):1362-3.
- Allen A, Siegfried E, Silverman R, et al. Significant absorption of topical tacrolimus in 3 patients with netherton syndrome. Arch Dermatol. 2001;137:747–750.
- Bens G, Boralevi F, Buzenet C, et al. Topical treatment of Netherton’s syndrome with tacrolimus ointment without significant systemic absorption. Br J Dermatol. 2003;224–226. England. DOI:10.1046/j.1365-2133.2003.05443.x
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57–62.
- Saif GB, Al-Khenaizan S. Netherton syndrome: successful use of topical tacrolimus and pimecrolimus in four siblings. Int J Dermatol. 2007;46:290–294.
- Henno A, Choffray A, De La Brassinne M. Amélioration par pimecrolimus topique de l’érythrodermie du syndrome de Netherton chez deux sœurs adultes. Ann Dermatol Venereol. 2006;133:71–72.
- Margolis JS, Abuabara K, Bilker W, et al. Persistence of mild to moderate atopic dermatitis. JAMA Dermatol. 2014;150:593–600.
- Kim M, Jung M, Hong S-P, et al. Topical calcineurin inhibitors compromise stratum corneum integrity, epidermal permeability and antimicrobial barrier function. Exp Dermatol. 2010;19:501–510.
- Ibbotson SH. A perspective on the use of NB-UVB phototherapy vs. PUVA photochemotherapy. Front Med. 2018;5:1–8.
- Wong T, Hsu L, Liao W. Phototherapy in psoriasis: a review of mechanisms of action. J Cutan Med Surg. 2013;17:6–12.
- Kaminska ECN, Ortel B, Sharma V, et al. Narrowband UVB phototherapy as a novel treatment for Netherton syndrome. Photodermatol Photoimmunol Photomed. 2012;28:162–164.
- Singer R, Çopur M, Yüksel EN, et al. Ichthyosis linearis circumflexa in a child. Response to narrowband UVB therapy. J Dermatol Case Rep. 2015; 9(4):110–2.
- Nagata T. Netherton’s syndrome which responded to photochemotherapy. Dermatology. 1980;161:51–56.
- Capezzera R, Venturini M, Bianchi D, et al. UVA1 phototherapy of netherton syndrome [1]. Acta Derm Venereol. 2004;84:69–70.
- Permatasari F, Zhou B, Luo D. Epidermal barrier: adverse and beneficial changes induced by ultraviolet B irradiation depending on the exposure dose and time (Review). Exp Ther Med. 2013;6:287–292.
- Hong SP, Kim MJ, Jung M-Y, et al. Biopositive effects of low-dose UVB on epidermis: coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J Invest Dermatol. 2008;128:2880–2887.
- Stern RS, Lunder EJ. Risk of squamous cell carcinoma and methoxsalen (psoralen) and UV-A radiation (PUVA). A meta-analysis. Arch Dermatol. 1998;134:1582–1585.
- Marcil I, Stern RS. Squamous-cell cancer of the skin in patients given PUVA and ciclosporin: nested cohort crossover study. Lancet. 2001;358:1042–1045.
- Ormerod AD. Topical tacrolimus and pimecrolimus and the risk of cancer: how much cause for concern? Br J Dermatol. 2005;153:701–705.
- Komatsu N, Tsai B, Sidiropoulos M, et al. Quantification of eight tissue kallikreins in the stratum corneum and sweat. J Invest Dermatol. 2006;126:927–931.
- Yu Y, Prassas I, Muytjens CMJ, et al. Proteomic and peptidomic analysis of human sweat with emphasis on proteolysis. J Proteomics. 2017;155:40–48.
- Shroot B. Further light is shed on topical therapy. J Invest Dermatol. 2003;121:xiii–xiv.
- Lundwall Å, Band V, Blaber M, et al. A comprehensive nomenclature for serine proteases with homology to tissue kallikreins. Biol Chem. 2006;387:637–641.
- Loessner D, Goettig P, Preis S, et al. Expert Opinion on therapeutic targets kallikrein-related peptidases represent attractive therapeutic targets for ovarian cancer. Expert Opin Ther Targets. 2018;22:745–763.
- Filippou PS, Ren AH, Soosaipillai A, et al. Kallikrein-related peptidases protein expression in lymphoid tissues suggests potential implications in immune response: KLK protein expression in lymphoid tissues. Clin Biochem. 2020;77:41–47.
- Shaw JLV, Diamandis EP. Distribution of 15 human kallikreins in tissues and biological fluids. Clin Chem. 2007;53:1423–1432.
- Diamandis EP, Yousef GM. Human tissue kallikrein gene family: a rich source of novel disease biomarkers. Expert Rev Mol Diagn. 2001;1:182–190.
- Prassas I, Eissa A, Poda G, et al. Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nat Rev Drug Discov. 2015;14:183–202.
- Avgeris M, Scorilas A. Expert opinion on therapeutic targets kallikrein-related peptidases (KLKs) as emerging therapeutic targets : focus on prostate cancer and skin pathologies. Expert Opin Ther Targets. 2016;20:801–818.
- Sotiropoulou G, Pampalakis G. Targeting the kallikrein-related peptidases for drug development. Trends Pharmacol Sci. 2012;33:623–634.
- Komatsu N, Takata M, Otsuki N, et al. Expression and localization of tissue kallikrein mRNAs in human epidermis and appendages. J Invest Dermatol. 2003;121:542–549.
- Komatsu N, Saijoh K, Toyama T, et al. Multiple tissue kallikrein mRNA and protein expression in normal skin and skin diseases. Br J Dermatol. 2005;153:274–281.
- Brattsand M, Egelrud T. Purification, molecular cloning, and expression of a human stratum corneum trypsin-like serine protease with possible function in desquamation. J Biol Chem. 1999;274:30033–30040.
- Hansson L. Cloning, expression, and characterization of stratum corneum chymotryptic enzyme. J Biol Chem. 1994;269:19420–19426.
- Stefansson K, Brattsand M, Ny A, et al. Kallikrein-related peptidase 14 may be a major contributor to trypsin-like proteolytic activity in human stratum corneum. Biol Chem. 2006;387:761–768.
- Oikonomopoulou K, DeAngelis RA, Chen H, et al. Induction of complement C3a receptor responses by kallikrein-related peptidase 14. J Immunol. 2013;191:3858–3866.
- Michael IP, Sotiropoulou G, Pampalakis G, et al. Biochemical and enzymatic characterization of human kallikrein 5 (hK5), a novel serine protease potentially involved in cancer progression. J Biol Chem. 2005;280:14628–14635.
- Brattsand M, Stefansson K, Lundh C, et al. A proteolytic cascade of kallikreins in the stratum corneum. J Invest Dermatol. 2005;124:198–203.
- Yoon H, Laxmikanthan G, Lee J, et al. Activation profiles and regulatory cascades of the human kallikrein-related peptidases. J Biol Chem. 2007;282:31852–31864.
- Sales KU, Masedunskas A, Bey AL, et al. Matriptase initiates activation of epidermal pro-kallikrein and disease onset in a mouse model of Netherton syndrome. Nat Genet. 2010;42:676–683. DOI:10.1038/ng.629
- Lin C-Y, Wang J-K, Johnson MD. The spatiotemporal control of human matriptase action on its physiological substrates: a case against a direct role for matriptase proteolytic activity in profilaggrin processing and desquamation. Hum Cell. 2020;33:459–469.
- Lai C-H, Chang S-C, Chen Y-J, et al. Matriptase and prostasin are expressed in human skin in an inverse trend over the course of differentiation and are targeted to different regions of the plasma membrane. Biol Open. 2016;5:1380–1387.
- Miyai M, Matsumoto Y, Yamanishi H, et al. Keratinocyte-specific mesotrypsin contributes to the desquamation process via kallikrein activation and LEKTI degradation. J Invest Dermatol. 2014;134:1665–1674.
- Borgoño CA, Michael IP, Shaw JLV, et al. Expression and functional characterization of the cancer-related serine protease, human tissue kallikrein 14. J Biol Chem. 2007;282:2405–2422.
- Debela M, Goettig P, Magdolen V, et al. Structural basis of the zinc inhibition of human tissue kallikrein 5. J Mol Biol. 2007;373:1017–1031.
- Debela M, Hess P, Magdolen V, et al. Chymotryptic specificity determinants in the 1.0 A structure of the zinc-inhibited human tissue kallikrein 7. Proc Natl Acad Sci U S A. 2007;104:16086–16091.
- Kishi T, Cloutier SM, Kündig C, et al. Activation and enzymatic characterization of recombinant human kallikrein 8. Biol Chem. 2006;387:723–731.
- Ogawa Y, Kinoshita M, Shimada S, et al. Zinc and skin disorders. Nutrients. 2018;10:199. [Internet]. https://pubmed.ncbi.nlm.nih.gov/29439479
- Ohman H, Vahlquist A. In vivo studies concerning a pH gradient in human stratum corneum and upper epidermis. Acta Derm Venereol. 1994;74:375–379.
- Fluhr JW, Elias PM. Stratum corneum pH: formation and function of the “acid mantle”. Exog Dermatol. 2002;1: 163–175. DOI:10.1159/000066140
- Sotiropoulou G, Pampalakis G, Diamandis EP. Functional roles of human Kallikrein-related peptidases. J Biol Chem. 2009;284:32989–32994.
- Eissa A, Amodeo V, Smith CR, et al. Kallikrein-related peptidase-8 (KLK8) is an active serine protease in human epidermis and sweat and is involved in a skin barrier proteolytic cascade. J Biol Chem. 2011;286:687–706.
- Goettig P, Magdolen V, Brandstetter H. Natural and synthetic inhibitors of kallikrein-related peptidases (KLKs). Biochimie. 2010;92:1546–1567.
- Komatsu N, Saijoh K, Jayakumar A, et al. Correlation between SPINK5 gene mutations and clinical manifestations in netherton syndrome patients. J Invest Dermatol. 2008;128:1148–1159.
- Meyer-Hoffert U, Wu Z, Kantyka T, et al. Isolation of SPINK6 in human skin: selective inhibitor of kallikrein-related peptidases. J Biol Chem. 2010;285:32174–32181.
- Brännström K, Ohman A, von Pawel Rammingen U, et al. Characterization of SPINK9, a KLK5-specific inhibitor expressed in palmo-plantar epidermis. Biol Chem. 2012;393:369–377.
- Brattsand M, Stefansson K, Hubiche T, et al. SPINK9: A selective, skin-specific kazal-type serine protease inhibitor. J Invest Dermatol. 2009;129:1656–1665.
- Heit C, Jackson BC, McAndrews M, et al. Update of the human and mouse SERPIN gene superfamily. Hum Genomics. 2013;7:1–14.
- Luo LY, Jiang W. Inhibition profiles of human tissue kallikreins by serine protease inhibitors. Biol Chem. 2006;387:813–816.
- Zhou GX, Chao L, Chao J. Kallistatin: a novel human tissue kallikrein inhibitor. Purification, characterization, and reactive center sequence. J Biol Chem. 1992;267:25873–25880.
- Scott FL, Sun J, Whisstock JC, et al. SerpinB6 is an inhibitor of kallikrein-8 in keratinocytes. J Biochem. 2007;142:435–442.
- Ulbricht D, Tindall CA, Oertwig K, et al. Kallikrein-related peptidase 14 is the second KLK protease targeted by the serpin vaspin. Biol Chem. 2018;399:1079–1084.
- Heiker JT, Klöting N, Kovacs P, et al. Vaspin inhibits kallikrein 7 by serpin mechanism. Cell Mol Life Sci. 2013;70:2569–2583.
- Clauss A, Lilja H, Lundwall A. The evolution of a genetic locus encoding small serine proteinase inhibitors. Biochem Biophys Res Commun. 2005;333:383–389.
- Wilkinson TS, Roghanian A, Simpson AJ, et al. WAP domain proteins as modulators of mucosal immunity. Biochem Soc Trans. 2011;39:1409–1415.
- Franzke CW, Baici A, Bartels J, et al. Antileukoprotease inhibits stratum corneum chymotryptic enzyme. Evidence for a regulative function in desquamation. J Biol Chem. 1996;271:21886–21890.
- Kalinina P, Vorstandlechner V, Buchberger M, et al. The whey acidic protein WFDC12 is specifically expressed in terminally differentiated keratinocytes and regulates epidermal serine-protease activity. J Invest Dermatol. 2020;S0022-202X(20):32252.
- Wiedow O, Schröder JM, Gregory H, et al. Elafin: an elastase-specific inhibitor of human skin. Purification, characterization, and complete amino acid sequence. J Biol Chem. 1990;265:14791–14795.
- de Veer SJ, Furio L, Harris JM, et al. Proteases and proteomics: cutting to the core of human skin pathologies. Proteomics - Clin Appl. 2014;8:389–402.
- de Veer SJ, Furio L, Harris JM, et al. Proteases: common culprits in human skin disorders. Trends Mol Med. 2014;20:166–178. DOI:10.1016/j.molmed.2013.11.005
- Chittock J, Cooke A, Lavender T, et al. Development of stratum corneum chymotrypsin-like protease activity and natural moisturizing factors from birth to 4 weeks of age compared with adults. Br J Dermatol. 2016;175:713–720.
- Diamandis EP, Yousef GM, Olsson AY. An update on human and mouse glandular kallikreins. Clin Biochem. 2004;37:258–260.
- Yvonne Olsson A, Lundwall Å. Organization and evolution of the glandular kallikrein locus in Mus musculus. Biochem Biophys Res Commun. 2002;299:305–311.
- Pavlopoulou A, Pampalakis G, Michalopoulos I, et al. Evolutionary history of tissue kallikreins. PLoS One. 2010;5(11):e13781.
- Furio L, Pampalakis G, Michael IP, et al. KLK5 inactivation reverses cutaneous hallmarks of Netherton Syndrome. PLoS Genet. 2015;11:e1005389. DOI:10.1371/journal.pgen.1005389
- Kasparek P, Ileninova Z, Zbodakova O, et al. KLK5 and KLK7 ablation fully rescues lethality of Netherton Syndrome-like phenotype. PLoS Genet. 2017;13(1):e1006566.
- Zingkou E, Pampalakis G, Charla E, et al. A proinflammatory role of KLK6 protease in Netherton syndrome. J Dermatol Sci. 2019;95:28–35.
- Klucky B, Mueller R, Vogt I, et al. Kallikrein 6 induces E-cadherin shedding and promotes cell proliferation, migration, and invasion. Cancer Res. 2007;67:8198–8206.
- Chiticariu E, Hohl D. Netherton Syndrome: insights into pathogenesis and clinical implications. J Invest Dermatol. 2020;140:1129–1130.
- Di WL, Larcher F, Semenova E, et al. Ex-vivo gene therapy restores LEKTI activity and corrects the architecture of netherton syndrome-derived skin grafts. Mol Ther. 2011;19:408–416.
- Gálvez V, Chacón-Solano E, Bonafont J, et al. Efficient CRISPR-Cas9-mediated gene ablation in human keratinocytes to recapitulate genodermatoses: modeling of Netherton Syndrome. Mol Ther - Methods Clin Dev. 2020;18:280–290.
- Wang S, Olt S, Schoefmann N, et al. SPINK5 knockdown in organotypic human skin culture as a model system for Netherton syndrome: effect of genetic inhibition of serine proteases kallikrein 5 and kallikrein 7. Exp Dermatol. 2014;23:524–526.
- Walker AL, Denis A, Bingham RP, et al. Design and development of a series of borocycles as selective, covalent kallikrein 5 inhibitors. Bioorg Med Chem Lett. 2019;29:126675. DOI:10.1016/j.bmcl.2019.126675
- Wågberg F, Leonardsson G Sixera Pharma AB, assignee. Benzoxazinone derivatives for treatment of skin diseases. World patent WO2015/112081A1. 2015 Jul 30.
- Sixera Pharma [Internet]. Stockholm: scientific Research ; 2018c [cited 2020 Jun 29]. https://sixerapharma.com/scientific-research/.2018
- Vinnova [Internet]. Stockholm: new treatment for Netherton syndrome. [ updated 2019 Nov 25; cited 2020 Jun 29]. https://www.vinnova.se/en/p/new-treatment-for-netherton-syndrome/.e.2019
- BridgeBio [Internet]. Palo Alto CA: the pipeline ; 2019c [cited 2020 Jun 29]. https://bridgebio.com/pipeline.2019
- United States Securities and Exchange Commission [Internet]. Washington DC: form S-1 registration statement BridgeBio Pharma Inc. [ updated 2019 May 24; cited 2020 Jun 29]. https://www.sec.gov/Archives/edgar/data/1743881/00011931251915710
- Tan X, Soualmia F, Furio L, et al. Toward the first class of suicide inhibitors of kallikreins involved in skin diseases. J Med Chem. 2015;58:598–612.
- Di Paolo CT, Filippou PS, Yu Y, et al. Screening of chemical libraries in pursuit of kallikrein-5 specific inhibitors for the treatment of inflammatory dermatoses. Clin Chem Lab Med. 2019;57:1737–1743.
- de Souza AS, Pacheco BDC, Pinheiro S, et al. 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7. Bioorg Med Chem Lett. 2019;29:1094–1098.
- Murafuji H, Sakai H, Goto M, et al. Discovery and structure-activity relationship study of 1,3,6-trisubstituted 1,4-diazepane-7-ones as novel human kallikrein 7 inhibitors. Bioorg Med Chem Lett. 2017;27:5272–5276.
- Murafuji H, Sugawara H, Goto M, et al. Structure-based drug design to overcome species differences in kallikrein 7 inhibition of 1,3,6-trisubstituted 1,4-diazepan-7-ones. Bioorg Med Chem. 2018;26:3639–3653.
- Freitas RF, Teixeira TSP, Barros TG, et al. Isomannide derivatives as new class of inhibitors for human kallikrein 7. Bioorg Med Chem Lett. 2012;22:6072–6075.
- Tiryakioğlu NO, Önal Z, Saygili SK, et al. Treatment of ichthyosis and hypernatremia in a patient with Netherton syndrome with a SPINK5 c.153delT mutation using kallikrein inhibiting ointment. Int J Dermatol. 2017;56:106–108.
- Elias PM. The how, why and clinical importance of stratum corneum acidification. Exp Dermatol. 2017;26:999–1003.
- Lau JL, Dunn MK. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg Med Chem. 2018;26:2700–2707.
- Lemmens-Gruber R, Kamyar MR, Dornetshuber R. Cyclodepsipeptides - potential drugs and lead compounds in the drug development process. Curr Med Chem. 2009;16:1122–1137.
- Wang X, Gong X, Li P, et al. Structural diversity and biological activities of cyclic depsipeptides from fungi. Molecules. 2018;23(1):169.
- Krastel P, Liechty B-M, SCHMITT E, et al. Krastel P, Liechty BM, Schmitt E, Schreiner EP, inventors; Novartis AG, assignee. Use of Cyclic Depsipeptides to Inhibit Kallikrein 7. World patent WO/2009/024528. 2009 Feb 26.
- LifeMax [Internet]. Redwood City CA: LM-030 for treatment of Netherton syndrome ; 2020c [cited 2020 Jun 29]. https://www.lifemaxhealthcare.com/lm-030.2020
- Luckett S, Garcia RS, Barker JJ, et al. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J Mol Biol. 1999;290:525–533.
- de Veer SJ, Furio L, Swedberg JE, et al. Selective substrates and inhibitors for kallikrein-related peptidase 7 (KLK7) shed light on KLK Proteolytic activity in the stratum corneum. J Invest Dermatol. 2017;137:430–439. DOI:10.1016/j.jid.2016.09.017
- de Veer SJ, Ukolova SS, Munro CA, et al. Mechanism-based selection of a potent kallikrein-related peptidase 7 inhibitor from a versatile library based on the sunflower trypsin inhibitor SFTI-1. Biopolymers. 2013;100:510–518.
- de Veer SJ, Swedberg JE, Brattsand M, et al. Exploring the active site binding specificity of kallikrein-related peptidase 5 (KLK5) guides the design of new peptide substrates and inhibitors. Biol Chem. 2016;397:1237–1249.
- de Veer SJ, Wang CK, Harris JM, et al. Improving the selectivity of engineered protease inhibitors: optimizing the P2 prime residue using a versatile cyclic peptide library. J Med Chem. 2015;58:8257–8268.
- Kale SS, Bergeron-Brlek M, Wu Y, et al. Thiol-to-amine cyclization reaction enables screening of large libraries of macrocyclic compounds and the generation of sub-kilodalton ligands. Sci Adv. 2019;5:1–10.
- Chen X, Leahy D, van Haeften J, et al. A versatile and robust serine protease inhibitor scaffold from Actinia tenebrosa. Mar Drugs. 2019;17(12):701.
- Vasileiou Z, Barlos KK, Gatos D, et al. Synthesis of the proteinase inhibitor LEKTI domain 6 by the fragment condensation method and regioselective disulfide bond formation. Biopolymers. 2010;94:339–349.
- Cloutier SM, Kündig C, Felber LM, et al. Development of recombinant inhibitors specific to human kallikrein 2 using phage-display selected substrates. Eur J Biochem. 2004;271:607–613.
- Deperthes D, Kundig C, Hovnanian A, et al. Deperthes D, Kundig C, Hovnanian A, Deraison C, inventors; Dermadis SA, INSERM, assignees. Use of serine protease inhibitors in the treatment of skin disease. World patent WO/2009/093119. 2009 Jul 30.
- Mazereeuw-Hautier J, Cope J, Ong C, et al. Topical recombinant alpha1-antitrypsin: a potential treatment for Netherton syndrome? Arch Dermatol. 2006;142:396–398.
- Lu RM, Hwang YC, Liu IJ, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1–30.
- Sexton DJ, Chen T, Martik D, et al. Specific inhibition of tissue kallikrein 1 with a human monoclonal antibody reveals a potential role in airway diseases. Biochem J. 2009;422:383–392.
- Scarisbrick IA, Yoon H, Panos M, et al. Kallikrein 6 regulates early CNS demyelination in a viral model of multiple sclerosis. Brain Pathol. 2012;22:709–722.
- Jones RGA, Martino A. Targeted localized use of therapeutic antibodies: a review of non-systemic, topical and oral applications. Crit Rev Biotechnol. 2016;36:506–520.
- Tsianakas A, Brunner PM, Ghoreschi K, et al. The single-chain anti-TNF-α antibody DLX105 induces clinical and biomarker responses upon local administration in patients with chronic plaque-type psoriasis. Exp Dermatol. 2016;25:428–433.
- Barde C, Laffitte E, Campanelli A, et al. Intralesional infliximab in noninfectious cutaneous granulomas: three cases of necrobiosis lipoidica. Dermatology. 2011;222:213–216.
- Streit M, Beleznay Z, Braathen LR. Topical application of the tumour necrosis factor-α antibody infliximab improves healing of chronic wounds. Int Wound J. 2006;3:171–179.
- Teich N, Klugmann T. Rapid improvement of refractory pyoderma gangrenosum with infliximab gel in a patient with ulcerative colitis. J Crohn’s Colitis. 2014;8:85–86.
- Burek-Kozlowska A, Morell A, Hunziker T. Topical immunoglobulin G in atopic dermatitis. Int Arch Allergy Immunol. 1994;104:104–106.
- Sotiropoulou G, Pampalakis G. Kallikrein-related peptidases: bridges between immune functions and extracellular matrix degradation. Biol Chem. 2010;391:321–331.
- Carrier Y, Ma HL, Ramon HE, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131:2428–2437.
- Gruver AL, Sempowski GD. Cytokines, leptin, and stress-induced thymic atrophy. J Leukoc Biol. 2008;84:915–923.
- Kondo S, Sauder DN. Tumor necrosis factor (TNF) receptor type 1 (p55) is a main mediator for TNF-alpha-induced skin inflammation. Eur J Immunol. 1997;27:1713–1718.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79–85.
- Taniguchi K, Yamamoto S, Hitomi E, et al. Interleukin 33 is induced by tumor necrosis factor alpha and interferon gamma in keratinocytes and contributes to allergic contact dermatitis. J Investig Allergol Clin Immunol. 2013;23:428–434.
- Harden JL, Krueger JG, Bowcock AM. The immunogenetics of Psoriasis: a comprehensive review. J Autoimmun. 2015;64:66–73.
- Ahmad S, Azid NA, Boer JC, et al. The key role of TNF-TNFR2 interactions in the modulation of allergic inflammation: a review. Front Immunol. 2018;9:2572.
- Monaco C, Nanchahal J, Taylor P, et al. Anti-TNF therapy: past, present and future. Int Immunol. 2015;27:55–62.
- Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12:49–62.
- Fontao L, Laffitte E, Briot A, et al. Infliximab infusions for netherton syndrome: sustained clinical improvement correlates with a reduction of thymic stromal lymphopoietin levels in the skin. J Invest Dermatol. 2011;131:1947–1950. DOI:10.1038/jid.2011.124
- Roda Â, Mendonça-Sanches M, Travassos AR, et al. Infliximab therapy for Netherton syndrome: a case report. JAAD Case Rep. 2017;3:550–552.
- Hernández MV, Meineri M, Sanmartí R. Skin lesions and treatment with tumor necrosis factor alpha antagonists. Reumatol Clínica 2013;9:53–61. (English Ed). doi:10.1016/j.reuma.2012.04.007
- Nakamura M, Lee K, Singh R, et al. Eczema as an adverse effect of anti-TNFα therapy in psoriasis and other Th1-mediated diseases: a review. J Dermatolog Treat. 2017;28:237–241.
- Billi AC, Gudjonsson JE. Interleukin-17 receptor D: an orphan receptor finds a home in the skin. Sci Immunol. 2019;4(36):eaax0687.
- Gaffen SL, Jain R, Garg AV, et al. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600.
- Li L, Huang L, Vergis AL, et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;120:331–342.
- Lin AM, Rubin CJ, Khandpur R, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011;187:490–500.
- Paller AS. Pathogenesis-Based therapy with repurposed biologics for monogenic inflammatory skin disorders. JAMA Dermatol. 2020. DOI:10.1001/jamadermatol.2020.1018
- Luchsinger I, Knöpfel N, Theiler M, et al. Secukinumab therapy for Netherton Syndrome. JAMA Dermatol. 2020;156:907–911. DOI:10.1001/jamadermatol.2020.1019
- Blanchard SK, Prose NS. Successful use of secukinumab in Netherton syndrome. JAAD Case Rep. 2020;6:577–578.
- Barbieux C, Bonnet Des Claustres M, de la Brassinne M, et al. Duality of Netherton syndrome manifestations and response to ixekizumab. S0190–9622(20)32224–6. J Am Acad Dermatol. 2020; DOI:10.1016/j.jaad.2020.07.054.
- Tait Wojno ED, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: room for discovery. Immunity. 2019;50:851–870.
- Swindell WR, Beamer MA, Sarkar MK, et al. RNA-seq analysis of IL-1B and IL-36 responses in epidermal keratinocytes identifies a shared MyD88-dependent gene signature. Front Immunol. 2018;9:1–20.
- Yawalkar N, Egli F, Brand CU, et al. Antigen-presenting cells and keratinocytes express interleukin-12 in allergic contact dermatitis. Contact Dermatitis. 2000;42:18–22.
- Yoon J, Leyva Castillo JM, Wang G, et al. IL-23 induced in keratinocytes by endogenous TLR4 ligands polarizes dendritic cells to drive IL-22 responses to skin immunization. J Exp Med. 2016;213:2147–2166.
- Kulig P, Musiol S, Freiberger SN, et al. IL-12 protects from psoriasiform skin inflammation. Nat Commun. 2016;7:13466.
- Teng MWL, Bowman EP, McElwee JJ, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015;21:719–729.
- Volc S, Maier L, Gritsch A, et al. Successful treatment of Netherton syndrome with ustekinumab in a 15-year-old girl. Br J Dermatol. 2020;183:165–167.
- Ngiow SF, Teng MWL, Smyth MJ. A balance of interleukin-12 and −23 in cancer. Trends Immunol. 2013;34:548–555.
- Young L, Czarnecki D. The rapid onset of multiple squamous cell carcinomas in two patients commenced on ustekinumab as treatment of psoriasis. Australas J Dermatol. 2012;53:57–60.
- Levin AA, Gottlieb AB. Specific targeting of interleukin-23p19 as effective treatment for psoriasis. J Am Acad Dermatol. 2014;70:555–561.
- Sofen H, Smith S, Matheson RT, et al. Guselkumab (an IL-23-specific mAb) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J Allergy Clin Immunol. 2014;133:1032–1040.
- Machado Á, Torres T. Spotlight on risankizumab and its potential in the treatment of plaque psoriasis: evidence to date. Psoriasis. 2018;8:83–92.
- Galluzzo M, D’adamio S, Bianchi L, et al. Tildrakizumab for treating psoriasis. Expert Opin Biol Ther. 2017;17:645–657.
- Junttila IS. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. 2018;9:888.
- Bieber T. Interleukin-13: targeting an underestimated cytokine in atopic dermatitis. Allergy Eur J Allergy Clin Immunol. 2020;75:54–62.
- Sastre J, Dávila I. Dupilumab: a new paradigm for the treatment of allergic diseases. J Investig Allergol Clin Immunol. 2018;28:139–150.
- FDA. US Food & Drug Administration [Internet]. Silver Spring, MD: FDA approves first treatment for chronic rhinosinusitis with nasal polyps; 2019[ updated 2019 Jun 26; cited 2020 Aug 15]. https://www.fda.gov/news-events/press-announcements/fda-appro
- Regeneron [Internet]. TARRYTOWN, N.Y. and PARIS: dupixent® (dupilumab) Approved for severe asthma by european commission. 2019 [ updated 2019 May 7; cited 2020 Aug 15]. https://investor.regeneron.com/index.php/node/22091/pdf.
- Steuer AB, Cohen DE. Treatment of Netherton syndrome with dupilumab. JAMA Dermatol. 2020. DOI:10.1001/jamadermatol.2019.4608
- Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol. 2003;3:721–732.
- Gomez G. Current strategies to inhibit high affinity FcεRI-mediated signaling for the treatment of allergic disease. Front Immunol. 2011;187:1–8.
- Genentech [Internet]. San Francisco, CA: FDA Approves Genentech’s Xolair® (omalizumab) for Allergic Asthma in Children. [ updated 2016 Jul 7; cited 2020 Aug 16]. https://www.gene.com/media/press-releases/14632/2016-07-07/fda-approves-genent
- Genentech [Internet]. San Francisco, CA: FDA Approves XOLAIR® (omalizumab) for subcutaneous use for people with chronic idiopathic urticarial (CIU), a form of chronic hives. [ updated 2014 Mar 21; cited 2020 Aug 16]. https://www.gene.com/me
- Gasser P, Eggel A. Targeting IgE in allergic disease. Curr Opin Immunol. 2018;54:86–92.
- Yalcin AD. A case of netherton syndrome: successful treatment with omalizumab and pulse prednisolone and its effects on cytokines and immunoglobulin levels. Immunopharmacol Immunotoxicol. 2016;38:162–166.
- Stryk S, Siegfried EC, Knutsen AP. Selective antibody deficiency to bacterial polysaccharide antigens in patients with Netherton syndrome. Pediatr Dermatol. 1999;16:19–22.
- Wasserman RL. Personalized Therapy: immunoglobulin replacement for antibody deficiency. Immunol Allergy Clin North Am. 2019;39:95–111.
- Small AM, Cordoro KM. Netherton syndrome mimicking pustular psoriasis: clinical implications and response to intravenous immunoglobulin. Pediatr Dermatol. 2016;33:e222–3.
- Zelieskova M, Banovcin P, Kozar M, et al. A novel SPINK5 mutation and successful subcutaneous immunoglobulin replacement therapy in a child with Netherton syndrome. Pediatr Dermatol. 2020;n/a:1–3.
- Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. 2018;9:1682.
- Nies JF, Panzer U. IL-17C/IL-17RE: emergence of a Unique Axis in TH17 Biology. Front Immunol. 2020;11:1–12.
- Foulkes AC, Warren RB. Brodalumab in psoriasis: evidence to date and clinical potential. Drugs Context. 2019;8:1–11.
- Sawyer L, Fotheringham I, Wright E, et al. The comparative efficacy of brodalumab in patients with moderate-to-severe psoriasis : a systematic literature review and network meta- analysis. J Dermatolog Treat. 2018;29:557–568.
- Guttman-Yassky E, Krueger JG. IL-17C: a unique epithelial cytokine with potential for targeting across the spectrum of atopic dermatitis and psoriasis. J Invest Dermatol. 2018;138:1467–1469.
- Johnston A, Fritz Y, Dawes SM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. 2013;190:2252–2262.
- Vandeghinste N, Klattig J, Jagerschmidt C, et al. Neutralization of IL-17C reduces skin inflammation in mouse models of psoriasis and atopic dermatitis. J Invest Dermatol. 2018;138:1555–1563.
- MorphoSys [Internet]. Planegg/Munich, Germany, and Mechelen, Belgium: OR106 Clinical Development in Atopic Dermatitis Stopped. [ updated 2019 Oct 28; cited 2020 Aug 16]. https://www.morphosys.com/media-investors/media-center/morphosys-ag-mo
- Guttman-Yassky E, Brunner PM, Neumann AU, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, phase 2a trial. J Am Acad Dermatol. 2018;78:872–881.
- Tsai YC, Tsai TF. Anti-interleukin and interleukin therapies for psoriasis: current evidence and clinical usefulness. Ther Adv Musculoskelet Dis. 2017;9:277–294.
- Gudjonsson JE, Johnston A, Ellis CN. Novel systemic drugs under investigation for the treatment of psoriasis. J Am Acad Dermatol. 2012;67:139–147.
- Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281:8–27.
- Werman A, Werman-Venkert R, White R, et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci U S A. 2004;101:2434–2439.
- Maier JA, Statuto M, Ragnotti G. Endogenous interleukin 1 alpha must be transported to the nucleus to exert its activity in human endothelial cells. Mol Cell Biol. 1994;14:1845–1851.
- Afonina IS, Müller C, Martin SJ, et al. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015;42:991–1004.
- Yao C, Karabasil MR, Purwanti N, et al. Tissue kallikrein mK13 is a candidate processing enzyme for the precursor of interleukin-1beta in the submandibular gland of mice. J Biol Chem. 2006;281:7968–7976.
- LaRock CN, Todd J, LaRock DL, et al. IL-1β is an innate immune sensor of microbial proteolysis. Sci Immunol. 2016;1(2):eaah3539.
- Swindell WR, Beamer MA, Sarkar MK, et al. RNA-Seq analysis of IL-1B and IL-36 responses in epidermal keratinocytes identifies a shared MyD88-dependent gene signature. Front Immunol. 2018;9:80.
- Mantovani A, Dinarello CA, Molgora M, et al. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity. 2019;50:778–795.
- Barker JN, Mitra RS, Griffiths CE, et al. Keratinocytes as initiators of inflammation. Lancet. (London, England) 1991;337: 211–214.doi: 10.1016/0140-6736(91)92168-2
- Kupper TS, Ballard DW, Chua AO, et al. Human keratinocytes contain mrna indistinguishable from monocyte interleukin lα and β mrna: keratinocyte epidermal cell-derived thymocyte-activating factor is identical to interleukin 1. J Exp Med. 1986;164:2095–2100.
- Wood LC, Jackson SM, Elias PM, et al. Cutaneous barrier perturbation stimulates cytokine production in the epidermis of mice. J Clin Invest. 1992;90:482–487.
- Klicznik MM, Szenes-Nagy AB, Campbell DJ, et al. Taking the lead – how keratinocytes orchestrate skin T cell immunity. Immunol Lett. 2018;200:43–51.
- Noske K. Secreted immunoregulatory proteins in the skin. J Dermatol Sci. 2018;89:3–10.
- Dinarello CA, Simon A, Van Der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652.
- Dinarello CA, van der Meer JWM. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25:469–484.
- Li J, Leyva-Castillo JM, Hener P, et al. Counterregulation between thymic stromal lymphopoietin– and IL-23–driven immune axes shapes skin inflammation in mice with epidermal barrier defects. J Allergy Clin Immunol. 2016;138:150–161.
- Leslie KS, Tripathi SV, Nguyen TV, et al. An open-label study of anakinra for the treatment of moderate to severe hidradenitis suppurativa. J Am Acad Dermatol. 2014;70:243–251.
- Tauber M, Viguier M, Alimova E, et al. Partial clinical response to anakinra in severe palmoplantar pustular psoriasis. Br J Dermatol. 2014;171:646–649.
- Viguier M, Guigue P, Pagès C, et al. Successful treatment of generalized pustular psoriasis with the interleukin-1-receptor antagonist Anakinra: lack of correlation with IL1RN mutations. Ann Int Med. United States. 2010;66–67. DOI:10.7326/0003-4819-153-1-201007060-00030
- Hüffmeier U, Wätzold M, Mohr J, et al. Successful therapy with anakinra in a patient with generalized pustular psoriasis carrying IL36RN mutations. Br J Dermatol. England. 2014;202–204. DOI:10.1111/bjd.12548
- Rossi-Semerano L, Piram M, Chiaverini C, et al. First clinical description of an infant with interleukin-36-receptor antagonist deficiency successfully treated with anakinra. Pediatrics. 2013;132(4):e1040-7.
- Ultsch M, Bevers J, Nakamura G, et al. Structural basis of signaling blockade by anti-IL-13 antibody lebrikizumab. J Mol Biol. 2013;425:1330–1339.
- Hanania NA, Korenblat P, Chapman KR, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir Med. 2016;4:781–796.
- Guttman-Yassky E, Blauvelt A, Eichenfield LF, et al. Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 Inhibitor, in adults with moderate to severe atopic dermatitis: a phase 2b randomized clinical trial. JAMA Dermatol. 2020;156:411–420.
- Popovic B, Breed J, Rees DG, et al. Structural characterisation reveals mechanism of IL-13-neutralising monoclonal antibody tralokinumab as inhibition of binding to IL-13R α 1 and IL-13R α 2. J Mol Biol. 2017;429:208–219.
- Brightling CE, Chanez P, Leigh R, et al. Efficacy and safety of tralokinumab in patients with severe uncontrolled asthma: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3:692–701.
- Wollenberg A, Howell MD, Guttman-Yassky E, et al. Treatment of atopic dermatitis with tralokinumab, an anti–IL-13 mAb. J Allergy Clin Immunol. 2019;143:135–141.
- LeoPharma [Internet]. Ballerup, Danemark: LEO Pharma announces positive top-line results for tralokinumab from three Phase 3 studies in adult patients with moderate-to-severe AD. [ updated 2019 Dec 11; cited 2020 Aug 18]. https://leo-pharma
- Boguniewicz M, Alexis AF, Beck LA, et al. Expert perspectives on management of moderate-to-severe atopic dermatitis: a multidisciplinary consensus addressing current and emerging therapies. J Allergy Clin Immunol Pract. 2017;5:1519–1531.
- Nygaard U, Vestergaard C, Deleuran M. Emerging treatment options in atopic dermatitis: systemic therapies. Dermatology. 2018;233:344–357.
- Shrimanker R, Borg K, Connolly C, et al. Late Breaking Abstract - Effect of timapiprant, a DP2 antagonist, on airway inflammation in severe eosinophilic asthma. Eur Respir J. 2019;54:RCT3784.
- Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16:676–689.
- Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. 2015;43:29–40.
- Konishi T, Tsuda T, Sakaguchi Y, et al. Upregulation of interleukin-33 in the epidermis of two Japanese patients with Netherton syndrome. J Dermatol. 2014;41:258–261.
- AnaptysBio[Internet]. San Diego. CA: Etokimab; c2020 . [cited 2020 Jul 02]. https://www.anaptysbio.com/pipeline/etokimab/
- Furue M, Yamamura K, Kido-Nakahara M, et al. Emerging role of interleukin-31 and interleukin-31 receptor in pruritus in atopic dermatitis. Allergy Eur J Allergy Clin Immunol. 2018;73:29–36.
- Dillon SR, Sprecher C, Hammond A, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–760.
- Ruzicka T, Hanifin JM, Furue M, et al. Anti–interleukin-31 receptor a antibody for atopic dermatitis. N Engl J Med. 2017;376:826–835.
- Silverberg JI, Pinter A, Pulka G, et al. Phase 2B randomized study of nemolizumab in adults with moderate-to-severe atopic dermatitis and severe pruritus. J Allergy Clin Immunol. 2020;145:173–182.
- Kabashima K, Furue M, Hanifin JM, et al. Nemolizumab in patients with moderate-to-severe atopic dermatitis: randomized, phase II, long-term extension study. J Allergy Clin Immunol. 2018;142:1121–1130.
- Németh T, Sperandio M, Mócsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. 2020;19:253–275.
- Stockley R, De Soyza A, Gunawardena K, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med. 2013;107:524–533.
- Mereo BioPharma [Internet]. London, UK: MPH-966 (ALVELESTAT) c2020. [cited 2020 Aug 08]. https://www.mereobiopharma.com/pipeline/mph-966-alvelestat/
- Sullivan GP, Henry CM, Clancy DM, et al. Suppressing IL-36-driven inflammation using peptide pseudosubstrates for neutrophil proteases. Cell Death Dis. 2018;9(3):378.
- Zani ML, Baranger K, Guyot N, et al. Protease inhibitors derived from elafin and SLPI and engineered to have enhanced specificity towards neutrophil serine proteases. Protein Sci. 2009;18:579–594.
- Oikonomopoulou K, Diamandis EP, Hollenberg MD, et al. Proteinases and their receptors in inflammatory arthritis: an overview. Nat Rev Rheumatol. 2018;14:170–180.
- Rattenholl A, Steinhoff M. Role of proteinase-activated receptors in cutaneous biology and disease. Drug Dev Res. 2003;59:408–416.
- Shpacovitch V, Feld M, Bunnett NW, et al. Protease-activated receptors: novel PARtners in innate immunity. Trends Immunol. 2007;28:541–550.
- Nystedt S, Emilsson K, Wahlestedt C, et al. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci. 1994;91(9208):LP– 9212. DOI:10.1073/pnas.91.20.9208
- Molino M, Barnathan ES, Numerof R, et al. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049.
- Ramachandran R, Mihara K, Chung H, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR 2). J Biol Chem. 2011;286:24638–24648.
- Oikonomopoulou K, Hansen KK, Saifeddine M, et al. Proteinase-activated receptors, targets for kallikrein signaling. J Biol Chem. 2006;281:32095–32112.
- Adams MN, Ramachandran R, Yau MK, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130:248–282.
- Henehan M, De Benedetto A. Update on protease-activated receptor 2 in cutaneous barrier, differentiation, tumorigenesis and pigmentation, and its role in related dermatologic diseases. Exp Dermatol. 2019;28:877–885.
- Wakita H, Furukawa F, Takigawa M. Thrombin and trypsin induce granulocyte-macrophage colony-stimulating factor and interleukin-6 gene expression in cultured normal human keratinocytes. Proc Assoc Am Physicians. 1997;109:190–207.
- Hou L, Kapas S, Cruchley AT, et al. Immunolocalization of protease-activated receptor-2 in skin: receptor activation stimulates interleukin-8 secretion by keratinocytes in vitro. Immunology. 1998;94:356–362.
- Iwakiri K, Ghazizadeh M, Jin E, et al. Human airway trypsin-like protease induces PAR-2-mediated IL-8 release in psoriasis vulgaris. J Invest Dermatol. 2004;122:937–944.
- Buddenkotte J, Stroh C, Engels IH, et al. Agonists of proteinase-activated receptor-2 stimulate upregulation of intercellular cell adhesion molecule-1 in primary human keratinocytes via activation of NF-kappa B. J Invest Dermatol. 2005;124:38–45.
- Aoki M, Yamaguchi R, Yamamoto T, et al. Granulocyte-macrophage colony-stimulating factor primes interleukin-13 production by macrophages via protease-activated receptor-2. Blood Cells Mol Dis. 2015;54:353–359.
- Yamaguchi R, Yamamoto T, Sakamoto A, et al. Mechanism of interleukin-13 production by granulocyte-macrophage colony-stimulating factor-dependent macrophages via protease-activated receptor-2. Blood Cells Mol Dis. 2015;55:21–26.
- Zhao J, Munanairi A, Liu X, et al. PAR2 mediates itch via TRPV3 signaling in keratinocytes. J Invest Dermatol. 2020;2:1–9.
- Seiberg M, Paine C, Sharlow E, et al. The protease-activated receptor 2 regulates pigmentation via keratinocyte-melanocyte interactions. Exp Cell Res. 2000;254:25–32.
- Ramachandran R, Noorbakhsh F, Defea K, et al. Targeting proteinase-activated receptors: therapeutic potential and challenges. Nat Rev Drug Discov. 2012;11:69–86.
- Zhu Y, Underwood J, Macmillan D, et al. Persistent kallikrein 5 activation induces atopic dermatitis-like skin architecture independent of PAR2 activity. J Allergy Clin Immunol. 2017;140:1310–1322.
- Böhm SK, Khitin LM, Grady EF, et al. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem. 1996;271:22003–22016.
- Frateschi S, Camerer E, Crisante G, et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat Commun. 2011;2:161.
- Barr TP, Garzia C, Guha S, et al. PAR2 pepducin-based suppression of inflammation and itch in atopic dermatitis models. J Invest Dermatol. 2019;139:412–421.
- Comeau MR, Ziegler SF. The influence of TSLP on the allergic response. Mucosal Immunol. 2010;3:138–147.
- Ziegler SF, Artis D. review Sensing the outside world : TSLP regulates barrier immunity. Nat Publ Gr. 2010;11:289–293.
- Simpson EL, Parnes JR, She D, et al. Tezepelumab, an anti-thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J Am Acad Dermatol. 2019;80:1013–1021.
- Marone G, Spadaro G, Braile M, et al. Tezepelumab: a novel biological therapy for the treatment of severe uncontrolled asthma. Expert Opin Investig Drugs. 2019;28:931–940.
- Gauvreau GM, O’Byrne PM, Boulet L-P, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. 2014;370:2102–2110.
- Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377:936–946.
- Venkataramani S, Low S, Weigle B, et al. Design and characterization of Zweimab and Doppelmab, high affinity dual antagonistic anti-TSLP/IL13 bispecific antibodies. Biochem Biophys Res Commun. 2018;504:19–24.
- Ito T, Wang Y-H, Duramad O, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202:1213–1223.
- Webb GJ, Hirschfield GM, Lane PJL. OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol. 2016;50:312–332.
- Ziegler SF, Liu Y-J. Thymic stromal lymphopoietin in normal and pathogenic T cell development and function. Nat Immunol. 2006;7:709–714.
- Guttman-Yassky E, Pavel AB, Zhou L, et al. GBR 830, an anti-OX40, improves skin gene signatures and clinical scores in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;144:482–493.
- Kvist-hansen A, Hansen PR, Skov L. Systemic Treatment of Psoriasis with JAK Inhibitors . Dermatol Ther (Heidelb). 2020;10:29–42.
- Damsky W, King BA. The promise of a new drug class. J Am Dermatol. 2017;76: 736–744. DOI:10.1016/j.jaad.2016.12.005
- Ciechanowicz P, Rakowska A, Sikora M, et al. JAK-inhibitors in dermatology: current evidence and future applications. J Dermatolog Treat. 2019;30:648–658.
- Khavari PA, Rollman O, Vahlquist A. Cutaneous gene transfer for skin and systemic diseases. J Intern Med. 2002;252:1–10.
- Roedl D, Oji V, Buters JTM, et al. rAAV2-mediated restoration of LEKTI in LEKTI-deficient cells from Netherton patients. J Dermatol Sci. 2011;61:194–198.
- Di W-L, Mellerio JE, Bernadis C, et al. Phase I study protocol for ex vivo lentiviral gene therapy for the inherited skin disease, Netherton syndrome. Hum Gene Ther Clin Dev. 2013;24:182–190.
- Di WL, Lwin SM, Petrova A, et al. Generation and clinical application of gene-modified autologous epidermal sheets in netherton syndrome: lessons learned from a phase 1 trial. Hum Gene Ther. 2019;30:1067–1078. DOI:10.1089/hum.2019.049
- Krystal Biotech [Internet]. Pittsburgh, PA: our targets; c2020. 2020 [cited 2020 Aug 08]. https://www.krystalbio.com/patients-and-families/our-targets/
- Agarwal P, Krishnan S, Freedman JC, inventors; Krystal Biotech Inc, assignee. Compositions and methods for the treatment of Netherton syndrome. US patent US 2020/0093874A1. 2020 Mar 26 .
- Geusens B, Sanders N, Prow T, et al. Cutaneous short-interfering RNA therapy. Expert Opin Drug Deliv. 2009;6:1333–1349.
- Leachman SA, Hickerson RP, Schwartz ME, et al. First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther. 2010;18:442–446.
- Zakrewsky M, Kumar S, Mitragotri S. Nucleic acid delivery into skin for the treatment of skin disease: proofs-of-concept, potential impact, and remaining challenges. J Control Release. 2015;219:445–456.
- Kigasawa K, Kajimoto K, Hama S, et al. Noninvasive delivery of siRNA into the epidermis by iontophoresis using an atopic dermatitis-like model rat. Int J Pharm. 2010;383:157–160.
- Hashim IIA, Motoyama K, Abd-ElGawad A-EH, et al. Potential use of iontophoresis for transdermal delivery of NF-κB decoy oligonucleotides. Int J Pharm. 2010;393:128–135.
- Desai PR, Marepally S, Patel AR, et al. Topical delivery of anti-TNFα siRNA and capsaicin via novel lipid-polymer hybrid nanoparticles efficiently inhibits skin inflammation in vivo. J Control Release. 2013;170:51–63.
- Bracke S, Carretero M, Guerrero-Aspizua S, et al. Targeted silencing of DEFB4 in a bioengineered skin-humanized mouse model for psoriasis: development of siRNA SECosome-based novel therapies. Exp Dermatol. 2014;23:199–201.
- Kim ST, Lee K-M, Park H-J, et al. Topical delivery of interleukin-13 antisense oligonucleotides with cationic elastic liposome for the treatment of atopic dermatitis. J Gene Med. 2009;11:26–37.
- Rosi NL, Giljohann DA, Thaxton CS, et al. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science. 2006;312(1027):LP– 1030. DOI:10.1126/science.1125559
- Giljohann DA, Seferos DS, Prigodich AE, et al. Gene regulation with polyvalent siRNA-nanoparticle conjugates. J Am Chem Soc. 2009;131:2072–2073.
- Cutler JI, Auyeung E, Mirkin CA. Spherical nucleic acids. J Am Chem Soc. 2012;134:1376–1391.
- Zheng D, Giljohann DA, Chen DL, et al. Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation. Proc Natl Acad Sci U S A. 2012;109:11975–11980.
- Lewandowski KT, Thiede R, Guido N, et al. Topically delivered tumor necrosis factor-α–targeted gene regulation for psoriasis. J Invest Dermatol. 2017;137:2027–2030.
- Dermelix Biotherapeutics. [Internet]. King of Prussia, PA: pipeline ; c2020. [cited 2020 Aug 04]. http://dermelix.com/pipeline/
- Zhvania P, Hoyle NS, Nadareishvili L, et al. Phage therapy in a 16-year-old boy with netherton syndrome. Front Med. 2017;4:1–5.
- Motta JP, Bermúdez-Humarán LG, Deraison C, et al. Food-grade bacteria expressing elafin protect against inflammation and restore colon homeostasis. Sci Transl Med. 2012;4(158):158ra144.
- Bermúdez-Humarán LG, Motta JP, Aubry C, et al. Serine protease inhibitors protect better than IL-10 and TGF-β anti-inflammatory cytokines against mouse colitis when delivered by recombinant lactococci. Microb Cell Fact. 2015;14:1–11.
- Azitra Inc [Internet]. Farmington, CT: our pipeline ; c2020. [cited 2020 Jul 07]. https://azitrainc.com/pipeline/