
ABSTRACT
Introduction: Choroideremia is an X-linked inherited retinal degeneration resulting from mutations in the CHM gene, encoding Rab escort protein-1 (REP1), a protein regulating intracellular vesicular transport. Loss-of-function mutations in CHM lead to progressive loss of retinal pigment epithelium (RPE) with photoreceptor and choriocapillaris degeneration, leading to progressive visual field constriction and loss of visual acuity. Three hundred and fifty-four unique mutations have been reported in CHM. While gene augmentation remains an ideal therapeutic option for choroideremia, other potential future clinical strategies may exist.
Areas covered: The authors examine the pathophysiology and genetic basis of choroideremia. They summarize the status of ongoing gene therapy trials and discuss CHM mutations amenable to other therapeutic approaches including CRISPR/Cas-based DNA and RNA editing, nonsense suppression of premature termination codons, and antisense oligonucleosides for splice modification. The authors undertook a literature search in PubMed and NIH Clinical Trials in October 2020.
Expert opinion: The authors conclude that AAV-mediated gene augmentation remains the most effective approach for choroideremia. Given the heterogeneity of CHM mutations and potential risks and benefits, genome-editing approaches currently do not offer significant advantages. Nonsense suppression strategies and antisense oligonucleotides are exciting novel therapeutic options; however, their clinical viability remains to be determined.
1. Introduction
1.1. Choroideremia: pathophysiology and clinical features
Choroideremia is an inherited retinal degeneration that affects approximately 1 in 50,000 people worldwide. It is caused by mutations in the CHM gene, located on Xq21.2 (OMIM *300390). CHM encodes Rab-escort protein 1 (REP1), a polypeptide involved in the prenylation pathway of Rabs, GTPase-associated proteins essential for normal intracellular vesicular transport. Binding of REP1 to its Rab facilitates the addition of a geranylgeranyl group to the Rab by Rab geranylgeranyltransferase (RabGGTase) in a prenylation reaction. REP1 is subsequently involved in delivery of the prenylated Rab to a specific membrane [Citation1]. Cellular models suggest that pathogenic mutations in REP1 result in aberrant binding to and thus under-prenylation of Rabs [Citation2,Citation3], an observation supported by animal models of choroideremia showing that Rab under-prenylation is associated with disordered phagocytic function of the retinal pigment epithelium (RPE), and subsequent RPE degeneration [Citation1,Citation4]. Whether retinal degeneration in choroideremia is primarily driven by RPE or photoreceptor loss is a source of some debate. Conditional knockout of REP1 in the RPE and photoreceptors in mice suggest that photoreceptor dysfunction and loss may occur independently of RPE dysfunction [Citation5], but RPE degeneration nonetheless accelerates photoreceptor loss [Citation6]. OCT studies in choroideremia patients indicate that photoreceptor loss generally follows RPE loss to give rise to stereotypical ‘islands’ of surviving retina, and choriocapillaris atrophy generally occurs later on in the disease process [Citation7]. Accordingly, structural changes typical of photoreceptor stress secondary to RPE disease have been observed in the form of outer retinal tubulations [Citation7,Citation8]. Adaptive optics imaging in choroideremia indicates that a degree of photoreceptor structural abnormality is present within the islands of surviving RPE [Citation9] and cone photoreceptors may be able to survive a short distance beyond the edge of RPE degeneration [Citation10]. However, it remains unclear to what extent these stressed or structurally abnormal photoreceptors are amenable to therapeutic rescue.
Although REP1 is ubiquitously expressed throughout the body, its deficiency through genetic mutations in CHM only manifests as disease in the eye. The lack of systemic effects is thought to be due to redundancy of Rab prenylation activity provided by REP2, a homologous protein encoded by the CHM-like (CHML) X-linked retrogene located on chromosome 1q42 [Citation11]. It remains unclear why this compensation mechanism is insufficient in the outer retina, leading to slow degeneration of the RPE and photoreceptors [Citation12]. It is established that certain Rabs (e.g. Rab27a) compete for REP1 in preference to REP2, and that a Rab ‘prenylation hierarchy’ exists [Citation13] where certain Rabs bind more efficiently to both REP1 and REP2, thus out-competing lower ranking Rabs which become underprenylated in the absence of REP1 [Citation2,Citation13,Citation14]. This, combined with the high phagocytic demands placed on RPE by continuous photoreceptor outer segment turnover, may lead to errors in vesicular trafficking and cumulative cellular toxicity [Citation15].
Choroideremia typically presents in males with nyctalopia during childhood, followed by progressive and disabling loss of peripheral vision beginning in the late teens. The area of surviving RPE generally follows a steady exponential decay with a half-life of around 5.5 years (95% CI: 5.0–6.1) [Citation12]. In end-stage disease, usually by the fifth to seventh decade of life, central vision becomes extinguished as the centripetal RPE degeneration encroaches on the fovea (). Female carriers of CHM mutations are usually asymptomatic although wide variation may be seen in retinal status and visual function [Citation8,Citation16]. This is likely the consequence of random inactivation of X chromosomes in females giving rise to mosaics of healthy and diseased RPE cells in the retina [Citation17]. As the majority of choroideremia patients harbor null mutations in CHM, there is limited evidence for phenotype-genotype correlation [Citation18]. However, rare cases of splicing defects have been reported in which residual CHM mRNA transcripts have been associated with unusually slow rates of disease progression [Citation19].
Figure 1. Representative left eye images of 32 year old male with CHM mutation. (a) Widefield optos color image showing baring of sclera and peripheral pigmentation. (b) Macular fundus autofluorescence showing the central area of relative RPE preservation. (c) OCT image showing loss of outer retina with central island of preservation. (d) Microperimetry data showing central preservation of retinal sensitivity with sharply demarcated sensitivity drop-off corresponding to island of surviving RPE
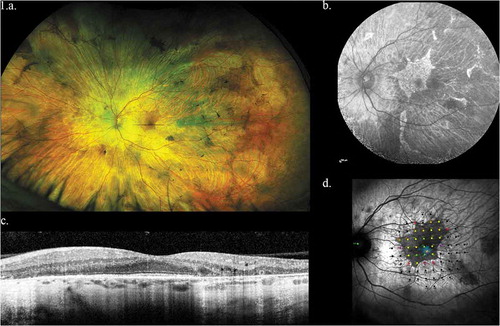
Choroideremia has several features that make it an attractive target for genetic therapies. The disease is monogenic with an easily recognizable phenotype and X-linked family history. The disease progression is relatively slow, thus providing a long therapeutic window for intervention. As a disease that affects the young, the potential benefits of treatment over a lifetime are substantial. As a disease that affects only the eye, a relatively immune privileged site, targeted local therapy would be feasible.
1.2. Potential therapeutic targets
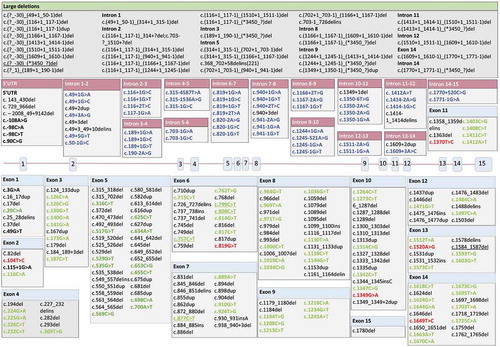
The CHM gene contains 15 exons (OMIM) and is 5,442 bp in length, with the last exon making up the majority of the transcript length, of which 3,450 bp encodes the 3ʹ-untranslated region (UTR) ()). It encodes REP1, a 653 amino acid (aa) protein. The Leiden Open Variation Database lists 628 CHM public variants, of which 545 are classed as pathological or likely pathological, and 354 are unique variants. Mutations have been reported within the promoter region [Citation20,Citation21], introns [Citation22,Citation23], and exons [Citation18]. The vast majority of the exonic variants are nonsense mutations which result in an absence of or truncation of the protein. Missense mutations associated with a clinical phenotype remain rare, but at least three have been identified. These have been associated with a reduction in REP1 expression, presumably due to destabilization of protein structure, or a functional reduction in prenylation activity [Citation24–26]. One theory advanced to explain this is that missense mutations in REP1 may be well tolerated, based on the fact that REP2 shares only around 75% amino acid identity with REP1 yet retains similar affinity for Rab binding [Citation1,Citation14]. It has been also been postulated that as a chaperone protein, REP1 folding may be influenced by its interaction with the Rab ligand rather than post-translational modification, thus rendering it more tolerant of single amino acid changes [Citation18].
Figure 2a. (a) Structure of the CHM gene annotated with pathogenic variants. Intronic mutations have been color coded with a pink header, exonic mutations with a blue header. Single-base substitutions are shown in bold: of these, single-base substitutions resulting in missense mutations are colored red; terminations are colored green, and splicing defects are colored purple. The five most common described variants are underlined (c.(?-30)_(*3450?)del (n = 20), c.757 C > T (n = 18), c.799 C > T (n = 16), c.1584_1587del (n = 11), 808 C > T and 877 C > T (n = 10 each)
Pathogenic CHM variants occur primarily in exonic DNA (66%), of which just over half are due to substitutions. Of these substitutions, the vast majority (over 90%) result in a premature termination codon (PTC). The next most common region for pathogenic mutations involve splicing/intronic regions (18%), followed by deletions (15%) which commonly result in no protein product being formed [Citation27] ()).
Figure 2b. (b) Classification of pathogenic variants: inner ring shows mutation location (exonic, intronic, multiple locations (comprising large or multiple indels or duplications); outer ring shows effect of mutation (premature termination codon, missense variant, no protein product formed, frameshift, unknown) Source: Leiden Open Variation Database (LOVD) [Citation27]
![Figure 2b. (b) Classification of pathogenic variants: inner ring shows mutation location (exonic, intronic, multiple locations (comprising large or multiple indels or duplications); outer ring shows effect of mutation (premature termination codon, missense variant, no protein product formed, frameshift, unknown) Source: Leiden Open Variation Database (LOVD) [Citation27]](/cms/asset/9f9457f3-67f8-422b-8e2e-0b5e42a5f46a/ieod_a_1882300_f0002b_oc.jpg)
The current arsenal of potential therapeutic options for choroideremia include gene augmentation, CRISPR/Cas-mediated base-editing, prime-editing, nonsense-suppression and antisense oligonucleotides. This review discusses the potential applications of each approach to choroideremia-associated targets, their impact, feasibility, and key advantages and disadvantages. The authors undertook a literature search in PubMed and NIH Clinical Trials between October 2020 and January 2021. Search terms used included choroideremia, CHM, REP1, AAV, AON, nonsense-suppression, CRISPR.
2. Gene augmentation therapy
Gene augmentation for choroideremia is an appealing therapeutic strategy. It is applicable to any CHM mutation, independent of its location. It is also applicable to large or multiple deletions, which would be less amenable to site-specific strategies such as base- or prime-editing. It is important to note that there has been no evidence for any toxicity or dominant negative effect for any of these abnormal protein products in female carriers: it is also clear from carriers that partial expression of wild-type REP1 (as may be achieved through gene augmentation) is generally sufficient to preserve visual function, although retinal morphology may be affected [Citation16,Citation28].
Another consideration is that there exists now considerable clinical experience with AAV-mediated transgene expression in a wide variety of inherited diseases. A search on ClinicalTrials.gov shows 234 registered trials involving AAV-delivered gene therapy, of which 47 are related to ophthalmic disease, ranging from Leber hereditary optic neuropathy to X-linked retinoschisis [Citation29]. In September 2019, NICE and the FDA approved voretigene neparvovec (Luxturna, Novartis), an AAV2-mediated gene augmentation therapy for Leber Congenital Amaurosis (LCA) secondary to biallelic mutations in RPE65. The length of the REP1 cDNA recommends it to this approach; at 1959 bp, it fits easily into an AAV vector with ample room for regulatory elements. Direct targeting of photoreceptors and RPE is possible by subretinal injection, and the surgical technique to achieve this with minimal retina trauma has been refined [Citation30]. Meanwhile, a phase 1 trial using an alternative approach of intravitreal injection of a modified AAV capsid (4D-110, Roche Pharma and 4DMT) is also currently recruiting participants [Citation31].
From earlier clinical trials, it is known that AAV2 effectively transduces and has high tropism for RPE and photoreceptors, and a number of ubiquitous and tissue-specific promoters are available to drive transgene expression in these cell types. A number of phase 1/2 clinical trials are in progress worldwide and reports are encouraging (). In November 2019, enrollment was completed for phase 3 clinical trials of timrepigene emparvovec (BIIB111, Biogen Inc, Cambridge, MA, USA), a multicentre randomized efficacy trial of high- and low-dose subretinal AAV2-REP1 taking place across the United States, Europe and South America, and preliminary data reporting is expected in 2021.
Table 1. Summary of trials involving REP1 replacement and their status
Although AAV-REP1 therapy shows extremely promising data, exploration of other therapeutic modalities remains worthwhile. Theoretically, integration of an AAV-delivered gene into the genome at an unspecified point could result in disruption of normal gene expression, although integration rates are low and evidence of integration events following AAV gene therapy is scant [Citation32,Citation33]. For two of the aforementioned three documented missense CHM mutations, REP1 transcript levels were unaffected and mutant proteins were detectable although diminished [Citation24,Citation25]. However, it should be noted that no dominant negative effect was observed in RPE cells carrying the p.Leu457Pro missense variant, which were amenable to rescue of prenylation activity by AAV-mediated gene augmentation [Citation24].
Alternative approaches being developed for choroideremia gene therapy include directed evolution of novel AAV serotypes delivered via an intravitreal approach that might have improved transduction compared to wild-type AAV2 vectors. This approach is being developed by Roche and 4D-MT (NCT04483440) [Citation31]. Compared with subretinal injection, intravitreal delivery of AAV would most likely require a dose of several log units higher with increased risk of inflammation, although administration would be easier. For choroideremia however, the RPE layer needs to be targeted. While structures in direct contact with the vitreous such as ganglion cells, Mueller cells and foveal photoreceptors in non-human primates (NHPs) may be successfully transduced by intravitreal vectors, no published data to date has shown efficient targeting of the RPE. In a preclinical study of intravitreal AAV evolved in nonhuman primates, data on two evolved vectors NHP#9 and NHP#26 were presented. The former was injected at 1.5 × 1012 viral genomes which is a log unit higher than the maximal AAV2 doses used in human clinical trials and failed to transduce RPE cells, despite good inner retinal expression. Similarly, no RPE cell transduction was observed with NHP#26 even when using immunohistochemistry to detect faint GFP expression [Citation46]. Since successful RPE transduction is almost certainly essential for treating choroideremia with gene replacement, intravitreal AAV is unlikely to deliver successful outcomes. Furthermore, intravitreal administration of AAV carries the potential development of immune and inflammatory responses which are detectable both locally and systemically, and could adversely affect visual outcomes [Citation47,Citation48]. Indeed, the NHP subjects in Byrne et al.’s paper were systemically immunosuppressed daily with subcutaneous ciclosporin [Citation46].
3. Genomic editing
3.1. Principles of CRISPR-Cas genome editing
The discovery of RNA-guided Cas9 endonucleases from prokaryotic Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) systems and their adaptation into powerful gene editing tools have revolutionized the field of molecular biology [Citation49]. CRISPR-Cas9 systems’ highly programmable and precise editing abilities have opened up a wide array of potential therapeutic genetic targets in human inherited diseases. Although the characterization of CRISPR biology and subsequent adaptation for gene editing has occurred only in this decade, the potential of CRISPR-based strategies for the treatment of inherited retinal diseases has already been rapidly and extensively realized. CRISPR/Cas9-based genome editing in mouse models has already been achieved in proof-of-principle studies as well as targeting of common mutations causing inherited retinal diseases [Citation50–54]. Successful pre-clinical genome editing of a deep-intronic mutation in CEP290, which causes LCA10, has led to the first FDA-approved CRISPR genome engineering clinical trial for an inherited retinal disease, and the first delivery into a patient was achieved in early 2020 [Citation52].
The CRISPR-Cas family originates from the bacterial innate immune system’s need to recognize and destroy intruder viral DNA and can be found in a wide variety of prokaryotic species. Family members differ in mechanisms of action, but all CRISPR-Cas systems require a target-specific CRISPR RNA (crRNA) to guide it to the target sequence, and (for type II systems) a trans-activating RNA (tracrRNA), which forms a scaffold structure to interact with the Cas protein. For genome editing applications, researchers have linked the naturally occurring crRNA and tracrRNA described above to form a single guide RNA (sgRNA). The Cas9:sgRNA complex scans DNA within the nucleus, searching first for the appropriate protospacer adjacent motif (PAM), a nucleotide sequence specific to each Cas family, which allows binding of the Cas protein. Subsequently, the sgRNA binds to a target sequence adjacent to the PAM, and the Cas endonuclease cleaves the nucleic acid to generate a double-strand break (DSB). In eukaryotes, Cas9-induced DSBs are repaired by DNA repair mechanisms [Citation55]. Commonly this occurs via error-prone non-homologous end joining (NHEJ), resulting in random insertions or deletions (indels) and disruption of the target gene [Citation56]. Alternatively, a repair template with homology to the target region can be delivered to stimulate error-free homology-directed repair (HDR), although at a lower efficiency than NHEJ-mediated repair. Nevertheless, the correction of a point-mutation by HDR has been shown to be highly inefficient, particularly in non-dividing cells, such as those present in the neuroretina. Furthermore, when Cas9 nuclease generates a DSB, this simultaneously generates undesired indels at a substantial frequency, thus introducing further unwanted mutations [Citation57].
3.2. CRISPR-based DNA base editing in choroideremia
To address the limitations of HDR-mediated gene editing, CRISPR-Cas-mediated single-base pair editing systems (or ‘base editing’ systems) have been devised which allow for targeted restoration of single-base mutations. Two classes of DNA base editors have been described to date: cytosine base editors and adenine base editors [Citation58,Citation59]. DNA base editors encompass two key components. The first is an inactivated Cas enzyme (or Cas nickase, nCas9) which retains its programmable DNA binding ability, but which has lost its ability to generate DSBs. The second is a single-stranded DNA-modifying enzyme (cytidine or adenine deaminase) fused to nCas9 for targeted nucleotide alteration. Collectively, all four transition mutations (A > G, C > T, G > A and T > C) can be installed with the available CRISPR/Cas base editor systems. Recently, Kurt et al. described the engineering of two novel base editor architectures that can efficiently induce targeted C-to-G base transversions [Citation60]. In addition, recent studies report dual-base editor systems for combinatorial editing in human cells. Together, these new base editors expand the range of DNA base editors to transversion mutations and may allow for targeting of more complex compound edits than are currently achievable by a single DNA base editor.
Analysis of publicly available variants in the Leiden Open Variation Database (LOVD) [Citation27] reveals that 52.8% of pathogenic variants in the CHM database are substitutions, with the remaining variants not amenable to base editing as they involve indels ()). Despite this, 30.8% of pathogenic variants were amenable to base editing with the commonly used editors capable of editing transition variants (A > G, C > T, G > A, A > G). Of these, C > T and G > A mutations were more common, with C > T substitutions resulting in the creation of a premature stop codon the most common (). Most of these mutations are unique within families, however, and there is no single mutation that is obviously common and easily targetable. The five most common pathogenic variants are presented in , and collectively account for around 11% of all pathogenic variants. These all arise from C > T transitions, creating a TGA (STOP) from a CGA (Arg) codon in exon 6 or 7. These mutations are amenable to editing with cytidine base editors or the recently developed RESCUE RNA editor (see below, section 3.4). Though the cost of individualized treatment may be high, it is worth considering that if a CRISPR base or prime editor can be delivered separately from its guide RNA, a single C > T base-editing construct can be used with separate, personalized guides, allowing greater therapeutic flexibility.
Figure 3. Potential genome editing targets in CHM. The inner ring shows type of mutation. The middle ring shows the single-base substitutions (which overwhelmingly produce terminations, as previously described) and other consequences of the mutations (Frameshifts, splice defects, no protein product formed, or unknown). The outer ring shows the number of potential base editing targets in CHM (red: currently restricted to transitions, which comprise approximately 30.8% of choroideremia-associated mutations; the remaining 69.2% are not amenable to base-editing methods): the number of potential prime editing (orange: prime editing opens up the potential number of targets up to 85.7% of CHM variants): and remaining uneditable mutations (yellow: large insertions, deletions and duplications) Source: Leiden Open Variation Database (LOVD) [Citation27]
![Figure 3. Potential genome editing targets in CHM. The inner ring shows type of mutation. The middle ring shows the single-base substitutions (which overwhelmingly produce terminations, as previously described) and other consequences of the mutations (Frameshifts, splice defects, no protein product formed, or unknown). The outer ring shows the number of potential base editing targets in CHM (red: currently restricted to transitions, which comprise approximately 30.8% of choroideremia-associated mutations; the remaining 69.2% are not amenable to base-editing methods): the number of potential prime editing (orange: prime editing opens up the potential number of targets up to 85.7% of CHM variants): and remaining uneditable mutations (yellow: large insertions, deletions and duplications) Source: Leiden Open Variation Database (LOVD) [Citation27]](/cms/asset/0b0cbc3b-996c-45cf-b57f-0ad59021b219/ieod_a_1882300_f0003_oc.jpg)
Table 2. Most common substitutions in CHM. The top five are all C to T transitions, which lead to the creation of a STOP codon
3.3. CRISPR-based prime-editing in choroideremia
Recently, a method to overcome the inability of base-editors to generate precise base-edits beyond the four transition mutations has been described by Anzalone et al. [Citation61]. Known as prime editing, this method enables the introduction of indels and all 12 base-to-base conversions (both transitions and transversions), as well as insertions up to 44 bp and deletions up to 80 bp. As with CRISPR-mediated base editing, prime editing does not involve the creation of DSBs. Prime editors use an engineered reverse transcriptase fused to nCas9 and a prime editing guide RNA (pegRNA). Here, the pegRNA differs significantly from regular sgRNAs and plays a major role in the system’s function. The pegRNA contains not only the sequence complementary to the target sequence that directs nCas9 to its target site but also an additional sequence spelling the desired sequence change, which is reverse transcribed into the genome. The first generation of prime editors (PE1) was comprised of Moloney murine leukemia virus reverse transcriptase (M-MLV RT) linked to the C-terminus of nCas9 and pegRNA, which was expressed on a second plasmid. The latest generation prime editor, using enhanced M-MLV RT, performed all possible transition and transversion mutations with average editing efficiencies of 33% (±7.9%) [Citation61].
As well as vastly expanding the number of potentially correctable pathogenic mutations in human DNA, prime editing has other advantages over previous CRISPR-mediated base editing approaches. Prime editors have less stringent PAM requirements due to the varied length of the reverse transcriptase template, and no ‘bystander’ editing. Prime editing could potentially address all pathogenic CHM mutations apart from the 14% comprising large deletions, insertions or duplications ().
Prime editing offers similar benefits as base editing but provides greater target flexibility. Though its efficiency is low, examination of REP1 mRNA transcript levels in choroideremia patients with rare slow-progressing phenotypes show they have overall mRNA levels of <1% of those of non-affected individuals, while patients with normal disease phenotypes had undetectable mRNA levels. This implies that even an editing efficiency of 1% of mRNA transcripts may give sufficient protein product to slow down disease progression [Citation19]. There is a paucity of long-term data, but prime editing has enormous potential in addressing the range of human heritable diseases [Citation62]. However, current base and prime editor sizes both total >6 kb, well beyond the packaging capacity of AAV. Finally, it is important to note that prime editing tools are still in their infancy, and additional in vivo characterization is needed before their broader use for targeting inherited retinal diseases. Indeed, the introduction of exogenous reverse transcriptase should be treated with caution due to potential enabling effects on dormant pro-viral or retro-elements.
3.4. CRISPR-RNA editing
Finally, CRISPR-directed RNA editing represents another novel approach to targeted correction of single nucleotide variants, in RNA rather than DNA [Citation54]. A wide variety of approaches have been developed to edit RNA in vitro. Each approach currently uses a variant of the Adenosine Deaminase Acting on RNA (ADAR), naturally expressed enzymes in human cells that undertake physiological RNA editing functions. These deaminases convert adenosine bases to inosine in RNA, which is read as a guanosine in cellular processes such as translation and splicing [Citation63]. This effectively creates an A > G edit in RNA and can be harnessed for the correction of G > A mutations. ADAR variants have also been engineered to create C > U edits: together, they can theoretically address up to 10% of known CHM mutations [Citation64,Citation65]. Harnessed for site-directed RNA editing, ADAR can be recruited to editing sites of interest by systems that link ADAR to an effector molecule and direct the ADAR-effector system with a gRNA to the base to be edited [Citation54]. Many effectors have been developed including those based on CRISPR-Cas13 [Citation65,Citation66] or Cas9 systems [Citation67], bacteriophage-derived λN peptide [Citation71] and BoxB system, aptamer-like systems such as the MS2 bacteriophage coat protein (MCP) or GluR2 system [Citation69]. Additionally, systems that deliver only a gRNA and use only endogenously expressed ADAR have been developed [Citation69–71], in contrast [Citation68] to other systems that require ADAR overexpression.
RNA editing provides a number of theoretical advantages for safety in clinical translation. RNA editing is not permanent, as edited mRNAs eventually decay. A self-limiting treatment has an inherent appeal in a disease that affects the young, particularly as there is a paucity of long-term in vivo data for all DNA and RNA editing applications, simply by virtue of their novelty. The disadvantage of a self-limiting treatment, of course, is that repeated treatments would be required to maintain the therapeutic effect. Alternatively, a method of continual ADAR and gRNA expression must be used to achieve ongoing on-target repair. A mismatch between ADAR and gRNA expression in such a system may result in an increase in off-target effects. On the other hand, with a self-limiting system, off-target effects are also not permanent. This may allow for reversible or titratable editing, or systems with an in-built ‘off’ safety switch.
While RNA editing is an exciting prospective tool to add to the gene therapy researcher’s toolbox, the technology is still in its infancy. Though truncated ADAR-Cas systems have been engineered to maintain editing efficacy while meeting AAV-packaging constraints, AAV-delivery of an ADAR-Cas system has yet to be achieved. Work on targeting human mutations transfected into HEK293 cells achieved at best 35% editing efficacy: typical efficacies were only ‘up to’ 28% [Citation66]. Alternate systems such as the GluR2 and MS2 systems are readily deliverable within AAV packaging constraints. Delivery of these systems via AAV in vivo to two mouse models have resulted in editing efficiencies of less than 5% [Citation69]. Further work is required to understand the impact of off-target RNA editing in the transcriptome. A functional RNA editor ready for in-human trials is not a near-future possibility.
4. Antisense oligonucleotides
In eukaryotic mRNA processing, splicing of pre-mRNA is directed by interactions between the spliceosome (a complex of proteins and small nuclear RNAs which facilitates intronic excision) and the 5ʹ donor, branch, and 3ʹ acceptor sites within each intron. Typically, the splice donor site (5ʹ end of the intron) includes a GU sequence, followed by a relatively unconserved section which is followed by the branchpoint and a polypyrimidine tract, a sequence rich in pyrimidines, and terminating in the acceptor site at the 3ʹ-end, usually an AG sequence. Mutations within these key areas of the intron can lead to the formation of a cryptic splice site, able to redirect the pre-mRNA’s interactions with the spliceosome and resulting in the insertion of an aberrant exon.
Splice-modifying antisense oligonucleotides (AON) are short nucleotide sequences designed to restore normal splicing by redirecting pre-mRNA/spliceosome interaction. AONs are designed either to bind to the cryptic splice site and prevent it from interacting with the spliceosome, or to bind to the aberrant exon and thus encourage skipping of said aberrant exon during splicing [Citation72]. An AON targeting the common deep-intronic mutation in CEP290 (c.2991 + 1655A>G, p.Cys998*) in LCA10 has been shown to be able to correct aberrant splicing in pre-clinical studies and is undergoing phase 1 clinical trial with a good safety profile and promising early efficacy data [Citation73–75]. The trial by Cideciyan et al. is particularly encouraging because the AON was delivered by intravitreal injections with effects appearing to last several months. Moreover, it affected functional and anatomical changes in photoreceptors of some treated eyes, with improved retinal structure by optical coherence tomography (OCT), improvements in full field stimulus testing, and improvement in visual acuity in one treated patient [Citation75].
Of the 500 or so pathogenic variants found in choroideremia, 100 are predicted to cause splicing abnormalities. However, the vast majority of these are single nucleotide substitutions in the 5ʹ splice donor or 3ʹ splice acceptor sites, and therefore not amenable to the AON mechanism of splice rescue. Few potential targets have been described [Citation76]. One attempt has been made to target the c.315–4587 T > A mutation, which creates a 98-bp pseudoexon insertion and loss of protein function. An AON was designed to target the pseudoexon and delivered in vitro to lymphoblast cells from two choroideremia patients. Disappointingly, despite showing almost full redirection of splicing to produce detectable levels of corrected transcript in both patient-derived lymphoblast cells, resultant REP1 expression was low. The authors have speculated that this may be due to unpredicted effects on splicing from AON binding, or possible initiation of transcript degradation caused by AON binding [Citation77].
5. Nonsense suppression strategies
Around 32% of all CHM mutations (and 49% of all CHM mutations within coding sequences) result in the formation of a premature termination codon without inducing frameshift or splicing defects. A strategy which can be applied to all PTC-generating mutations therefore has great appeal and applicability.
The generation of a PTC can result in either nonsense-mediated decay of the resultant mRNA transcript, or, if it escapes decay, the production of a truncated and nonfunctional protein product. In nonsense-mediated decay, activity is triggered by the relative location of the PTC within the mRNA sequence to the binding location of a protein complex, the exon-junction complex (EJC). The EJC normally binds approximately 20–24 nucleotides upstream of an mRNA splice-site. This forms a mature ribonuclear complex facilitating normal interaction with the ribosome. If a PTC occurs 50–55 nucleotides upstream of an EJC, this allows the EJC to bind to a kinase-associated protein complex containing the component UF1. Binding triggers release of factors from the complex leading to deadenylation, decapping and exonuclease activity with subsequent degradation of the ribonuclear complex.
If an mRNA transcript containing a premature PTC escapes nonsense-mediated decay, during translation the PTC results in termination of the protein, leading to a truncated protein product. Normal binding of tRNA with its matching site on mRNA takes place within the ribosome ‘A’ site during translation. A cognate tRNA matches three of its mRNA base pairs, while a near-cognate tRNA matches only two. The normal levels of near-cognate tRNA matches compared to cognate tRNA are less than 0.1% in normal translation [Citation78]. When a PTC exists, no cognate tRNA exists: instead, eukaryotic release factor 1 (eRF1) binds to the stop codon at the ‘A’ site and initiates release of the polypeptide from the ribosomal complex. Nonsense suppression therapy works by promoting read-through of transcripts with a PTC by promoting binding of near-cognate tRNAs at the PTC instead of eRF1. In clinical usage, aminoglycoside antibiotics exploit this mechanism by binding to bacterial ribosomes and causing fatal translational errors. Eukaryotic cells have greater resistance to the substitution of near-cognate tRNAs due to differences in ribosomal structure. Nonetheless, aminoglycosides such as gentamicin, paromomycin, streptomycin among others promote read-through at eukaryotic PTC sites. Moosajee et al. showed that in a zebrafish model of choroideremia, where homozygous REP1 knockout caused by chmru848 usually confers embryonic lethality, administration of gentamicin and paromomycin improved read-through and conferred a 1.5- to 1.7-fold improvement in survival [Citation79].
Use of aminoglycosides is, however, limited by systemic toxicity and retinal toxicity. Synthesis of novel aminoglycosides and aminoglycoside-like molecules has aimed to address this issue, leading to the discovery of a small-molecule PTC124 (Ataluren) which has been shown to increase transcriptional read-through without concomitant toxicity. Delivery of Ataluren topically to the eye has been optimized and it has little retinal toxicity compared with traditional aminoglycosides. Both Ataluren and a similar molecule based on Ataluren, PTC414, has been shown in human CHMY42X/y fibroblasts to increase prenylation activity by 45% and 36%: Ataluren has also shown amelioration of ophthalmic and systemic abnormalities in the embryonic chmru848 zebrafish [Citation80].
Further improvements on efficacy can be achieved by increasing the number of potential transcripts available for read-through promotion. Depending on the position of the PTC, mRNA transcripts may be more or less subject to nonsense-mediated decay, resulting in significant variation of available transcripts between individuals: therefore read-through strategies are highly dependent on mutation location. Downregulating nonsense-mediated decay via inhibition of UF1 leads to an increased number of surviving mRNA transcripts as potential read-through promotion targets.
Nonsense-suppression strategies are theoretically appealing as they can be delivered systemically and non-invasively, and have wide-ranging therapeutic targets beyond the eye. There are currently 34 trials registered for Ataluren (TranslarnaTM) on the Clinical Trials database, including a phase 2 trial for nonsense mutations in aniridia (NCT02647359). Unlike any targeted therapy delivered by subretinal injection, treatment is not limited spatially in the eye. However, previous clinical trials in cystic fibrosis [Citation81] and Duchenne’s muscular dystrophy [Citation82] have shown only small treatment effects limited to rigidly defined patient subgroups. By its nature, nonsense-suppression strategies are nonspecific and must promote near-cognate tRNA binding throughout all translation events, potentially increasing production of other anomalous proteins. Furthermore, the applicability of nonsense-suppression strategies may be much narrower than the number of nonsense mutations in CHM. Administration of Ataluren to patient-derived fibroblasts carrying a c.772A>T substitution leading to p.Lys258* failed to show a statistically significant trend in improving prenylation profile compared to non-treated fibroblasts, and did not increase levels of REP1 mRNA or detectable REP1 protein [Citation83]. The authors’ in silico modeling suggested that the preferred amino acid substitutions provided by Ataluren-induced enhanced read-through were still damaging to REP1 function, and proposed that patients may require mutational screening and modeling before initiation of therapy.
Currently, therefore, while nonsense-suppressing strategies show in vitro and some in vivo promise, much work remains before it can be a viable therapeutic approach in choroideremia.
6. Expert opinion
The first gene therapy treatment for choroideremia was performed in a 57-year-old man 10 years ago in a clinical trial that used AAV2 to deliver the CHM gene by subretinal injection. Since then, the early results have led to a multinational phase III clinical trial involving hundreds of patients across many countries worldwide. During this time, progress has been made with other molecular therapies, such as CRISPR and RNA editing and alternate AAV vectors have also been developed. It is therefore prudent to look back and ask if AAV2 gene replacement is still the preferred option against these developments.
The research so far has a strong emphasis on AAV2-REP1 gene replacement therapy, and with good reason, because it is supported by comprehensive pre-clinical data. Gene augmentation via REP1 replacement has a strong theoretical basis, because virtually all mutations reported to date have been null. REP1 fits well within an AAV vector and preclinical studies have shown strong expression of the transgene within RPE and retina [Citation84]. Though the intervention is invasive and subretinal injection in the degenerate retina of choroideremia is technically challenging, only a single intervention is likely to be required for long-lasting effects. Though there is the theoretical possibility of a mutant truncated protein competing with the transgene product for Rab prenylation, this effect has not been seen in vitro [Citation24].
None of the novel evolved AAV capsids to date has shown efficient targeting of the retinal pigment epithelium after intravitreal injection and so the subretinal route of administration is likely to remain the optimal method of gene delivery for the foreseeable future. While the focus on technological innovation over the last 10 years has been on molecular therapies, it should be noted that surgical techniques have also improved considerably during this time and there are now many surgeons with expertise in subretinal gene therapy worldwide.
While base editing and, more pertinently, prime editing have potential applications to a significant number of mutations in choroideremia, an individualized editing approach seems to offer few advantages over gene augmentation in this disease. Although base and prime editing offer unique advantages for correction of the endogeneous transcript, these are particularly useful for diseases where the gene length prohibits packaging in an AAV, or where the mutated protein product has a toxic or inhibitory effect. However, this does not appear to be a significant problem in choroideremia.
Two much more interesting approaches with potential are nonsense suppression strategies and antisense oligonucleotides. Nonsense suppression strategies are an extremely appealing idea since they can be delivered orally, they apply to and they are applicable to a large number of mutations in choroideremia. However, clinical trials in other diseases have shown only limited success, they may have more limited applicability than first appears, and they require long-term administration for effect. Newer formulations of nonsense suppression strategy drugs are more tolerated and have fewer toxic side effects than the drugs from which they are developed, but nonetheless long-term safety data are lacking. However, as companies continue systemically to create and test new molecules, this remains an avenue to watch. Antisense oligonucleotides have shown great promise in LCA with sustained effects, and may be ideal for deep-intronic splice defects. They are delivered by intravitreal injection, which is well tolerated by patients, and retain the gene expression under its endogenous promoter. Unfortunately, however, choroideremia presents few suitable mutation hotspots for targeting using the AON approach. Therefore it is likely that, for the foreseeable future, gene augmentation remains the standard approach to treating choroideremia.
Article highlights
• Choroideremia is a loss of function disease. The majority of mutations in choroideremia are truncating, nonsense mutations leading to loss of protein product.
• Gene replacement therapy is the most logically appealing form of therapy for choroideremia. AAV2-REP1 is currently in phase 3 trials and remains the most promising treatment likely to be available in the near future.
• CRISPR-mediated base and prime editing are exciting new therapeutic options with widespread applicability, programmability and flexibility. Over 50% of mutations in choroideremia are single-base substitutions: of the remainder, only 14% (comprising large deletions, duplications or insertions) are not amenable to prime editing. Nonetheless, due to the heterogeneity of mutations in CHM, currently, they do not represent a cost-effective alternative to gene replacement.
• RNA editing, antisense oligonucleotides and nonsense suppression strategies are therapies that do not rely on permanent editing of the genome. However, so far, there is a lack of long-term data for these therapies, and there is also a lack of potential therapeutic sites for antisense oligonucleotides.
This box summarizes key points contained in the article.
Declaration of interest
RE MacLaren is a consultant to Biogen and receives research funding for clinical trials involving choroideremia gene therapy. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter discussed in this work.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Preising MN, Ayuso C. Rab escort protein 1 (REP1) in intracellular traffic: a functional and pathophysiological overview. Ophthalmic Genet. 2004;25:101–110.
- Seabra MC, Ho YK, Anant JS. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem. 1995;270:24420–24427.
- Seabra MC, Brown MS, Goldstein JL. Retinal degeneration in choroideremia: deficiency of Rab geranylgeranyl transferase. Science. 1993;259:377–381.
- Wavre-Shapton ST, Tolmachova T, da Silva ML, et al. Conditional ablation of the choroideremia gene causes age-related changes in mouse retinal pigment epithelium. PLoS One. 2013;8:1–11.
- Tolmachova T, Anders R, Abrink M, et al. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest. 2006;116:386–394.
- Tolmachova T, Wavre-Shapton ST, Barnard AR, et al. Retinal pigment epithelium defects accelerate photoreceptor degeneration in cell type-specific knockout mouse models of choroideremia. Investig Ophthalmol Vis Sci. 2010;51:4913–4920.
- Xue K, Oldani M, Jolly JK, et al. Correlation of optical coherence tomography and autofluorescence in the outer retina and choroid of patients with choroideremia. Investig Ophthalmol Vis Sci. 2016;57:3674–3684.
- Paavo M, Carvalho JRL, Lee W, et al. Patterns and intensities of near-infrared and short-wavelength fundus autofluorescence in choroideremia probands and carriers. Investig Ophthalmol Vis Sci. 2019;60:3752–3761.
- Gill JS, Moosajee M, Dubis AM. Cellular imaging of inherited retinal diseases using adaptive optics. Eye. 2019;33:1683–1698.
- Foote KG, Rinella N, Tang J, et al. Cone structure persists beyond margins of short-wavelength autofluorescence in choroideremia. Investig Ophthalmol Vis Sci. 2019;60:4931–4942.
- Cremers FPM, Armstrong SA, Seabra MC, et al. REP-2, a Rab escort protein encoded by the choroideremia-like gene. J Biol Chem. 1994;269:2111–2117.
- Aylward JW, Xue K, Patrício MI, et al. Retinal degeneration in choroideremia follows an exponential decay function. Ophthalmology. 2018;125:1122–1124.
- Köhnke M, Delon C, Hastie ML, et al. Rab GTPase prenylation hierarchy and its potential role in choroideremia disease. PLoS One. 2013;8:1–11.
- Tanaka D, Kameyama K, Okamoto H, et al. Caenorhabditis elegans Rab escort protein (REP-1) differently regulates each Rab protein function and localization in a tissue-dependent manner. Genes Cells. 2008;13:1141–1157.
- Strunnikova NV, Barb J, Sergeev YV, et al. Loss-of-function mutations in Rab escort protein 1 (REP-1) affect intracellular transport in fibroblasts and monocytes of choroideremia patients. PLoS One. 2009;4:e8402.
- Jauregui R, Park KS, Tanaka AJ, et al. Spectrum of disease severity and phenotype in choroideremia carriers. Am J Ophthalmol. 2019;207:77–86.
- Carrel L, Willard HF. Heterogeneous gene expression from the inactive X chromosome: an X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others. Proc Natl Acad Sci U S A. 1999;96:7364–7369.
- Simunovic MP, Jolly JK, Xue K, et al. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Investig Ophthalmol Vis Sci. 2016;57:6033–6039.
- Fry LE, Patrício MI, Williams J, et al. Association of messenger RNA level with phenotype in patients with choroideremia: potential implications for gene therapy dose. JAMA Ophthalmol. 2020;138:128–135.
- Radziwon A, Arno GK, Wheaton D, et al. Single-base substitutions in the CHM promoter as a cause of choroideremia. Hum Mutat. 2017;38:704–715.
- Vaché C, Torriano S, Faugère V, et al. Pathogenicity of novel atypical variants leading to choroideremia as determined by functional analyses. Hum Mutat. 2019;40:31–35.
- Contestabile MT, Piane M, Cascone NC, et al. Clinical and genetic studies in a family with a new splice-site mutation in the choroideremia gene. Mol Vis. 2014;20:325–333.
- Garcia-Hoyos M, Lorda-Sanchez I, Gómez-Garre P, et al. New type of mutations in three Spanish families with choroideremia. Investig Ophthalmol Vis Sci. 2008;49:1315–1321.
- Torriano S, Erkilic N, Faugère V, et al. Pathogenicity of a novel missense variant associated with choroideremia and its impact on gene replacement therapy. Hum Mol Genet. 2017;26:3573–3584.
- Esposito G, De Falco F, Tinto N, et al. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with rab geranylgeranyl transferase. Hum Mutat. 2011;32:1460–1469.
- Sergeev YV, Smaoui N, Sui R, et al. The functional effect of pathogenic mutations in Rab escort protein 1. Mutat Res - Fundam Mol Mech Mutagen. 2009;665:44–50.
- Fokkema IFAC, Taschner PEM, Schaafsma GCP, et al. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–563.
- Edwards TL, Groppe M, Jolly JK, et al. Correlation of retinal structure and function in choroideremia carriers. Ophthalmology. 2015;122(6):1274-6.
- Clinical Trials NIH. [ cited 2021 Jan 6]. Available from: https://www.clinicaltrials.gov/
- Xue K, Groppe M, Salvetti AP, et al. Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye. 2017;31:1308–1316.
- Clinical Trials NIH Identifier NCT04483440 [Internet]. 2020 [cited 2021 Jan 5]. p. 1. Available from: https://clinicaltrials.gov/ct2/show/study/NCT04483440.
- MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial fi ndings from a phase 1/2 clinical trial. Lancet. 2014;383:1129–1137.
- Colella P, Ronzitti G, Mingozzi F.. Emerging issues in AAV-mediated in vivo gene therapy. Mol Ther - Methods Clin Dev. 2018;8:87–104.
- Edwards TL, Jolly JK, Groppe M, et al. Visual acuity after retinal gene therapy for choroideremia. N Engl J Med. 2016;374:1996–1998.
- Xue K, Jolly JK, Barnard AR, et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat Med. 2018;24:1507–1512.
- Clinical trials NIH Record NCT02341807. 2015 [cited 2020 Oct 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT02341807
- Dimopoulos IS, Hoang SC, Radziwon A, et al. Two-year results after aav2-mediated gene therapy for choroideremia: the Alberta experience. Am J Ophthalmol. 2018;193:130–142.
- Lam BL, Verriotto J, Gregori N, et al. Choroideremia gene therapy phase II clinical trial: 6-month results. Invest Ophthalmol Vis Sci. 2017;58:3386.
- Lam BL, Davis JL, Gregori NZ, et al. Choroideremia gene therapy phase 2 clinical trial: 24-month results. Am J Ophthalmol. 2019;197:65–73.
- Fischer MD, Ochakovski GA, Beier B, et al. Efficacy and safety of retinal gene therapy using adeno-associated virus vector for patients with choroideremia: a randomized clinical trial. JAMA Ophthalmol. 2019;137:1247–1254.
- Fischer MD, Ochakovski GA, Beier B, et al. Changes in retinal sensitivity after gene therapy in choroideremia. RETINA. 2020;40(1):160–168.
- Jolly JK, Xue K, Edwards TL, et al. Characterizing the natural history of visual function in choroideremia using microperimetry and multimodal retinal imaging. Investig Ophthalmol Vis Sci. 2017;58:5575–5583.
- Clinical Trials NIH Record NCT03507686 [Internet]. 2017 [cited 2020 Oct 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT03507686.
- Clinical Trials NIH Record NCT03496012 [Internet]. 2017 [cited 2020 Oct 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT03496012.
- Clinical Trials NIH Record NCT03584165 [Internet]. 2018 [cited 2020 Oct 30]. Available from: https://clinicaltrials.gov/ct2/show/NCT03584165.
- Byrne LC, Day TP, Visel M, et al. In vivo directed evolution of AAV in the primate retina. bioRxiv. 2019;1–12.
- Wassmer S, Comander J, Carvalho L, et al. 80. Delayed inflammatory response to intravitreal AAV gene transfer in non-human primates. Mol Ther. 2016;24:S35.
- Timmers AM, Newmark JA, Turunen HT, et al. Ocular inflammatory response to intravitreal injection of adeno-associated virus vector: relative contribution of genome and capsid. Hum Gene Ther. 2020;31:80–89.
- Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821.
- Giannelli SG, Luoni M, Castoldi V, et al. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum Mol Genet. 2018;27:761–779.
- Hu S, Du J, Chen N, et al. In vivo CRISPR/Cas9-mediated genome editing mitigates photoreceptor degeneration in a mouse model of X-linked retinitis pigmentosa. Investig Ophthalmol Vis Sci. 2020;61:1–10.
- Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. 2019;25:229–233.
- Peddle CF, Maclaren RE. The application of CRISPR/CAS9 for the treatment of retinal diseases. Yale J Biol Med. 2017;90:533–541.
- Fry LE, Peddle CF, Barnard AR, et al. RNA editing as a therapeutic approach for retinal gene therapy requiring long coding sequences. Int J Mol Sci. 2020;21. DOI:10.3390/ijms21030777
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–350.
- Mao Z, Bozzella M, Seluanov A, et al. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7:2902–2906.
- Song F, Stieger K. Optimizing the DNA donor template for homology-directed repair of double-strand breaks. Mol Ther Nucleic Acids. 2017;7:53–60.
- Komor AC, Kim YB, Packer MS, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424.
- Komor AC, Zhao KT, Packer MS, et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci Adv. 2017;3:eaao4774.
- Kurt IC, Zhou R, Iyer S, et al. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat Biotechnol. 2020;39:41–46.
- Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157.
- Kantor A, McClements ME, MacLaren RE. CRISPR-Cas9 DNA base-editing and prime-editing. Int J Mol Sci. 2020;21:6240.
- Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17:83–96.
- Huang X, Lv J, Li Y, et al. Programmable C‐to‐U RNA editing using the human APOBEC 3A deaminase. EMBO J. 2020;39(22):e104741.
- Abudayyeh OO, Gootenberg JS, Franklin B, et al. A cytosine deaminase for programmable single-base RNA editing. Science. 2019;538:eaax7063.
- Cox DBT, Gootenberg JS, Abudayyeh OO, et al. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–1027.
- Marina RJ, Brannan KW, Dong KD, et al. Evaluation of engineered CRISPR-Cas-mediated systems for site-specific RNA editing. Cell Rep. 2020;33:108350.
- Montiel-Gonźalez MF, Vallecillo-Viejo IC, Rosenthal JJC. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016;44:1–12.
- Katrekar D, Chen G, Meluzzi D, et al. In vivo RNA editing of point mutations via RNA-guided adenosine deaminases. Nat Methods. 2019;16:239–242.
- Qu L, Yi Z, Zhu S, et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat Biotechnol. 2019;37:1059–1069.
- Merkle T, Merz S, Reautschnig P, et al. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat Biotechnol. 2019;37:133–138.
- Xue K, MacLaren RE. Antisense oligonucleotide therapeutics in clinical trials for the treatment of inherited retinal diseases. Expert Opin Investig Drugs. 2020;29:1163–1170.
- Collin RW, Den Hollander AI, Der Velde-Visser SDV, et al. Antisense oligonucleotide (AON)-based therapy for leber congenital amaurosis caused by a frequent mutation in CEP290. Mol Ther Nucleic Acids. 2012;1:e14.
- Gerard X, Perrault I, Hanein S, et al. AON-mediated exon skipping restores ciliation in fibroblasts harboring the common Leber congenital amaurosis CEP290 mutation. Mol Ther Nucleic Acids. 2012;1:e29.
- Cideciyan AV, Jacobson SG, Drack AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. 2019;25:225–228.
- Van Den Hurk JAJM, Van De Pol DJR, Wissinger B, et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Hum Genet. 2003;113:268–275.
- Garanto A, van der Velde-visser SD, Cremers FPM, et al. Antisense oligonucleotide-based splice correction of a deep-intronic mutation in chm underlying choroideremia. In: Ash JD, Anderson RE, LaVail MM, et al., editors. Retinal degeneration Disease. Cham: Springer International Publishing; 2018. 83–89.
- Richardson R, Smart M, Tracey-White D, et al. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp Eye Res. 2017;155:24–37.
- Moosajee M, Gregory-Evans K, Ellis CD, et al. Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease. Hum Mol Genet. 2008;17:3987–4000.
- Moosajee M, Tracey-White D, Smart M, et al. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsensemediated zebrafish model. Hum Mol Genet. 2016;25:3416–3431.
- Kerem E, Konstan MW, De Boeck K, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2:539–547.
- McDonald CM, Campbell C, Torricelli RE, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1489–1498.
- Torriano S, Erkilic N, Baux D, et al. The effect of PTC124 on choroideremia fibroblasts and iPSC-derived RPE raises considerations for therapy. Sci Rep. 2018;8:1–15.
- Tolmachova T, Tolmachov OE, Barnard AR, et al. Functional expression of Rab escort protein 1 following AAV2-mediated gene delivery in the retina of choroideremia mice and human cells ex vivo. J Mol Med. 2013;91:825–837.