
ABSTRACT
Introduction: Cardiac transthyretin (ATTR) amyloidosis is a progressive and fatal infiltrative cardiomyopathy (ATTR-CM) characterized by congestive cardiac failure, often with preserved left ventricular ejection fraction, and significant risk of conduction disease. Diagnosis is often delayed or missed due to poor specificity of echocardiography and the historical requirement for a histological diagnosis, frequently an endomyocardial biopsy.
Areas covered: Following a detailed literature review focusing on peer reviewed articles (Pubmed, Cochrane Library, Google Scholar), from 1995 to 2020, alongside international diagnostic guidelines and expert opinion in the field, this article will explore the current non-invasive diagnostic criteria for ATTR-CM including the role of transthoracic echocardiography, cardiac MRI, bone scintigraphy, and assessment for exclusion of a clonal dyscrasia.
Expert opinion: ATTR-CM is an emerging and increasingly diagnosed cause of heart failure, particularly in the elderly. Promising novel therapies make accurate and swift diagnosis of the disease vital. With the increasing use of cardiac MRI to investigate cardiomyopathy and repurposing of technetium-labeledbone scintigraphy, clinicians are now often able to diagnose ATTR-CM without recourse to an endomyocardial biopsy.
1. Introduction
Systemic amyloidosis is a rare disease caused by the extracellular deposition of amyloid, a fibrillar material, derived from a variety of precursor proteins, that aggregate with a highly abnormal cross B sheet confirmation [Citation1,Citation2]. Amyloid deposition is associated with disruption of tissue structure and disturbance of organ function, which if left unchecked, invariably leads to organ failure [Citation3,Citation4]. Cardiac amyloidosis (CA) classically manifests with restrictive cardiomyopathy, is a fatal condition, associated with significant morbidity and is often challenging to diagnose. In the majority of cases the amyloid fibril precursor protein is either a monoclonal immunoglobulin light chain (AL) [Citation5] or transthyretin (ATTR); the latter unmutated, wild-type transthyretin amyloidosis (wtATTR), or mutated, variant transthyretin amyloidosis (vATTR) [Citation6–8].
The cardiac phenotype typically includes excessive ventricular wall thickening leading to diastolic dysfunction and subsequent congestive cardiac failure, often complicated by syncope and arrhythmias. Extra-cardiac manifestations are common in systemic AL amyloidosis due to infiltration of other visceral organs by amyloid. In patients with vATTR the clinical phenotype can be dominated by cardiomyopathy (vATTR-CM), polyneuropathy (vATTR-PN) or may include both cardiomyopathy and polyneuropathy to varying degrees (vATTR-mixed).
Until recently, diagnosis of systemic amyloidosis was focused upon biopsy of a clinically affected or ‘target’ organ and staining with Congo red dye demonstrating pathognomonic green birefringence when visualised under cross-polarizedlight. Nonetheless, when there is a clinical suspicion of systemic amyloidosis, a ‘screening’ biopsy from subcutaneous fat, salivary gland, or rectum yields the diagnosis in 50 to 80%of patients with systemic AL amyloidosis [Citation9]. Unfortunately in patients with ATTR amyloidosis, screening biopsies demonstrate a lower yield thus frequently resulting in the need for an endomyocardial biopsy (EMB) to confirm the diagnosis [Citation10]. EMBare not routinely performed outside specialist centres in the U.Kdue to the need for technical expertise which can often lead to a delay in diagnosis [Citation11]. Risks include myocardial perforation and cardiac tamponade, the latter of which can be fatal.
CA has been recently recognised as an underdiagnosed cause of heart failure and in combination with advances in imaging modalities including repurposed bone scintigraphy and cardiac magnetic resonance imaging (CMR) over the last ten years, has enabled earlier diagnosis, improved understanding of pathogenesis, and the capacity to track disease in response to novel amyloid-specific therapies.
2. Background
2.1. Methodology
This article explores the available literature on ATTR amyloidosis focusing on the noninvasivenon-invasive diagnosis of ATTR-CM. A focused and detailed literature search was performed concentrating on peer reviewedpeer-reviewed articles, of sufficient power and relevance, on scientific databases (Pubmed, Cochrane LibraryLibrary, and Google Scholar) from 1995 onwards alongside; book chapters, international diagnostic guidelines as well as expert opinion in the field of ATTR-CM. This article will focus on the key elements pertaining to the current noninvasivenon-invasive diagnostic criteria for ATTR-CM including the prevalence and phenotype of the disease, role of transthoracic echocardiography, cardiac MRI, bone scintigraphy, and assessment for exclusion of a clonal dyscrasia.
2.2. Prevalence
Cardiac ATTR amyloid deposits have been reported in autopsy series, in up to a quarter of individuals over the age of 80 years [Citation12], although often the quantity of amyloid was small [Citation13] and the presence of ATTR cardiomyopathy was not demonstrated. Nonetheless, postmortempost-mortem studies have shown that cardiac amyloid deposition is commoner in patients with heart failure and preserved ejection fraction (HFpEF), than in an age-matched autopsy group without heart failure, with a more balanced gender split than is reported in clinical practice.
Diagnoses of wtATTR-CM have risen several foldin recent decades, with prevalence estimates of 10–16% in elderly patients presenting with HFpEF [Citation14,Citation15]. wtATTR-CM has a male preponderance and presents with a predominant cardiac phenotype although can often be preceded by lumbar canal stenosis, carpal tunnel syndrome, and/or tendinopathy [Citation8,Citation16]. This is in stark contrast to the phenotype of vATTR amyloidosis which classically presents at a younger age with a varied and often mixed presentation of peripheral neuropathy, autonomic neuropathy, and/or cardiomyopathy [Citation17]. There are 132 known causative TTR mutations [Citation18], the commonest encoding V122I (p. V142I) which is present in 3.43% of US African Americans, resulting in a clinical phenotype which closely mimics wtATTR-CM [Citation19]. It is estimated that approximately 2 million people in the US are carriers of this variant, and thus at risk of developing ATTR-CM. Whilst patients with vATTR due to certain mutations may experience disabling autonomic neuropathy, cardiac involvement remains the key prognostic determinant in ATTR amyloidosis, with a median survival of 3–6 years from diagnosis [Citation20].
3. Imaging
3.1. Echocardiography
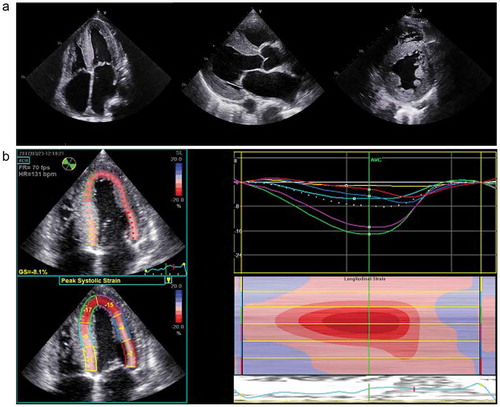
Echocardiography, whilst the most accessible and commonly used tool for investigating heart failure, is neither specific nor sensitive for the diagnosis of CA [Citation21]. Amyloidosis is characterized by thickened biventricular walls including left ventricular (LV) wall thickness greater than 12 mm and small, non-dilated ventricles (). There is a propensity towards a symmetrical increase in LV wall thickness in AL CA, while ATTR-CM has been reported to manifest with a more asymmetrical pattern [Citation22,Citation23], with a sigmoid septal morphology in 70% and septal curvature inversion in up to 30% of cases [Citation22]. Echocardiographic features of CA include thickened and at times sparkling appearance of the interatrial septum and valves as well as the classical ‘speckled’ myocardium. Extracellular amyloid infiltration leads to stiffening, impaired relaxation, and dysfunction of the ventricles, which in combination with atrial infiltration by amyloid, can lead to atrial dilatation, reduced blood flow, and a higher risk of thrombus formation [Citation24–27]. Despite CA being categorised as a cause of HFpEF, there is typically both systolic and diastolic dysfunction [Citation28]. Ejection fraction is a poor marker of systolic function in CA as it reflects radial contraction which is often not affected until the later stages of disease. Longitudinal function is classically impaired prior to radial contraction in CA and indices of longitudinal function such as longitudinal strain (LS) measurement by tissue Doppler and echocardiographic speckle tracking are important tools for diagnosing CA, demonstrating the distinctive appearance of a ‘bulls eye pattern’ due to apical sparing () as well as helping to distinguish CA from other hypertrophic cardiomyopathies [Citation29]. Diastolic function is almost always impaired in CA and can span from impaired relaxation to restrictive filling defects [Citation30]. Other markers of diastolic dysfunction can additionally be used to increase suspicion of CA, with TDI of the mitral annulus often being less than 6 cm/s [Citation31].
Figure 1. Transthoracic echocardiogram (TTE) of a patient with cardiac ATTR CM. TTE demonstrating concentric left ventricular hypertrophy (left to right) 4 Chamber view, parasternal and parasternal short axis view.4 Chamber global longitudinal strain demonstrating a ‘bull’s eye pattern’, characteristic of cardiac amyloidosis
3.2. Cardiac magnetic resonance imaging (CMR)
A now well-established imaging modality, particularly for investigation of cardiomyopathy, CMR provides unparalleled accuracy on cardiac morphology as well as myocardial tissue characterisation (). The deposition of amyloid fibrils leads to expansion of extracellular volume, which is well visualised by delayed or ‘late gadolinium enhancement’ (LGE) following administration of gadolinium-based contrast agents. Gadolinium passively accumulates extracellularly, giving rise to diffuse subendocardial or transmural LGE in CA and abnormal myocardial and blood pool gadolinium kinetics, described over 15 years ago [Citation32]. There are three main patterns of LGE recognised; none (), sub-endocardial () and transmural LGE (), with transmurality showing good correlation with the extent of myocardial infiltration [Citation33].
Figure 2. Cardiac MRI of a patient with ATTR CM (left to right) steady-state free precession cine; four chamber, two chamber, three chamber and short axis view demonstrating concentric LV hypertrophy
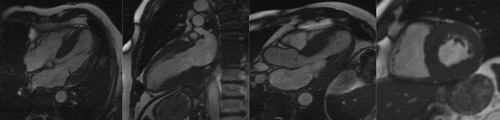
Figure 3. Cardiac MRI with phase sensitive inversion recovery reconstruction late gadolinium enhancement images demonstrating A) No LGE B) Sub-endocardial LGE and C) transmural LGE

An important consideration when using gadolinium-based contrast agents (GBCA) is the association with nephrogenic systemic fibrosis (NSF), a grave and potentially fatal condition. Whilst the risk of developing NSF is strongly related to baseline renal excretory function (the highest risk associated with an eGFR < 30 mL/min), the chemical structure of the contrast agent contributes to the risk of NSF. American College of Radiology guidelines recommend the preferential use of Group II agents in patients with advanced CKD, emphasising the importance of a calculated assessment of both the benefits and risks of administrating GBCA against the diagnostic challenges of performing a non-contrast scan. Whilst initially, the belief was that the gadolinium ion remained in a chelated state after intravenous administration, multiple studies have shown the presence of gadolinium deposits, in patients with normal renal function [Citation34], in a range of tissues including neural matter (dentate nucleus, thalamus, pons, and globus pallidus) [Citation35–37] and bone tissue [Citation38]. To date, the clinical significance of this remains unclear; furthermore an additional challenge of LGE is that it in light of its non-quantitative nature, its use to track disease over time remains limited.
T1 mapping which acts by direct quantification of an intrinsic myocardial signal, the longitudinal relaxation time has been used to overcome the limitations of GBCA. Pre-contrast or native myocardial T1 informs clinicians of disease severity by assessing both the degree of amyloid infiltration alongside both diastolic and systolic impairment ()[Citation39]. Key benefits of native myocardial T1 are the accuracy with which it can diagnose ATTR-CM as well as its role as an early disease marker, which often precedes left ventricular hypertrophy or LGE [Citation39,Citation40].
Figure 4. Cardiac MRI short axis steady-state free precession cine (left to right) with corresponding native myocardial T1 map, corresponding myocardial T2 map, corresponding phase sensitive inversion recovery reconstruction late gadolinium enhancement image and corresponding ECV map
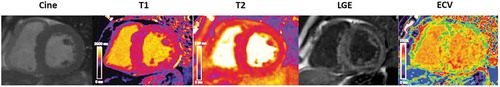
Extracellular volume (ECV) allows quantification of amyloid deposition in the myocardium and has been shown to correlate well with other markers of disease severity in both ATTR-CM and AL-CM [Citation22,Citation41].The ECV is globally increased, with values frequently >40% in ATTR-CM (). Novel features of ECV measurement in CA include its distinctive ability to diagnose early disease, quantify the burden of amyloid deposition, and thus track disease change over time [Citation42], a phenomenon recently reported with novel small interfering RNA therapies in ATTR-CM [Citation43].
In conclusion, characterisation of myocardial tissue by CMR in CA,provides clinicians with a detailed understanding of the multiple and varied disease processes that exist in CA, challenging the notion that CA’s main pathology is that of tissue deposition. Exemplary of this, T2 relaxation time is a time constant representing the decay of transverse magnetization and detects oedema in a range of pathologies including myocardial infarction, myocarditis, and Takotsubo cardiomyopathy [Citation44]. This has helped our understanding of CA as a heterogenouscondition with a range of contributing pathologies and was demonstrated by the discovery that T2 levels were found to be higher in patients with untreated AL CA compared with treated AL and ATTR-CM (); thereby demonstrating the potentially important pathophysiological and prognostic role of oedema [Citation45].
3.3. Radionuclide ‘bone’ scintigraphy
It is well recognised that radionuclide ‘bone’ scintigraphy with technetium labelled bisphosphonates localizes to cardiac amyloid deposits, although the reason for this remains unclear [Citation46]. 99mTc-labeled 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD), 99mTc-labeled pyrophosphate (PYP)PYP), and 99mTc-labeled-hydroxymethylene diphosphonate (HMDP) are both extremely sensitive (99%) and specific (86%) at detecting cardiac ATTR amyloid [Citation47]and may have a role in differentiating it from other cardiomyopathies with similar echocardiographic features, such as hypertrophic cardiomyopathy [Citation48–52]. Indeed, radionuclide ‘bone’ scintigraphy may even identify cardiac ATTR amyloid deposits early in the course of the disease, sometimes preceding abnormalities onechocardiography or CMR [Citation40,Citation53]. Whilst its use has been reported for the detection of ATTR amyloidosis among patients with known HFpEF [Citation54], cardiac localization is reported in a proportion of patients with cardiac AL amyloidosis, and whilst usually low grade, it may be grade 2 or 3 such that bone scintigraphy alone is unable to establish a diagnosis of ATTR CA and exclude AL CA [Citation51].
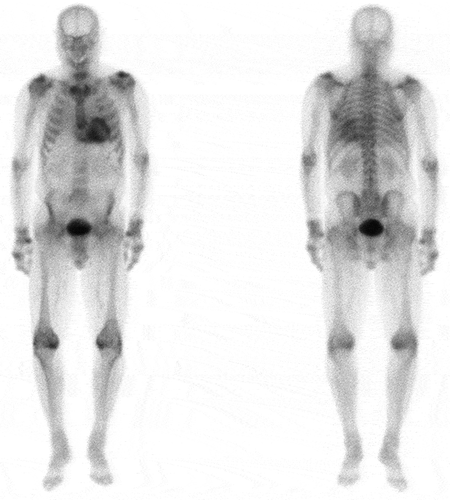
The role of bone scintigraphy as one component of an algorithm-based method for non-biopsy diagnosis of ATTR CM was highlighted in a seminal multicentre study. This diagnostic algorithm has been widely adopted in clinical practice, but needs to be followed to the letter in order for a secure diagnosis of ATTR-CM to be established [Citation47]. In a patient with heart failure and an echocardiogram or CMR suggesting amyloid cardiomyopathy, if the 99mTc-PYP/99mTc-DPD/99mTc-HMDP scan is reported to be grade 2 or 3 () and there is no evidence of a monoclonal protein by serum free light chain assay (i.e. the kappa/lambda ratio is within the normal polyclonal range) or by both serum and urine immunofixation, ATTR CM can be diagnosed without a biopsy (specificity and positive predictive value >98%) [Citation55]. There are a number of challenges with the non-biopsy diagnostic criteria for ATTR-CM including the ‘normal’ polyclonal serum free light chain ratio in the context of CKD which may be up to 3.1 [Citation56]. Similarly, there have been very occasional reports of both false negative bone scans (usually in association with particular and extremely rare pathogenic TTR variants) and false positive bone scans (possibly due to prior myocardial infarction or cardiac contusions). Overall however, the non-biopsy diagnostic algorithm for ATTR-CM appears to be extremely sensitive and specific for cardiac ATTR amyloidosis. Bone scintigraphy, if used in conjunction with the above tests for a monoclonal protein, also has a role in excluding cardiac amyloidosis although cannot do so entirely. However, in patients in whom there is no evidence of a monoclonal protein by serum free light chain assay together with negative serum and negative urine immunofixation, the absence of cardiac uptake on 99mTc-PYP (99m-Technetium labeledlabelled pyrophosphate, 99mTc-DPDDPD, or 99mTc-HMDP (99m-Technetium labeledlabelled hydroxymethylene diphosphonate) makes CA unlikely, although cannot exclude rare forms of cardiac amyloidosis such as apolipoprotein A-IV or apolipoprotein A-I amyloidosis. Similarly, if a patient has evidence of a plasma cell dyscrasia in the context of grade 2 or 3 cardiac uptake on bone scintigraphy, the differential diagnosis includes both cardiac AL and cardiac ATTR amyloidosis, such that definitive histological identification of the amyloid fibril protein by immunohistochemistry or proteomic analysis is required to establish the final diagnosis (). Likewise, the presence of low grade (grade 1) uptake on a 99mTc-PYP/99mTc-DPD/99mTc-HMDP scan requires further elucidation of the amyloid fibril protein and may be associated with cardiac AL, cardiac AApoAIAApoAI, or cardiac AA amyloidosis [Citation47,Citation57,Citation58].
Figure 5. A) 99mTc labeledlabelled DPD scintigraphy demonstrating Perugini Grade 2 cardiac uptake in a patient with ATTR-CM
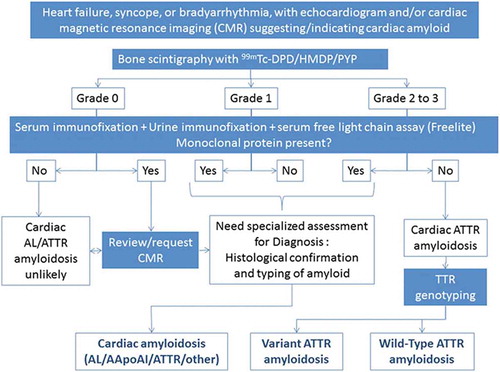
The use of radionuclide ‘bone’ scintigraphy to assess extra-cardiac involvement is a recently discovered and evolving phenomenon in patients with systemic amyloidosis. A typical pattern of muscle and soft-tissue uptake of 99mTc-DPD has been reported [Citation58] and has been further characterised by the evidence of amyloid deposition (by soft tissue biopsy) in a series of positive patients [Citation59]. Lung uptake may be present on 99mTc-HMDP scintigraphy [Citation60], although the clinical implications of such findings remain poorly understood.
4. Investigations for Clonal Disease
In systemic AL amyloidosis, deposits are comprised of monoclonal immunoglobulin light chains, driven by an often subtle underlying clonal dyscrasia. Thus in any patient suspected of systemic amyloidosis, a complete clonal assessment should be undertaken. Whilst monoclonal proteins can be detected in the serum and/or urine by electrophoresis and immunofixation (IFE) it is imperative that serum free light chain assay is also tested in all suspected patients since in up to 20% of patients with systemic AL amyloidosis; serum or urine IFE alone may not detect a monoclonal protein. Nonetheless it has been demonstrated that fully quantitative high sensitivity serum free light chain immunoassay (Freelite) has improved the sensitivity of detection of an underlying clone [Citation61]. The presence of a monoclonal protein is defined as an abnormal FLC ratio (<0.26 or > 1.65 in the context of a normal eGFR, and >3.1 in the context of advanced CKD) on serum freelite assay, or the presence of a monoclonal band on IFE of serum or urine. Recent data indicates that <1% of patients with systemic AL amyloidosis have no identifiable clonal marker on the basis of these three assays [Citation47]. Furthermore, the incidence of monoclonal gammopathy of unknown significance (MGUS) has been estimated to be ~5% in patients aged greater than 70 years and 7.5% in those 85 years or older [Citation62], which is particularly relevant in patients with wtATTR-CM who are often in their seventh or eighth decade and in whom the clonal disease may be incidental to the ATTR amyloid rather than underlie AL amyloid. For this reason, all patients with high grade cardiac uptake on bone scintigraphy who have a clonal disease should have the amyloid type confirmed histologically, often via endomyocardial biopsy, in order to prevent inappropriate delivery of potentially toxic chemotherapy for presumed cardiac AL amyloidosis to patients who in fact have ATTR-CM [Citation63].
5. Conclusion
In summary, cardiac ATTR amyloidosis can be reliably diagnosed in the absence of histology provided that all of the following criteria are met;
Heart failure with an echocardiogram or CMR that is consistent with or suggestive of amyloidosis
Grade 2 or 3 cardiac uptake on a radionuclide scan, using either 99mTc-DPD, 99mTc-PYPPYP, or 99mTc-HMDP
Absence of a detectable monoclonal protein despite serum and urine IFE, and sFLC (Freelite) assay
Histological confirmation and typing of amyloid should be sought in all cases of suspected cardiac amyloidosis in which these criteria are not met, ideally with an endomyocardial biopsy.
6. Expert Opinion
Whilst improved diagnostic techniques including work up for a clonal disorder, cardiac imaging, and repurposed scintigraphic assessments have made the non-invasive diagnosis of ATTR-CM easier, it remains a rare and challenging disease to diagnose. Misdiagnosis most commonly takes the form of incorrect typing of amyloid such that chemotherapy is inappropriately administered for mistakenly identified cardiac AL amyloidosis or alternatively, not administered for mistakenly identified ATTR-CM. In patients with a clonal disorder and high-grade cardiac uptake on radionuclide imaging, histological typing of amyloid, usually from an endomyocardial biopsy, remains essential.
Typically, wtATTR-CM (the commonest presentation of ATTR-CM) is a disease of elderly Caucasian men, who often give a history of sporting excellence or activity, with a preceding history of carpal tunnel syndrome or less commonly, spinal stenosis. Whilst vATTR secondary to V1221 presents with a similar clinical phenotype to wtATTR, the key differentiator is that of African or Afro-Caribbean heritage and less of a male preponderance. Presence of the V1221 allele has been shown to be a substantial risk factor for the development of ATTR-CM [Citation64]. This is in contrast to cardiac AL amyloidosis which is present in up to 60% of patients diagnosed with systemic AL amyloidosis but is often accompanied by extra-cardiac manifestations including soft tissue amyloid (manifesting as macroglossia) as well as proteinuric renal insufficiency and tends to manifest with a more severe and symptomatic amyloid cardiomyopathy. Despite this, differentiating between ATTR-CM and AL-CM in the absence of a histological diagnosis can be made more challenging by a number of factors. One such factor which frequently accompanies ATTR-CM is CKD. Progressive renal impairment is typically associated with disproportionate retention of kappa as opposed to lambda LC such that the normal polyclonal FLC ratio may be up to 3.1 in advanced CKD (eGFR <30 ml/min). Correcting the normal range for FLC ratio in CKD can help avoid the need for unnecessary endomyocardial biopsies [Citation56].
ATTR-CM is an emerging disease which may very well be a common cause of HFpEF in the elderly. Whilst not explored in this article, there have been a number of recent advances in treatment options for patients with ATTR-CM (both wild-type and variant) highlighting further the importance of accurate and rapid diagnosis of the disease.
In the coming years, with the increased use of both CMR and radionuclide ‘bone’ scintigraphy to diagnose and characterise cardiomyopathies and increasing awareness of cardiac amyloidosis within cardiology and geriatric communities alike, our prediction is that many more patients will attain an accurate and non-invasive diagnosis of ATTR-CM with only the more complex and challenging cases such as those who require an endomyocardial biopsy being referred to expert centres.
Article highlights
ATTR-CM is a progressive and fatal infiltrative cardiomyopathy typically manifesting with LV thickening and diastolic HF.
wtATTR-CM classically presents in elderly Caucasian men in their sixth, seventh or eighth decade with a preceding history of carpal tunnel syndrome and/or spinal stenosis.
Diagnosis is often missed or delayed due to the poor specificity of echocardiography and the absence of cardiac histology.
A combination of CMR, repurposed radionuclide ‘bone’ scintigraphy, and biochemical analyses permit non-invasive diagnosis of ATTR-CM with high specificity.
Recent advances in disease modifying therapies make the accurate and rapid diagnosis of ATTR-CM imperative.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers onf this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Lachmann HJ, Hawkins PN. Systemic amyloidosis. Curr Opin Pharmacol. 2006;6(2):214–220.
- Merlini G. Systemic amyloidosis: are we moving ahead? Neth J Med.The Netherlands Journal of Medicine. 2004;62(4):104–105.
- Gillmore JD, Hawkins PN. Pathophysiology and treatment of systemic amyloidosis. Nat Rev Nephrol. 2013;9(10):574–586.
- Pepys MB. Amyloidosis. In: Frank MM, Austen KF, Claman HN, et al., editors. Samter’s Immunologic Diseases. Fifth ed. Boston: Little, Brown and Company; 1994. p. 637–655.
- Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45–59.
- Benson MD. The hereditary amyloidoses. Best Pract Res Clin Rheumatol. 2003;17:909–927.
- Ruberg FL, Berk JL. Transthyretin (TTR) Cardiac Amyloidosis. Circulation. 2012;126(10):1286–1300.
- Pinney JH, Whelan CJ, Petrie A, et al. Senile Systemic Amyloidosis: clinical Features at Presentation and Outcome. J Am Heart Ass. 2013;2:2.
- Ansari-Lari MA, Ali SZ. Fine-needle aspiration of abdominal fat pad for amyloid detection: a clinically useful test? Diagn Cytopathol. 2004;30:178–181.
- Fine NM, Arruda-Olson AM, Dispenzieri A, et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol. 2014;113(10):1723–1777. .
- Maurer MS. Noninvasive Identification of ATTRwt cardiac amyloid: the re-emergence of nuclear cardiology. Am J Med. 2015;128(12):17.
- Cornwell GG 3rd, Murdoch WL, Kyle RA, et al. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75:618–623.
- Tanskanen M, Peuralinna T, Polvikoski T, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232–239. .
- Castano A, Manson DK, Maurer MS, et al. Transthyretin cardiac amyloidosis in older adults: optimizing cardiac imaging to the corresponding diagnostic and management goal. Curr Cardiovasc Risk Rep. 2017;11:11.
- Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–2594. .
- Carr AS, Pelayo-Negro AL, Evans MR, et al. A study of the neuropathy associated with transthyretin amyloidosis (ATTR) in the UK. J Neurol NeurosurgJournal of Neurology, Neurosurgery & Psychiatry. 2016;87(6):620–627. .
- Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 20132006;3696(92):819–829. .
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat.Human Mutation. 2014;35(9):E2403–E2412. .
- Jacobson DR, Alexander AA, Tagoe C, et al. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans African–Americans. Amyloid. 2015;22(3):171–174. .
- Castano A, Drachman BM, Judge D, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163–178. .
- Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005;112(13):2047–2060.
- Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic Resonance in Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol. 2017;70(4):466–477. .
- Gonzalez-Lopez E, Gagliardi C, Dominguez F, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J. 2017;38(24):1895–1904. .
- Murphy L, Falk RH. Left atrial kinetic energy in AL amyloidosis: can it detect early dysfunction? Am J Cardiol. 2000;86(2):244–246.
- Modesto KM, Dispenzieri A, Cauduro SA, et al. Left atrial myopathy in cardiac amyloidosis: implications of novel echocardiographic techniques. EurEuropean Heart J.Journal. 2005;26(2):173–179. .
- Feng D, Edwards WD, Oh JK, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116(21):2420–2426. .
- Martinez-Naharro A, Gonzalez-Lopez E, Corovic A, et al. High prevalence of intracardiac thrombi in cardiac amyloidosis. J Am Coll Cardiol. 2019;73(13):1733–1734.
- Chacko L, Martone R, Bandera F, et al. Echocardiographic phenotype and prognosis in transthyretin cardiac amyloidosis. Eur Heart J. 2020;41(14):1439–1447.
- Martinez-Naharro A, Hawkins PN, Fontana M. Cardiac amyloidosis. Clin Med (Lond).Clinical Medicine. 2018;18(Suppl 2):s30–s35.
- Falk RH, Quarta CC. Echocardiography in cardiac amyloidosis. Heart Fail Rev. 2015;20(2):125–131.
- Porciani MC, Lilli A, Perfetto F, et al. Tissue Doppler and strain imaging: a new tool for early detection of cardiac amyloidosis. Amyloid. 2009;16(2):63–70.
- Maceira AM, Joshi J, Prasad SK, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2005;111:186–193.
- Fontana M, Pica S, Reant P, et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2015;132(16):1570–1579.
- McDonald RJ, Levine D, Weinreb J, et al. Gadolinium retention: a research roadmap from the 2018 NIH/ACR/RSNA Workshop on Gadolinium Chelates. Radiology. 2018;289(2):517–534.
- McDonald RJ, McDonald JS, Kallmes DF, et al. Intracranial gadolinium deposition after contrast-enhanced MR Imaging. Radiology 2015;275(3):772–782.
- Kanda T, Fukusato T, Matsuda M, et al. Gadolinium-based contrast agent accumulates in the brain even in subjects without severe renal dysfunction: evaluation of autopsy brain specimens with inductively coupled plasma mass spectroscopy. Radiology 2015;276(1):228–232.
- Stojanov DA, Aracki-Trenkic A, Vojinovic S, et al. Increasing signal intensity within the dentate nucleus and globus pallidus on unenhanced T1W magnetic resonance images in patients with relapsing-remitting multiple sclerosis: correlation with cumulative dose of a macrocyclic gadolinium-based contrast agent, gadobutrol. Eur Radiol. 2016;26(3):807–815.
- Murata N, Gonzalez-Cuyar LF, Murata K, et al. Macrocyclic and other non-group 1 gadolinium contrast agents deposit low levels of gadolinium in brain and bone tissue: preliminary results from 9 patients with normal renal function. Invest Radiol. 2016;51(7):447–453.
- Karamitsos TD, Piechnik SK, Banypersad SM, et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging. 2013;6(4):488–497.
- Fontana M, Banypersad SM, Treibel TA, et al. Native T1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):157–165.
- Banypersad SM, Sado DM, Flett AS, et al. Quantification of myocardial extracellular volume fraction in systemic al amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. 2012;6(1):34-39.
- Martinez-Naharro A, Kotecha T, Norrington K, et al. Native T1 and extracellular volume in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2019;12(5):810–819.
- Fontana M, Martinez-Naharro A, Chacko L, et al. Reduction in CMR derived extracellular volume with patisiran indicates cardiac amyloid regression. Jacc Cardiovasc Imaging. 2021;14(1):189–199.
- Ferreira VM, Piechnik SK, Robson MD, et al. Myocardial tissue characterization by magnetic resonance imaging: novel applications of T1 and T2 mapping. J Thorac Imaging. 2014;29(3):147–154.
- Kotecha T, Martinez-Naharro A, Treibel TA, et al. Myocardial edema and prognosis in amyloidosis. J Am Coll Cardiol. 2018;71(25):2919–2931.
- Ali A, Turner DA, Rosenbush SW, et al. Bone scintigram in cardiac amyloidosis: a case report. Clin Nucl Med. 1981;6(3):105–108.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412.
- Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076–1084.
- Rapezzi C, Guidalotti P, Salvi F, et al. Usefulness of Tc-99m-DPD scintigraphy in cardiac amyloidosis. J Am Coll Cardiol. 2008;51(15):1509–1510.
- Rapezzi C, Quarta CC, Guidalotti PL, et al. Usefulness and limitations of 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging. 2011;38(3):470–478.
- Bokhari S, Castano A, Pozniakoff T, et al. 99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6(2):195–201.
- Quarta CC, Guidalotti PL, Longhi S, et al. Defining the diagnosis in echocardiographically suspected senile systemic amyloidosis. JACC Cardiovasc Imaging. 2012;5(7):755–758.
- Glaudemans AW, Van Rheenen RW, Van Den Berg MP, et al. Bone scintigraphy with (99m)technetium-hydroxymethylene diphosphonate allows early diagnosis of cardiac involvement in patients with transthyretin-derived systemic amyloidosis. Amyloid. 2014;21(1):35–44.
- Castano A, Bokhari S, Maurer MS. Unveiling wild-type transthyretin cardiac amyloidosis as a significant and potentially modifiable cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2595–2597.
- Maurer MS, Bokhari S, Damy T, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12(9):e006075.
- Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol. 2008;3(6):1684–1690.
- Falk RH, Lee VW, Rubinow A, et al. Sensitivity of technetium-99m-pyrophosphate scintigraphy in diagnosing cardiac amyloidosis. Am J Cardiol. 1983;51(5):826–830.
- Hutt DF, Quigley A-M, Page J, et al. Utility and limitations of 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in systemic amyloidosis. Eur Heart J Cardio Imag. 2014;15(11):1289–1298.
- Hutt DF, Fontana M, Burniston M, et al. Prognostic utility of the Perugini grading of 99mTc-DPD scintigraphy in transthyretin (ATTR) amyloidosis and its relationship with skeletal muscle and soft tissue amyloid. Eur Heart J Cardiovasc Imaging. 2017;18(12):1344–1350.
- Cappelli F, Gallini C, Costanzo EN, et al. Lung uptake during 99mTc-hydroxymethylene diphosphonate scintigraphy in patient with TTR cardiac amyloidosis: an underestimated phenomenon. Int J Cardiol. 2018;254:346–350.
- Lachmann HJ, Gallimore R, Gillmore JD, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol. 2003;122(1):78–84.
- Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369.
- Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–1791.
- Gillmore JD, Hawkins PN. V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(18):1769.