
ABSTRACT
Introduction
Primary hyperoxaluria type 1 (PH1) is an inborn error of glyoxylate metabolism whereby excessive endogenous oxalate production can result in kidney failure, systemic oxalosis and early death. The approval of the first therapeutic agent to alter the treatment pathway for PH1 – lumasiran, a novel RNA interference-based drug – represents a new era in the management of patients with PH1.
Areas covered
A description of PH1, its pathophysiology, clinical characteristics and available treatment strategies is provided, with a focus on substrate reduction therapy and the development of lumasiran for pediatric and adult patients with PH1.
Expert opinion
Until very recently, treatment options for patients with PH1 have been burdensome, ineffective in preventing disease progression or associated with high morbidity. Lumasiran targets hepatic glycolate oxidase, the enzyme responsible for the synthesis of the oxalate precursor, glyoxylate. Administered subcutaneously monthly initially and then quarterly, it has been found to significantly lower urinary and plasma oxalate levels, potentially reduce the development of nephrocalcinosis and halt disease progression. For the first time, patients with PH1 have a treatment that is well tolerated, effective, and administered in a way that does not adversely impact the quality of life of those affected by this devastating disease.
1. Introduction
Primary hyperoxaluria type 1 (PH1) is a rare inborn error of glyoxylate metabolism, caused by a deficiency of the peroxisomal enzyme alanine-glyoxylate aminotransferase (AGT) that results in excessive oxalate production [Citation1,Citation2]. The incidence of PH1 is estimated at approximately 1:120,000 live births, and prevalence at 1–3 per million population in Europe and North America [Citation3,Citation4], although frequency estimates are likely to be skewed due to underdiagnosis [Citation5,Citation6]. Clinical presentation, even within affected families, can vary widely and range from occasional symptomatic urolithiasis to nephrocalcinosis and end-stage renal disease with systemic involvement [Citation7,Citation8].
In addition to PH1, at least two other types of primary hyperoxaluria, PH2 and PH3, have been genetically confirmed with deficiencies in the cytosolic glyoxylate reductase/hydroxypyruvate reductase [Citation9] and mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzymes, respectively [Citation10,Citation11]. PH1 is the most common (accounting for around 80% of cases) and the most severe type of primary hyperoxaluria [Citation12,Citation13], and therefore the most clinically relevant. Its importance is further underscored by the fact that it is responsible for 1–2% of pediatric patients with kidney failure in Europe and North America [Citation14–16], and up to 17% in certain populations with a high rate of consanguinity [Citation17,Citation18].
Until recently, treatment strategies have focused on minimizing calcium oxalate deposition to slow disease progression through hyperhydration, high-dose pyridoxine and/or citrate therapy [Citation1]. These are burdensome for patients and do not prevent progressive kidney failure, although pyridoxine therapy may delay or avert renal impairment in responsive patients [Citation19,Citation20]. Combined liver and kidney transplantation is recommended for patients with stage 4 chronic kidney disease [Citation1], which although potentially curative, is associated with significant mortality risk and a lifelong requirement for immunosuppressants [Citation21–23].
In late 2020, lumasiran (Oxlumo™, Alnylam Pharmaceuticals, Inc.) became the first drug to be approved in the USA and Europe for the treatment of PH1 [Citation24,Citation25]. The availability of this subcutaneously administered RNA interference (RNAi) therapeutic agent is transforming the way patients with PH1 are being managed, potentially offering the opportunity to improve both disease control and quality of life. This article reviews the underlying pathophysiology of PH1 and aims to explain why a novel targeted approach such as lumasiran is much needed, providing evidence for its efficacy and improving outcomes in patients with this devastating disease.
2. Pathophysiology
2.1. Normal glyoxylate metabolism
Glyoxylate is generated during glycine and hydroxyproline catabolism that occur in the peroxisome and mitochondrion, respectively. In humans, glycolate is an important precursor [Citation26], with peroxisomal glyoxylate produced from the activity of glycolate oxidase (GO, a flavin mononucleotide-dependent-α-hydroxy acid oxidase encoded by the HAO1 gene). AGT, a pyridoxal 5ʹ phosphate dependent enzyme, catalyzes the metabolism of peroxisomal glyoxylate by transferring the amino group from alanine to glyoxylate to produce pyruvate and glycine. Alternatively, excess peroxisomal glyoxylate is transported to the cytoplasm, where it is either reduced to glycolate by glyoxylate reductase hydroxypyruvate reductase (GRHPR) or oxidized to oxalate by L-lactate dehydrogenase (LDH). Oxalate cannot be further metabolized and is excreted in urine.
2.2. Glyoxylate metabolism in PH1
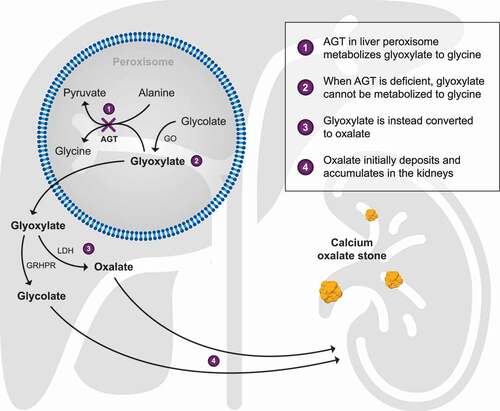
For patients with PH1, the deficiency in AGT activity means that glyoxylate cannot efficiently convert to glycine but instead it is oxidized to oxalate by cytosolic lactate dehydrogenase (). Consequently, urinary oxalate excretion is very high, typically >1 mmol/1.73 m2 per day (normal <0.5 mmol/1.73 m2 per day) [Citation8], and since oxalate is poorly soluble, calcium oxalate crystals form within the tubular renal lumen when the urine becomes supersaturated [Citation7,Citation8,Citation27,Citation28]. Some crystals aggregate to form stones, while others adhere to, and are internalized into tubular epithelial cells, with subsequent extrusion into the interstitial region, causing an inflammatory reaction and nephrocalcinosis [Citation8,Citation29].
Figure 1. Consequences of the block (X) in glyoxylate metabolism when AGT is deficient
2.3. Genetic features
AGT deficiency in PH1 is due to genetic mutations in the AGXT gene that are inherited in an autosomal recessive manner [Citation30]. To date, more than 200 disease-causing mutations along with many polymorphic variants have been described (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk). Common mutations in the AGXT gene include c.508 G > A (p.G170R), c33_34insC (p.K12QfsX156), and c.731 T > C (p.1244 T) [Citation8], accounting for at least one of the two affected alleles in more than 60% of patients with PH1 [Citation31]. Most mutations result in AGT mistargeting, low catalytic activity, and reduced or negligible AGT expression [Citation32–34]. Interestingly, the c.508 G > A mutation is found in conjunction with a polymorphic allele c.32 C > T (p.Pro1 1Leu) and the combination leads to a functional protein expressed in the mitochondrion. However, in the mitochondrion AGT is metabolically inefficient but can be rescued to a certain extent by the presence of pyridoxine, a co-factor for AGT [Citation35,Citation36]. As well as an enzymatic phenotype–genotype correlation, there appears to be significant clinical phenotype–genotype associations, with, for example, age at onset of symptoms and age at end-stage renal failure correlating with the mutation type [Citation20,Citation37].
3. Clinical characteristics
The clinical presentation of PH1 is heterogeneous. The median age at onset of symptoms is 4–6 years but ranges from infancy to after the sixth decade of life [Citation5,Citation6,Citation20,Citation37]. Most patients present with symptoms related to recurrent urolithiasis, with or without nephrocalcinosis; however, some present with a severe, early-onset form in infancy that is often characterized by a failure to thrive because of end-stage renal disease [Citation38]. At the other end of the spectrum, some individuals remain asymptomatic or minimally symptomatic into their sixth decade of life and beyond [Citation13], although more than half of these patients present with end-stage renal failure at the time of diagnosis [Citation5]. Occasionally, a diagnosis is made following failure of kidney transplantation [Citation39,Citation40], a situation that should be entirely avoidable.
The natural history of PH1 is one of progressive decline in renal function as calcium oxalate continues to deposit in kidney tissue, and complications such as obstruction and infection occur. Once glomerular filtration rate falls below 30–40 mL/min/1.73 m2 body surface area, renal oxalate excretion becomes compromised to a point where plasma concentrations exceed the supersaturation threshold (>30 mol/L) [Citation7,Citation8,Citation28]. At this stage, systemic deposition of calcium oxalate (oxalosis) occurs in extra-renal tissues, including the bone, eyes, vasculature, heart, skin, and central nervous system, among others (see ) [Citation8,Citation41–44]. Systemic complications cause high morbidity, severely impacting patient quality of life, and, if untreated or treated late, result in early death.
Table 1. Frequency of organ involvement due to systemic oxalosis
4. Treatment
Treatments for PH1 focus on preserving renal function by minimizing oxalate production and deposition, with the aim of preventing associated complications.
4.1. Conservative therapies
For many years, the standard of care for patients with preserved renal function has been conservative therapy. This includes aggressive hydration (≥3 L/m2/24 h) to maintain a high urine output, oral potassium citrate to inhibit calcium oxalate crystallization, and pyridoxine treatment to lower urine oxalate excretion [Citation1]. For adequate preservation of renal function, early diagnosis and prompt initiation of such therapies are critical [Citation45].
However, hyperhydration is burdensome, especially for young children, some of whom may require nasogastric or gastrostomy fluid delivery if they are unable to adhere to therapy, while only a small minority of patients with susceptible genotypes (c.508 G > A [p.G170R], and c.454 T > A [p.F153I]), respond to pyridoxine treatment [Citation19,Citation36]. Although patients may benefit in the short term, conservative therapy cannot stop progression to end-stage renal disease over time.
4.2. Dialysis and transplantation
To date, for patients with advanced or end-stage kidney disease, combined liver and kidney transplantation has been the treatment of choice [Citation1]. Dialysis is ineffective for this group of patients as it cannot overcome the rate of oxalate production [Citation27,Citation46]. However, dialysis is required in practice as a bridge to transplantation, where most patients need intensive therapy. Hemodialysis removes oxalate more efficiently than peritoneal dialysis [Citation47] but significant oxalate rebound occurs within 24 h to 80% of pre-dialysis levels [Citation48]. A combination of intermittent daily hemodialysis and overnight peritoneal dialysis was previously used to improve oxalate removal, but home hemodialysis, including overnight dialysis, is now the recommended approach [Citation49]. While liver transplantation is potentially curative, it is associated with many challenges: worldwide, there is an organ shortage, delay in access to transplantation; undertaking the procedure places a significant physical, psychological, and financial burden on patients and their families, and is associated with significant morbidity and mortality. The latest updates from the OxalEurope registry report data from 244 patients who underwent transplantation (including 196 combined and sequential liver-kidney transplantations) between 1978 and 2019. The median age at transplantation was 16.8 years, and in all groups, the 10-year survival rate was 80% [Citation50]. This high mortality at a young age indicates the severity of PH1, and while ethically justified, it is hard to contemplate transplantation to be an optimal approach for such a young patient population based on the existing evidence.
4.3. Targeted therapy
To address the unmet need for an efficacious and durable treatment for PH1 (and the other types of PH), novel therapeutic approaches are being developed. These are aimed at different targets, either focused on reducing oxalate levels or minimizing renal damage subsequent to calcium oxalate crystallization, and include strategies such as gene, protein and cell therapies, small drug administration (chaperones and enzyme inhibitors), and the use of oxalate-degrading bacteria and enzymes. Substrate reduction therapy has been used successfully in other inborn errors of metabolism [Citation49,Citation51], and in theory, depleting glyoxylate, the main precursor of oxalate, by inhibiting one or more of the glyoxylate biosynthetic enzymes should be of therapeutic benefit in patients with PH1. GO was identified as a candidate target [Citation52] – not only is it chemically upstream from AGT, where the gene defect responsible for PH1 is located (), but any subsequent buildup of glycolate is readily excreted in urine at high concentrations without causing kidney damage or other toxicity [Citation52]. Other successful enzyme targets in PH1 include lactate dehydrogenase A (the major hepatic form) but are beyond the scope of this review [Citation53–55].
Figure 2. By silencing HAO1 and depleting GO, lumasiran inhibits oxalate production
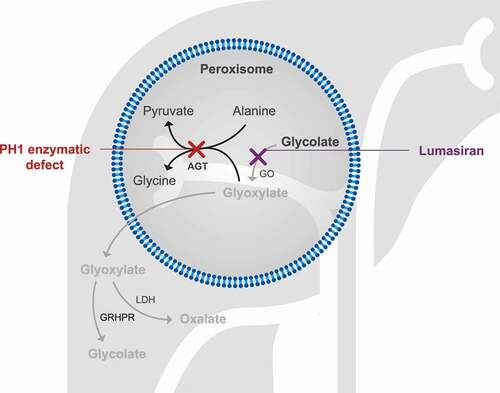
GO can be targeted in different ways to potentially treat PH1. Chemical inhibition of its active site has been investigated and although some candidate molecules have been identified, none have advanced to clinical trial [Citation56]; RNAi therapy has proven more successful. This new class of medicines harnesses the natural biological process of RNAi that is present in most eukaryotic cells to silence genes [Citation57,Citation58]. Lumasiran, developed by Alnylam Pharmaceuticals, is an RNAi therapy that works by suppressing the expression of the hydroxyacid oxidase 1 (HAO1) gene encoding GO, and thus acts independently of the underlying disease-causing AGT mutation. It is one of four currently approved RNAi-based therapies and the only licensed therapy for PH1 [Citation24,Citation25].
4.3.1. Lumasiran properties and mechanism of action
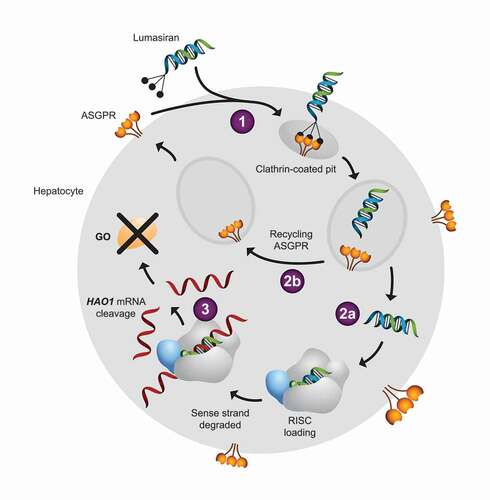
Lumasiran is a synthetic double-stranded small interfering RNA (siRNA), covalently linked to a triantennary N-acetylgalactosamine (GalNAc) [Citation59,Citation60]. It uses the endogenous RNAi pathway to target the 3ʹ untranslated region of hepatic HAO1 mRNA, resulting in its degradation (). In this manner, lumasiran prevents the translation of the corresponding GO protein. The presence of the GalNAc moiety conjugated to the sense strand of the siRNA promotes hepatic uptake via the asialoglycoprotein receptors (ASGPR), which have high affinity for GalNAc residues and are highly expressed on surface of hepatocytes [Citation61]. Binding of the GalNAc ligand of lumasiran to ASGPR triggers receptor-mediated endocytosis, allowing delivery of the siRNA to the cytoplasm for incorporation into the RNA-induced silencing complex. ASGPR is subsequently recycled to the cell surface for successive uptake of circulating lumasiran. Within the RNA-induced silencing complex, the antisense strand of lumasiran binds to the complementary sequence of the HAO1 mRNA, resulting in its specific and highly efficient cleavage. The lack of intact HAO1 mRNA prevents the synthesis of glycolate oxidase, thereby suppressing oxalate production because of the reduction in its precursor, glyoxylate.
Figure 3. Mechanism of action of lumasiran within hepatocytes. Lumasiran is a GalNAc-double-stranded RNA conjugate. (1) The GalNAc residues of lumasiran bind to ASGPR receptors, leading to endocytosis of the double-stranded RNA. Within the cytoplasm, (2a) the double-stranded RNA undergoes endocytic escape and is loaded into the RISC complex while (2b) ASGPR is recycled and returns to the cell surface for another round of lumasiran uptake. (3). RISC is guided to the HAO1 mRNA by homology to the RNA strand, cleaving the HAO1 RNA, and preventing translation to GO enzyme
As an RNAi-based therapy, lumasiran is an attractive option for PH1. The fact that the metabolic defect in PH1 occurs in the liver is a critical advantage for this modality. In general, delivering molecules to the liver is relatively easy, but it is the opportunity to leverage the abundance of ASGPRs to 1) selectively deliver siRNAs into hepatocytes; 2) limit off-target effects, and 3) mediate multiple rounds of GalNAc-siRNA conjugate uptake (assumed to be non-saturable at exposures in the therapeutic range), which makes lumasiran suitable for PH1. The proof-of-concept that HAO1-targeted siRNAs can reduce oxalate production was initially demonstrated preclinically in healthy rodents and animal models of PH1 [Citation62–64]. Are there consequences associated with an intervention involving lifelong knockdown of HAO1 for patients? At present, it appears unlikely. In rare cases of biallelic loss of function variants of HAO1, individuals do not have a disease phenotype despite high serum and urine glycolate levels [Citation65,Citation66], supporting the long-term tolerability of this approach.
4.3.2. Pharmacokinetic and pharmacodynamic properties
Following subcutaneous administration, pharmacokinetic studies have shown that lumasiran is rapidly absorbed, with a median time to reach maximum plasma concentrations of 4 h (range 0.5–12 h), and remains detectable up to 24–48 h post dose [Citation59,Citation60]. At clinically relevant concentrations in healthy volunteers, around 77–85% of lumasiran is bound to protein, and in adult patients the population estimate for the apparent central volume of distribution is 4.9 L. Lumasiran is rapidly eliminated from the plasma and taken up by the liver, with urinary excretion accounting for only 7–26% of the administered dose. The rapid targeted uptake by the liver results in a correspondingly rapid decline in plasma lumasiran levels, and there is no accumulation in the plasma after repeated once monthly or quarterly administration [Citation59,Citation60]. In the liver, lumasiran has a long half-life that allows it to maintain the pharmacodynamic effect of the drug over both dosing intervals. Consequently, plasma concentrations do not reflect the extent or duration of its activity; the mean terminal plasma half-life is 5.2 h. Lumasiran is metabolized by endo- and exonucleases to shorter length oligonucleotides but, according to in vitro studies, it does not undergo metabolism by CYP450 enzymes, inhibit or induce them, or modulate drug transporters [Citation59,Citation60]. Steady-state pharmacokinetics are not influenced by age, race or sex of the patient, mild/moderate renal impairment (estimated glomerular filtration rate [eGFR] ≥30 to <90 mL/min/1.73 m2) or mild/moderate hepatic impairment (bilirubin ≤1 x upper limit of normal [ULN]; aspartate aminotransferase >1 x ULN; bilirubin >1.0–1.5 x ULN), implying that lumasiran is suitable for most patient populations [Citation59,Citation60].
Dose-dependent increases in plasma glycolate levels are observed in healthy volunteers, providing an informative measure of target engagement. Median time to recovery (plasma glycolate <20% above baseline or ≥ ULN) after a single dose has been shown to be 134.5 (range 57–225) days at 3 mg/kg and 169 (range 113–242) days at 6 mg/kg [Citation67]. Urinary glycolate shows a similar response. No effect can be seen with the lowest dose of 0.3 mg/kg [Citation67]. In patients with PH1, dose-dependent reductions in urinary and plasma oxalate levels are observed. The reduction in 24-h urinary oxalate excretion is rapid and sustained over time, reaching within normal (≤0.46 mmol/24 h/1.73 m2) or near normal (≤1.5 x ULN, ≤0.69 mmol/24 h/1.73 m2) ranges [Citation67]. The effect of lumasiran at the recommended dosing regimens is rapid and can be observed within 2 weeks after the first dose, and maximal reductions in urinary and plasma oxalates are observed by the end of the loading dose phase and sustained with the maintenance dose regimen.
The incidence of treatment-emergent antibodies is low (6%) with low antibody titers (1:50). Their presence is transient and does not influence the pharmacokinetics and pharmacodynamics of lumasiran. Similarly, concomitant administration of pyridoxine does not affect the pharmacokinetics, pharmacodynamics or dosage of lumasiran [Citation60], therefore offering the possibility of combination treatment.
4.3.3. Clinical evidence for lumasiran
The clinical efficacy of lumasiran has been established in five clinical trials in 98 patients with PH1 [Citation67–73].
A phase 1/2 study evaluated multiple doses of lumasiran in a single-blind, placebo-controlled trial (NCT02706886) involving 20 adult and pediatric patients with PH1 who were randomized 3:1 to receive doses at 1 or 3 mg/kg per month or 3 mg/kg quarterly, or placebo [Citation67]. Following treatment (days 1–197), the mean maximum reduction in 24-h urinary oxalate excretion levels was 75% (range 43–92) from baseline (1.69 mmol/24 h/1.73 m2) with 15/20 (75%) patients achieving 24-h urinary oxalate excretion within the normal range (≤0.46 mmol/24 h/1.73 m2). Among patients receiving 3 mg/kg monthly or quarterly doses of lumasiran, 11/12 (92%) achieved 24-h urinary oxalate levels within the normal range. Data available for some patients (n = 9) showed that the mean maximum reduction in plasma oxalate concentrations was 74% (range 38–94) from baseline [Citation67]. Lumasiran was generally well tolerated in patients with PH1. Most adverse events (AEs) were mild to moderate in severity, with abdominal pain (18%), headache (18%), rhinitis (12%), nephrolithiasis (12%), and cough (12%) the most reported. There was one severe AE in the placebo group and no severe AEs in the lumasiran group. Two patients had AEs related to lumasiran, which consisted of mild or moderate transient injection-site reactions [Citation67]. Following study completion, all 20 patients were enrolled in a long-term, open-label extension Phase 2 study for continued dosing (NCT03350451). Further follow-up of patients, dosed in the OLE for a median of 15 (range: 11–22) months, showed that 17/18 (94%) achieved normal or near normal (≤1.5 x ULN) levels of urinary oxalate levels. Sustained reductions in urinary oxalate:creatinine ratios were observed across the different dosing regimens, reaching a mean maximal reduction in urinary oxalate:creatinine ratio of 77.5% (range 55.3–95.8, n = 20). Mean eGFR values were stable over time. Plasma oxalate levels were also decreased (mean maximum reduction 55.2%, n = 19), while plasma and urinary glycolate increased and later stabilized, consistent with the effect of lumasiran on GO. The most common drug-related AEs reported were mild, transient injection-site reactions. No discontinuations, treatment-related SAEs or AEs of nephrolithiasis were reported during this period, consolidating the safety of up to 22 months of exposure to lumasiran [Citation68,Citation69].
ILLUMINATE-A was a randomized, double-blind, placebo-controlled trial (NCT03681184) of lumasiran treatment in 39 patients ≥6 years of age with PH1 and an eGFR ≥30 mL/min/1.73 m2 [Citation70]. Patients received three loading doses of 3 mg/kg lumasiran (n = 26) or placebo (n = 13) administered once a month, followed by quarterly maintenance doses of 3 mg/kg lumasiran or placebo beginning 1 month after the last loading dose. A rapid and sustained reduction in 24-h urinary oxalate excretion was observed in patients treated with lumasiran, with decreases seen at month 1 and steady-state reached by the end of the loading-dose phase. The least squares (LS) mean percent change in 24-h urinary oxalate from baseline to Month 6 in those receiving lumasiran was –65.4% (95% confidence interval [CI]: –71.3 to –59.5) compared with those receiving placebo –11.8% (95% CI: –19.5 to –4.1), resulting in a between-group LS mean difference of –53% (95% CI: –62.3 to –44.8; p < 0.001). This treatment effect was observed across all prespecified subgroups, including baseline 24-h urinary oxalate excretion, baseline pyridoxine use, and baseline eGFR. By month 6, 52% (95% CI: 31–72) of patients treated with lumasiran achieved a normal (≤0.514 mmol/24 h/1.73 m2) 24-h urinary oxalate level compared with 0% (95% CI: 0–25) of patients receiving placebo (p = 0.001). Plasma oxalate levels also declined in lumasiran-treated patients; the LS mean percentage change from baseline to month 6 was –39.8% (–45.8 to –33.8) in the lumasiran group compared with –0.3% (–9.1 to 8.5) in the placebo group, and LS mean difference of –39.5% (95% CI: –50.1 to –28.9; p < 0.001). The eGFR remained stable in both groups during the 6-month period of treatment. Plasma glycolate levels initially increased and then plateaued in lumasiran-treated patients, consistent with a reduction in hepatic GO production due to the action of lumasiran [Citation70]. Overall, 85% and 69% of patients who received lumasiran and placebo, respectively, reported AEs. All AEs were mild or moderate in severity and no SAEs or deaths were reported; the most common AEs reported were related to injection-site reactions. These occurred more frequently in the lumasiran compared with placebo group (38% vs 0%, respectively) but were mild and transient and did not result in treatment discontinuation [Citation70]. One patient in the lumasiran group discontinued treatment because of fatigue and disturbance in attention. The parent of a second young patient stopped their participation due to needle phobia and inability to comply with protocol-specific testing.
The effect of lumasiran in patients <6 years of age with PH1 and an eGFR >45 ml/min/1.73 m2 for those ≥12 months of age or a normal serum creatinine for patients <12 months of age was evaluated in the single-arm phase 3 ILLUMINATE-B trial (NCT03905694) [Citation71,Citation72]. Similar to the ILLUMINATE-A trial, patients (n = 18) received 3 x monthly, then monthly or quarterly doses of lumasiran. The median age and body weight of patients was 50 (range 3–72) months and 14.5 kg (range 6.2–24.3), respectively. Lumasiran led to an LS mean reduction from baseline of 72.0% in spot urinary oxalate:creatinine ratio, averaged over months 3–6 [Citation72]. Urinary oxalate levels were reduced by 84.2% in patients <10 kg (n = 3), by 69.1% in patients 10 to <20 kg (n = 12), and by 69.7% in patients ≥20 kg (n = 3). At month 6, 9/18 (50%) patients achieved urinary oxalate excretion ≤1.5 x ULN, with 1 (6%) patient achieving normal (≤0.514 mmol/24 h/1.73 m2) urinary oxalate levels at this time point. The LS mean percentage change in plasma oxalate levels from baseline to month 6 (average of months 3–6) was –31.7% (95% CI: –39.5 to –23.9). The eGFR, calculated only in patients ≥12 months of age, remained stable from baseline to month 6. Of the 14 (78%) patients with baseline nephrocalcinosis, three had bilateral improvement, five had unilateral improvement, and no patients worsened after 6 months of treatment. There were no deaths, discontinuations or withdrawals, or SAEs. The most common treatment-related AE was injection-site reactions in 3/18 (17%) patients, all considered mild and transient [Citation72].
Most recently, lumasiran has been evaluated in patients with PH1 of all ages with severe renal impairment (eGFR ≤45 mL/min/1.73 m2 or elevated serum creatinine for patients <12 months of age) [Citation73]. The ILLUMINATE-C trial (NCT0452200) was a single-arm, open-label phase 3 study involving 21 patients, 15 of whom required hemodialysis. All patients were treated with 3 x monthly doses followed by ongoing monthly or quarterly doses. Top-line results released thus far showed that, at month 6, lumasiran treatment resulted in a substantial reduction in plasma oxalate in both dialysis-dependent and -independent patients [Citation73]. In addition, positive results across key secondary endpoints including measures of urinary oxalate (in dialysis-independent patients) were observed. There were no deaths or treatment-related SAEs, but two treatment-related discontinuations due to AEs were reported, although neither were drug related. The most common AEs were injection-site reactions, reported in 5/21 (23.8%) patients, all of which were mild [Citation73].
4.3.4. Lumasiran in clinical practice
Lumasiran is currently indicated for the treatment of PH1 to reduce urinary oxalate levels in pediatric and adult patients. The recommended dosing regimen consists of loading doses (once monthly for three doses) followed by maintenance doses (<10 kg body weight, once monthly; ≥10 kg body weight, once quarterly) administered subcutaneously, with dosing based on actual body weight (see ) [Citation59,Citation60].
Table 2. Recommended dosing regimen based on body weight
5. Conclusion
PH1 is an ultra-rare disease in which excessive oxalate production leads to compromised kidney function. Reducing oxaluria and oxalemia with appropriate therapeutic strategies are critical for preventing or reducing renal and systemic complications. Until recently, treatment options were limited to conservative therapies that are burdensome for patients and do not prevent disease progression, frequent dialysis that is largely ineffective in removing sufficient amounts of oxalate, and combined liver and kidney transplantation; a procedure, which although potentially curative, is associated with high morbidity and reliant on organ availability. Lumasiran, an RNAi therapeutic, is the first treatment to be approved for PH1. Its action, by targeting GO to inhibit hepatic oxalate production, directly addresses the underlying cause of PH1. Evidence to date from the ILLUMINATE-A, -B and -C trials show that lumasiran is associated with a substantial reduction in urinary oxalate levels in patients with PH1 aged between 3 months and 60 years and is well tolerated. However, longer-term data are needed to determine how effective lumasiran is in halting the course of the disease and in preventing serious kidney and systemic complications, as well as the burden and potential consequences of potentially lifelong treatment. Ongoing trials will also determine whether lumasiran is effective in those who already have kidney failure.
6. Expert opinion
New therapies that address the underlying cause of PH1 and have the potential for a favorable impact on disease manifestations have long been sought after. The availability of lumasiran in the USA and Europe represents a significant step change in the management of PH1, and further supports the notion that RNAi-based therapeutics are poised to become a standard modality of pharmacotherapy.
One of the challenges of using siRNAs is their delivery to their intended target. Initial advances in siRNA delivery were made using multicomponent systems such as lipid nanoparticles and polymers. While useful, multicomponent systems are encumbered by the need for intravenous administration and, in the case of lipid nanoparticles, steroid pre-medication to mitigate infusion-related reactions. Lumasiran utilizes a single-component siRNA delivery system by conjugating to GalNAc and exploiting the ASGPR system to increase the efficiency of delivery to hepatocytes. From the patient perspective, this delivery system is amenable to subcutaneous administration and therefore suitable for those who might be expected to have low adherence (e.g. adolescents) or difficulty with regular intravenous therapies (e.g. infants). Another benefit of siRNA therapeutics is that they are administered infrequently (quarterly for lumasiran after three loading doses are complete), can be administered in the home environment by a health care professional, and have the potential to perhaps be self-administrable in the future. For patients with PH1, their caregivers and families, this would be a welcome change from the burden associated with the frequency of conservative therapies and dialysis.
Successful knockdown of GO by lumasiran, and therefore reduction in hepatic oxalate production, is currently measured through plasma and urinary oxalate levels. However, accurate measurement of plasma and urine oxalate are not without their challenges: urine oxalate requiring a 24-h collection that may be difficult in infants and young children, and in practice, differences in sample preservation and analytical methods can cause variability in results between centers performing the tests. To negate the analytical problems, individual biological variability should be established by collecting three or four 24-h samples prior to the start of treatment, and all samples from individual patients should be analyzed in the same laboratory. The use of spot urine, or samples collected over a short timeframe, reporting the oxalate-to-creatinine ratio must be considered as a tool in longer term follow-up. However, it is important to remember that normal ranges are considerably higher in pediatric patients than in adults and vary in different age brackets. A reduction in calcium oxalate deposits, assessed by ultrasonography, computerized tomography or magnetic resonance, could also be used to measure treatment response.
If lumasiran significantly and consistently reduces urine oxalate as it currently appears to do, then provided it is started early in the treatment pathway, it is likely to obviate the need for combined liver–kidney transplantation. On the other hand, if lumasiran is commenced after established renal impairment, its role may be to avoid the need for liver transplantation by allowing dialysis to remove systemic oxalate until it is safe to proceed with isolated renal transplantation. Where possible, the first scenario would be the preferred way of utilizing lumasiran; however, delays in diagnosis remain a matter of concern worldwide and considerable efforts are still needed to increase awareness and facilitate early and accurate diagnosis of PH1.
Data from the ongoing Phase 2 OLE and extension period of the ILLUMINATE-A and ILLUMINATE-B studies are much anticipated as they will determine the long-term safety and clinical outcomes of lumasiran including rate of kidney stone events, changes in nephrocalcinosis and eGFR, and impact on quality of life. If initial data on the reduction in the development of nephrocalcinosis are confirmed, this will have an important impact on the progression of chronic kidney disease. More comprehensive data from ILLUMINATE-C will provide insight on whether lumasiran could have an impact on patients with severe renal impairment, including those on dialysis, with a role either to prevent the need for dialysis, or to proceed to isolated renal transplantation for those on dialysis.
A major drawback of lumasiran is its perceived high cost, which, together with the need for repeated dosing, means that the annual treatment costs are significant and beyond the reach of most patients worldwide. However, dialysis of the frequency required in attempting to manage oxalosis is also very expensive. Taken together with the costs of transplantation and associated immunosuppressive drugs, four annual doses of lumasiran may well be considered cost-effective if lumasiran is confirmed to prevent or potentially reverse systemic oxalosis in patients with renal impairment, removing the need for organ transplantation. The collection of real-world data is critical to better understand how lumasiran performs in practice and its value in the treatment of PH1.
Drug summary box
Drug name: lumasiran
Phase: III Indication: Primary Hyperoxaluria Type 1
Date and location of ODD: US FDA February 2016; EMA March 2016 Pharmacology description/MoA: RNA interference
Route of administration: Subcutaneous injection
Pivotal trials: ILLUMINATE-A: Garrelfs SF et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med. 2021 Apr 1;384(13):1216-26. ILLUMINATE-B: ILLUMINATE-B, a phase 3 open-label study to evaluate lumasiran, an RNAi therapeutic, in young children with primary hyperoxaluria type 1 (PH1) [Abstract]. J Am Soc Nephrol. 2020;31:515
Article highlights
PH1 is responsible for 1–2% of pediatric patients with kidney failure in Europe and North America, and of considerable clinical importance despite its rarity
Conservative therapies do not prevent disease progression and liver transplantation has been the only potentially curative option
The latest approach to the treatment of PH1 is substrate reduction therapy with the aim of reducing the production of glyoxylate, the precursor to oxalate
Lumasiran, a subcutaneously administered RNAi therapeutic targeting glycolate oxidase, is transforming the management of patients with PH1
Declaration of interest
S Hulton has conducted research on behalf of Dicerna and Alnylam and has received consultancy fees/honoraria from pharmaceutical companies Dicerna, Alnylam, and Chiesi within the last 3 years. She received no personal funding for this paper. The scientific team at Alnylam Pharmaceuticals GmBH were given the opportunity to check trial data factual accuracy but did not contribute to the content of the manuscript. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Writing assistance was provided by Maggie Lai of Comradis Limited, funded by Alnylam Pharmaceuticals GmBH.
Additional information
Funding
References
- Cochat P, Hulton SA, Acquaviva C, et al. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012 May;27(5):1729–1736.
- Purdue PE, Takada Y, Danpure CJ. Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J Cell Biol. 1990 Dec;111(6 Pt 1):2341–2351.
- Cochat P, Deloraine A, Rotily M, et al. Epidemiology of primary hyperoxaluria type 1. Societe de Nephrologie and the Societe de Nephrologie Pediatrique. Nephrol Dial Transplant. 1995;10(Suppl 8):3–7.
- Van Woerden CS, Groothoff JW, Wanders RJ, et al. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant. 2003 Feb;18(2):273–279.
- Van der Hoeven SM, Van Woerden CS, Groothoff JW. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults: results of the Dutch cohort. Nephrol Dial Transplant. 2012 Oct;27(10):3855–3862.
- Hopp K, Cogal AG, Bergstralh EJ, et al. Phenotype-genotype correlations and estimated carrier frequencies of primary hyperoxaluria. J Am Soc Nephrol. 2015 Oct;26(10):2559–2570.
- Harambat J, Fargue S, Bacchetta J, et al. Primary hyperoxaluria. Int J Nephrol. 2011;2011:864580.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009 Jun;75(12):1264–1271.
- Cramer SD, Ferree PM, Lin K, et al. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet. 1999 Oct;8(11):2063–2069.
- Belostotsky R, Seboun E, Idelson GH, et al. Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet. 2010 Sep 10;87(3):392–399.
- Belostotsky R, Pitt JJ, Frishberg Y. Primary hyperoxaluria type III: a model for studying perturbations in glyoxylate metabolism. J Mol Med (Berl). 2012 Dec;90(12):1497–1504.
- Salido E, Pey AL, Rodriguez R, et al. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta. 2012 Sep;1822(9):1453–1464.
- Cochat P, Rumsby G, Ingelfinger JR. Primary hyperoxaluria. N Engl J Med. 2013 Aug 15;369(7):649–658.
- Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005 May-Jun;25(3):290–296.
- Wuhl E, Van Stralen KJ, Wanner C, et al. Renal replacement therapy for rare diseases affecting the kidney: an analysis of the ERA-EDTA registry. Nephrol Dial Transplant. 2014 Sep;29(Suppl 4):iv1–8.
- Harambat J, Van Stralen KJ, Espinosa L, et al. Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol. 2012 Mar;7(3):458–465.
- Abumwais JQ. Etiology of chronic renal failure in Jenin district, Palestine. Saudi J Kidney Dis Transpl. 2012 Jan;23(1):158–161.
- Kamoun A, Chakroun D, Bejaoui M. Primary hyperoxaluria: a frequent cause of renal failure in Tunisian childhood. Pediatr Nephrol. 1991Nov;5(6):762. PMID: 1768592.
- Van Woerden CS, Groothoff JW, Wijburg FA, et al. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int. 2004 Aug;66(2):746–752.
- Mandrile G, Van Woerden CS, Berchialla P, et al. Data from a large European study indicate that the outcome of primary hyperoxaluria type 1 correlates with the AGXT mutation type. Kidney Int. 2014 Dec;86(6):1197–1204.
- Metry EL, Van Dijk LMM, Peters-Sengers H, et al. Transplantation outcomes in patients with primary hyperoxaluria: a systematic review. Pediatr Nephrol. 2021 Aug;36(8):2217–2226.
- Horoub R, Shamsaeefar A, Dehghani M, et al. Liver transplant for primary hyperoxaluria type 1: results of sequential, combined liver and kidney, and preemptive liver transplant. Exp Clin Transplant. 2021 May;19(5):445–449.
- Compagnon P, Metzler P, Samuel D, et al. Long-term results of combined liver-kidney transplantation for primary hyperoxaluria type 1: the French experience. Liver Transpl. 2014 Dec;20(12):1475–1485.
- European Commission. Commission implementing decision of 19.11.20 granting marketing authorisation under Regulation (EC) No 726/2004 of the European Parliament and of the Council for Oxlumo-lumasiran, an orphan medicinal product for human use; 2020 [cited 2021 Sept 13]. Available from: https://ec.europa.eu/health/documents/community-register/html/h1496.htm
- US Food and Drug Administration (FDA). FDA approves first drug to treat rare metabolic disorder; 2020 [cited 2021 Sept 13]. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-treat-rare-metabolic-disorder?utm_medium=email&utm_source=govdelivery
- Holmes RP, Assimos DG. Glyoxylate synthesis, and its modulation and influence on oxalate synthesis. J Urol. 1998 Nov;160(5):1617–1624.
- Hulton SA. The primary hyperoxalurias: a practical approach to diagnosis and treatment. Int J Surg. 2016 Dec;36(Pt D):649–654.
- Marangella M, Petrarulo M, Vitale C, et al. Serum calcium oxalate saturation in patients on maintenance haemodialysis for primary hyperoxaluria or oxalosis-unrelated renal diseases. Clin Sci (Lond). 1991 Oct;81(4):483–490.
- Schepers MS, Van Ballegooijen ES, Bangma CH, et al. Oxalate is toxic to renal tubular cells only at supraphysiologic concentrations. Kidney Int. 2005 Oct;68(4):1660–1669.
- Williams EL, Acquaviva C, Amoroso A, et al. Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Hum Mutat. 2009 Jun;30(6):910–917.
- Rumsby G, Williams E, Coulter-Mackie M. Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias. Kidney Int. 2004 Sep;66(3):959–963.
- Danpure CJ, Cooper PJ, Wise PJ, et al. An enzyme trafficking defect in two patients with primary hyperoxaluria type 1: peroxisomal alanine/glyoxylate aminotransferase rerouted to mitochondria. J Cell Biol. 1989 Apr;108(4):1345–1352.
- Fargue S, Lewin J, Rumsby G, et al. Four of the most common mutations in primary hyperoxaluria type 1 unmask the cryptic mitochondrial targeting sequence of alanine: glyoxylateaminotransferase encoded by the polymorphic minor allele. J Biol Chem. 2013 Jan 25;288(4):2475–2484.
- Danpure CJ. Primary hyperoxaluria: from gene defects to designer drugs?. Nephrol Dial Transplant. 2005 Aug;20(8):1525–1529.
- Fargue S, Rumsby G, Danpure CJ. Multiple mechanisms of action of pyridoxine in primary hyperoxaluria type 1. Biochim Biophys Acta. 2013 Oct;1832(10):1776–1783.
- Monico CG, Rossetti S, Olson JB, et al. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 2005 May;67(5):1704–1709.
- Harambat J, Fargue S, Acquaviva C, et al. Genotype-phenotype correlation in primary hyperoxaluria type 1: the p.Gly170Arg AGXT mutation is associated with a better outcome. Kidney Int. 2010 Mar;77(5):443–449.
- Soliman NA, Nabhan MM, Abdelrahman SM, et al. Clinical spectrum of primary hyperoxaluria type 1: experience of a tertiary center. Nephrol Ther. 2017 May;13(3):176–182.
- Naderi G, Tabassomi F, Latif A, et al. Primary hyperoxaluria type 1 diagnosed after kidney transplantation: the importance of pre-transplantation metabolic screening in recurrent urolithiasis. Saudi J Kidney Dis Transpl. 2015 Jul-Aug;26(4):783–785.
- Sikora P, Zaniew M, Grenda R, et al. Still diagnosed too late and under-recognized? The first comprehensive report on primary hyperoxaluria in Poland. Pol Arch Intern Med. 2020 Dec 22;130(12):1053–1063.
- Mookadam F, Smith T, Jiamsripong P, et al. Cardiac abnormalities in primary hyperoxaluria. Circ J. 2010 Nov;74(11):2403–2409.
- Leumann E, Hoppe B. The primary hyperoxalurias. J Am Soc Nephrol. 2001 Sep;12(9):1986–1993.
- Birtel J, Herrmann P, Garrelfs SF, et al. The ocular phenotype in primary hyperoxaluria type 1. Am J Ophthalmol. 2019 Oct;206:184–191.
- El Hage S, Ghanem I, Baradhi A, et al. Skeletal features of primary hyperoxaluria type 1, revisited. J Child Orthop. 2008 Jun;2(3):205–210.
- Fargue S, Harambat J, Gagnadoux MF, et al. Effect of conservative treatment on the renal outcome of children with primary hyperoxaluria type 1. Kidney Int. 2009 Oct;76(7):767–773.
- Marangella M, Petrarulo M, Cosseddu D, et al. Oxalate balance studies in patients on hemodialysis for type I primary hyperoxaluria. Am J Kidney Dis. 1992 Jun;19(6):546–553.
- Hoppe B, Graf D, Offner G, et al. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr Nephrol. 1996 Aug;10(4):488–492.
- Yamauchi T, Quillard M, Takahashi S, et al. Oxalate removal by daily dialysis in a patient with primary hyperoxaluria type 1. Nephrol Dial Transplant. 2001 Dec;16(12):2407–2411.
- Plumb TJ, Swee ML, Fillaus JA. Nocturnal home hemodialysis for a patient with type 1 hyperoxaluria. Am J Kidney Dis. 2013 Dec;62(6):1155–1159.
- Metry EL, Garrelfs SF, Peters-Sengers H, et al. Long-term transplantation outcomes in patients with primary hyperoxaluria type 1 included in the European hyperoxaluria consortium (OxalEurope) registry. Kidney Int. 2021;In press.
- Gambello MJ, Li H. Current strategies for the treatment of inborn errors of metabolism. J Genet Genomics. 2018 Feb 20;45(2):61–70.
- Martin-Higueras C, Luis-Lima S, Salido E. Glycolate oxidase is a safe and efficient target for substrate reduction therapy in a mouse model of primary hyperoxaluria type I. Mol Ther. 2016 Apr;24(4):719–725.
- Lai C, Pursell N, Gierut J, et al. Specific inhibition of hepatic lactate dehydrogenase reduces oxalate production in mouse models of primary hyperoxaluria. Mol Ther. 2018 Aug 1;26(8):1983–1995.
- Hoppe B, Koch A, Cochat P, et al. Safety, pharmacodynamics, and exposure-response modeling results from a first in human phase 1 study of nedosiran in primary hyperoxaluria. Kidney Int. 2021 Sep 2. DOI:https://doi.org/10.1016/j.kint.2021.08.015.
- Ariceta G, Barrios K, Brown BD, et al. Hepatic lactate dehydrogenase A: an RNA interference target for the treatment of all known types of primary hyperoxaluria. Kidney Int Rep. 2021 Apr;6(4):1088–1098.
- Rumsby G, Hulton SA. From pathogenesis to novel therapies in primary hyperoxaluria. Expert Opin Orphan Drugs. 2019;7(2):57–66.
- Hu B, Zhong L, Weng Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. 2020 Jun 19;5(1):101.
- Damase TR, Sukhovershin R, Boada C, et al. The limitless future of RNA therapeutics. Front Bioeng Biotechnol. 2021;9:628137.
- European Medicines Agency. Oxlumo 94.5 mg/0.5 mL solution for injection: summary of product characteristics; 2020 [cited 2021 Sept 13]. Available from: https://ec.europa.eu/health/documents/community-register/2020/20201119149780/anx_149780_en.pdf
- Alnylam. Oxlumo (lumasiran) injection, for subcutaneous use: US prescribing information; 2020 [cited 2021 Sept 13]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214103lbl.pdf
- Roggenbuck D, Mytilinaiou MG, Lapin SV, et al. Asialoglycoprotein receptor (ASGPR): a peculiar target of liver-specific autoimmunity. Auto Immun Highlights. 2012 Dec;3(3):119–125.
- Liebow A, Li X, Racie T, et al. An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J Am Soc Nephrol. 2017 Feb;28(2):494–503.
- Li X, Knight J, Fargue S, et al. Metabolism of (13) C5-hydroxyproline in mouse models of primary hyperoxaluria and its inhibition by RNAi therapeutics targeting liver glycolate oxidase and hydroxyproline dehydrogenase. Biochim Biophys Acta. 2016 Feb;1862(2):233–239.
- Dutta C, Avitahl-Curtis N, Pursell N, et al. Inhibition of glycolate oxidase with dicer-substrate siRNA reduces calcium oxalate deposition in a mouse model of primary hyperoxaluria type 1. Mol Ther. 2016 Apr;24(4):770–778.
- McGregor TL, Hunt KA, Yee E, et al. Characterising a healthy adult with a rare HAO1 knockout to support a therapeutic strategy for primary hyperoxaluria. Elife. 2020 Mar 24;9. DOI:https://doi.org/10.7554/eLife.54363
- Frishberg Y, Zeharia A, Lyakhovetsky R, et al. Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J Med Genet. 2014 Aug;51(8):526–529.
- Frishberg Y, Deschenes G, Groothoff JW, et al. Phase 1/2 study of lumasiran for treatment of primary hyperoxaluria type 1: a placebo-controlled randomized clinical trial. Clin J Am Soc Nephrol. 2021 May 13;16(7):1025–1036.
- Frishberg Y, Hulton SA, Cochat P, et al. Results from the ongoing Phase 2 open label extension study of lumasiran, an investigational RNAi therapeutic in patients with primary hyperoxaluria type 1 [Abstract]. J Am Soc Nephrol. 2020;31:520–521.
- Frishberg Y, Hulton SA, Cochat G, et al. Results from the ongoing phase 2 open-label extension study of lumasiran, an investigational RNAi therapeutic, in patients with primary hyperoxaluria type 1 (PH1); 2020 [cited 2021 Sept 13]. Available from: https://www.alnylam.com/wp-content/uploads/2020/10/Luma-ASN-2020_Phase-2-OLE.pdf
- Garrelfs SF, Frishberg Y, Hulton SA, et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med. 2021 Apr 1;384(13):1216–1226.
- Deschenes G, Cochat G, Magen D, et al. ILLUMINATE-B, a phase 3 open-label study to evaluate lumasiran, an RNAi therapeutic, in young children with primary hyperoxaluria type 1 (PH1) [Abstract]. J Am Soc Nephrol. 2020;31:515.
- Deschenes G, Cochat P, and Magen D, et al. ILLUMINATE-B, a Phase 3 open-label study to evaluate lumasiran, an RNAi therapeutic in young children with primary hyperoxaluria type 1. 2020 [cited 13 Sept 2021]. Available from: https://www.alnylam.com/wp-content/uploads/2020/10/Luma-ASN-2020_ILLUMINATE-B-6m.pdf .
- Alnylam. Alnylam reports positive topline results from ILLUMINATE-C phase 3 study of lumasiran in patients with advanced primary hyperoxaluria type 1; 2021 [ cited 2021 Sept 13]. Available from: https://investors.alnylam.com/press-release?id=25891