
ABSTRACT
Mucosal epithelium maintains tissue homeostasis through many processes, including epithelial barrier function, which separates the environment from the tissue. The barrier hypothesis of type 2 inflammatory disease postulates that epithelial and epidermal barrier dysfunction, which cause inappropriate exposure to the environment, can result in allergic sensitization and development of type 2 inflammatory disease. The restoration of barrier dysfunction once it's lost, or the prevention of barrier dysfunction, have the potential to be exciting new therapeutic strategies for the treatment of type 2 inflammatory disease. Neutrophil-derived Oncostatin M has been shown to be a potent disrupter of epithelial barrier function through the induction of epithelial-mesenchymal transition (EMT). This review will discuss these events and outline several points along this axis at which therapeutic intervention could be beneficial for the treatment of type 2 inflammatory diseases.
The barrier hypothesis
The role of mucosal epithelium is to maintain tissue homeostasis and defense. It achieves this through mucociliary clearance, anti-microbial peptide production, maintenance of air-surface liquid, regulation of ion permeability, and physical barrier function.Citation1 Epithelial barrier function is necessary for tissue homeostasis because it acts as a first line of defense against pathogens, allergens and environmental factors through the creation of a physical separation between the environment and the tissue. Epithelial barrier function is largely determined by the structure of intercellular epithelial junctions, including tight junctions, adherens junctions and desmosomes.Citation2 When the epithelial barrier is breached, pathogens, allergens and environmental factors can synergize with alarmins and pathogen- or danger-associated molecular patterns (PAMPs and DAMPs) that transit across the barrier and/or are released in response to cellular damage, resulting in the activation of innate and adaptive immune responses.Citation3 The immune system's ability to neutralize the pathogenic stimulus and repair the resulting tissue damage is necessary for the restoration of the tissue to a normal homeostatic state. However, if barrier function is not restored, it may result in the development of an unnecessary chronic inflammatory state within the tissue and potential allergic sensitization via continued exposure facilitated passage of allergens across the defective barrier. The barrier hypothesis postulates that epithelial or epidermal barrier dysfunction thereby precedes the development of allergic sensitization and type 2 inflammatory disease.
Mucosal epithelium refers to epithelium from the airways, gastrointestinal tract, eyes and genitourinary tract, among others. Differentiated epithelium has a polarized structure where undifferentiated basal cells lie along the basement membrane and differentiated cells line the apical surface. This review will focus on airways and esophageal epithelium. Airways epithelium has a pseudostratified columnar structure, consisting of at least 3 common cell types, basal cells, ciliated cells, and goblet cells. Basal cells are undifferentiated progenitor cells that are attached to the basement membrane and express keratin 5, which can be used as a marker of undifferentiated epithelium.Citation4 These cells will proliferate and migrate to cover a wound in the event of injury or infection.Citation5 Ciliated and goblet cells are the differentiated cells within airway epithelium which lie along the apical surface of the epithelial cell layer. These cells express keratin 8, which can be used as a marker of differentiated airway epithelial cells.Citation6 Goblet cells make mucins, which are released into the airway lumen to trap pathogens, allergens and other environmental factors that may otherwise result in injury or immune response. Ciliated cells contain cilia, comprised of acetylated α-tubulin, which beat in a coordinated fashion to clear the airways of the mucus and its trapped contents.Citation7 Together these differentiated airway epithelial cells are responsible for ion transport, maintenance of air-surface liquid, mucus production, mucociliary clearance and barrier function.Citation5,8 Esophageal epithelium has a stratified squamous structure that consists of several layers, in which cells become more differentiated closer to the apical layers.Citation9 The stratum basale comprises basal cells and lies along the basement membrane; these cells are undifferentiated progenitor cells.Citation10 The most apical layer is the stratum corneum, which comprises dead, flattened cells that have lost their nuclei and organelles. While the stratum corneum of epidermis is keratinized, mucosal stratified squamous epithelium, as in the esophagus, has a non-keratinized stratum corneum.Citation11
The barrier function of airways and esophageal epithelium is largely determined through the apical junction complex, which includes tight junctions and adherens junctions.Citation5,12 Tight junctions localize to the apical edge of the epithelial layer, just adjacent to the lumen,Citation13 and are comprised of intercellular proteins that self-adhere in the intercellular space, effectively sealing that area and regulating paracellular flux. These proteins include occludin, junctional adhesion molecule (JAM) family proteins, and claudin family proteins.Citation14 These protein complexes are tethered to the actin cytoskeleton through adaptor proteins including the zonula occludens (ZO) protein family. The tight junction and actin cytoskeleton form a tight band along the apical edge of the cell layer that is necessary for proper barrier function.Citation15,16 Adherens junctions localize to the basal border of the tight junction complex and are also tethered to the actin cytoskeleton.Citation5 These junctions consist of the intercellular self-adherent protein, E-cadherin, that maintains calcium dependent adhesion between epithelial cells. Adherens junctions also are comprised of accessory proteins, α-catenin, cateninδ1, and β-catenin, which collectively can act as a transcription factor upon disassembly of the junction.Citation17-21
This junctional structure is necessary for proper epithelial barrier function and junctional failure can lead to disease. For example, asthma is classically thought to involve a predisposition or skewing to type 2 inflammation, versus type 1 inflammation, that promotes persistent eosinophilic inflammation of the airways and subsequent tissue remodeling. However, this type 2 skewing model fails to fully explain many clinical features of asthma, including the heterogeneity of asthma phenotypes and the lack of association between airway eosinophilia and airway hyper-responsiveness.Citation22-26 The barrier hypothesis could help to explain some disease features that the type 2 skewing model does not because epithelial barrier dysfunction is proposed to be upstream of the generation of an immune response and could potentially result in type 1 or type 2 inflammation. Epithelial dysfunction has been demonstrated in asthma patients compared with control patients. Asthma patients have decreased expression of tight and adherens junction proteins and their epithelia have decreased barrier function as assessed by transepithelial electrical resistance (TEER) and dextran permeability across monolayers.Citation27-29 Additionally, reduced caveolin-1 expression in airway epithelium from asthma patients was shown to associate with decreased adherens junction expression, and promoted epithelial expression of the type 2-skewing cytokines TSLP and IL-33, suggesting that barrier dysfunction may initiate type 2 inflammation.Citation27 Thus, therapeutic intervention focused on either preventing barrier dysfunction, or restoring barrier once it is lost, may be effective in the treatment of type 2 inflammatory diseases, including asthma, chronic rhinosinusitis (CRS), eosinophilic esophagitis (EoE), and potentially allergic rhinitis and atopic dermatitis.
Type 2 inflammation has been shown to be important for immunity to helminths and toxins.Citation30,31 More recent studies have also shown that Type 2 immune responses are also important for the establishment of general tissue homeostasis.Citation32-34 Common features of type 2 inflammation include the expression of classical type 2 cytokines, IL-4, IL-5, and IL-13, tissue eosinophilia, mast cell activation and fibrosis. Prolonged type 2 inflammation can further promote or perpetuate allergic sensitization to otherwise innocuous food or aeroantigens that would normally be tolerated. Dysfunction of epithelial maintenance and repair of barrier has been implicated in the pathogenesis of many type 2 inflammatory diseases, including asthma, atopic dermatitis, eosinophilic esophagitis (EoE), and chronic rhinosinusitis (CRS).Citation35-38 Atopic dermatitis patients have been shown to express decreased levels of the tight junction protein, claudin 1 which negatively correlated with serum IgE and total eosinophil counts.Citation39 Recent genome-wide association studies (GWAS) have identified loss-of-function single nucleotide polymorphisms (SNPs) in the filaggrin gene that correlate with increased risk for the development of atopic dermatitis, allergic asthma, food allergy, and allergic rhinitis.Citation38,40-42 This observation is perhaps the best evidence so far in support of the barrier hypothesis. Filaggrin is a structural protein that is important for proper differentiation of the epidermal stratum corneum, which is the keratinized outer layer of the epidermis that is crucial for establishment of barrier function.Citation43,44 These loss-of-function SNPs result in impaired barrier function in the skin, leading to chronic inflammation and the potential generation of allergic sensitization.Citation45 Interestingly, airway epithelia do not express filaggrin, suggesting that the development of allergic asthma may be preceded by atopic dermatitis and subsequent cutaneous sensitization to aeroallergens or food allergens that drive disease at mucosal surfaces.Citation40,42 These observations and others provide evidence for the atopic march, which describes a characteristic progression of allergic disease in children that usually begins with the development of atopic dermatitis and allergic sensitization in infancy followed by food allergy in early childhood and finally allergic rhinitis and asthma later in childhood.Citation46
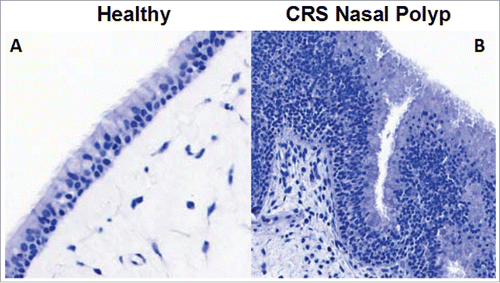
Healthy bronchial, nasal and esophageal epithelia are highly organized and polarized structures; healthy nasal epithelium is shown in . However, epithelia from asthma, CRS and EoE patients have some common changes in structural elements including an expansion of basal cells, or acanthosis, associated with a loss of differentiation throughout the cell layer; acanthosis in nasal epithelium from a CRS patient is also shown in . It is particularly striking that the morphology of the epithelia in these 3 different diseases has some similarities, potentially suggesting common mechanisms of epithelial dysfunction.Citation47,48 Many of these structural abnormalities are believed to lead to barrier dysfunction, and subsequent development of allergic sensitization, type 2 inflammatory disease or the exacerbation of established type 2 inflammation. A central theme of this review is that the barrier-disrupting cytokine, Oncostatin M (OSM), may play a role in mucosal allergic diseases affecting several different organs. Therapeutic interventions aimed at preventing barrier loss or restoring barrier function, either through targeting OSM or other mediators, could offer exciting alternative strategies for treating patients with type 2 inflammatory disease.
Figure 1. Epithelial morphology is abnormal in nasal polyps from CRS patients. Healthy nasal epithelium is a highly organized structure with undifferentiated progenitor cells along the basement membrane, and the differentiated cells, ciliated and goblet cells along the apical edge (A). However, in CRS the epithelium often has a large expansion of basal cells and is highly disorganized, which could lead to barrier dysfunction (B).
Oncostatin M and epithelial barrier function
OSM is a member of the IL-6 family of cytokines, which includes IL-11, IL-31 and leukemia inhibitory factor (LIF).Citation49 OSM has been shown to be expressed by many cell types of the haematopoietic lineage, including T cells, neutrophils, mast cells, macrophages and eosinophils.Citation50-53 OSM is not known to be expressed by epithelium, fibroblasts, or smooth muscle, which all express the 2 forms of the OSM receptor.Citation54 Human OSM signals through 2 heterodimeric receptors, both of which utilize glycoprotein 130 (gp130) for signaling. The type 1 OSM receptor is composed of the LIF receptor (LIFR) and gp130, and the type 2 OSM receptor is composed of the OSM receptor β-chain (OSMR) and gp130.Citation55,56 Of note, murine OSM does not bind the type 1 receptor, which is something to be considered regarding the implications of research done on OSM in mouse models.Citation57 The transcription factor STAT3 has been shown to be very important for epithelial homeostasis and repair.Citation58 Oncostatin M (OSM) is a potent activator of STAT3 and also activates other transcription factors, including STAT1, STAT5, PI3K, and the MAPK pathway through multiple tyrosine kinases including, janus kinase 1 (JAK1), janus kinase 2 (JAK2), or tyrosine kinase 2 (Tyk2).Citation49,59-64 OSM has been shown to have a wide range of physiologic effects, including in vitro inhibition of tumor growth, promotion of in vivo tumor invasiveness and metastasis, maintenance of bone metabolism, generation of joint inflammation and regulation of the hepatic acute phase response.Citation65-70 However, this review will focus on the effects of OSM that are related to epithelial repair and homeostasis, and inflammation.
OSM has been shown to be important in the repair of many tissue types. Biegel et al. have shown that OSM was important for regrowth after injury and that it decreased apoptosis in a STAT3 dependent manner in a colon carcinoma cell line. They also demonstrate elevated OSMR in inflamed colon biopsies from Crohn's disease and ulcerative colitis patients, which led them to suspect that OSM was important for epithelial repair and that it may be a potential drug target.Citation71 In a chronic diabetes cutaneous wound model, the treatment of wounds with recombinant OSM decreased expression of proinflammatory cytokines, IL-1β and TNF, and improved wound closure during the early inflammatory phase of repair.Citation72 In a model of cardiac repair, OSM mediated the dedifferentiation of cardiomyocytes, which is necessary for proper repair of cardiac injury.Citation73 Additionally, in a mouse demyelination spinal cord injury model, OSM induced many processes that are involved in repair of demyelination injuries including the mobilization of oligodendrocyte precursors, differentiation of oligodendrocytes, and the production of myelin.Citation74 These data suggest that OSM can play an important role in normal homeostatic tissue repair.
Epithelial damage repair occurs in 2 basic steps. First, basal cells proliferate and migrate into the wound to cover the damaged area, and second, the progenitor cells become contact inhibited, stop proliferating and differentiate back into ciliated or goblet cells. Intercellular junctions are then formed to reestablish barrier function in the repaired tissue.Citation75 The mechanism of OSM mediated barrier dysfunction in mucosal epithelium in vitro is currently unknown. OSM has been shown to be important for epithelial repair, as discussed above and OSM may be one signal that initiates the first step of repair following cellular damage through the induction of basal cell proliferation and migration to cover the wound. However, in vivo in a diseased state, either high levels of OSM, or the absence of a signal to initiate the second step of the repair process, could potentially prevent the transition to the second step of repair, leading to chronic proliferation of basal cells to create a state where differentiation and the generation of barrier function does not occur.
While OSM has been shown to be important for tissue repair, it has also been shown to have pathogenic effects in both type 1 and type 2 inflammation. In the context of type 1 inflammation, OSM treatment of dendritic cells (DC) induced polarization of Th1 cells through the induction of IL-12,Citation76 and Th1 cells can produce OSM.Citation77 OSM has been shown to play an important role in the autoimmune disease rheumatoid arthritis (RA). OSM is a biomarker of active RA compared with both quiescent RA and healthy control patients and it mediates its effects in RA through the modulation of matrix metalloproteinases.Citation78,79 OSM has also been shown to increase the expression of OSMR and IL-1R1 in synovial fibroblasts, thereby augmenting the pathogenic effects of OSM and IL-1 in RA.Citation80 In addition, OSM was sufficient to induce CCL13, a chemokine that is elevated in synovial fibroblasts of RA patients.Citation68
In the context of type 2 inflammation, previous studies have reported elevated levels of OSM in sinus tissue from allergic rhinitis patients, psoriatic skin and sputum of asthmatic patients with irreversible airflow obstruction.Citation81-83 Our group has shown elevated OSM in nasal polyps from CRS patients, in tissue biopsies and in induced sputum from asthma patients, and in biopsies from EoE patients compared with controls.Citation84,85 In a mouse model, intratracheal administration of adenoviral expressed OSM was shown to be sufficient to induce robust type 2 inflammation in the lung, without any specific antigen challenge.Citation86,87 Studies using reconstituted epidermis have shown that OSM treatment decreased expression of filaggrin, a molecule that is important for barrier function in keratinocytes, suggesting that it could also contribute to barrier dysfunction in the skin.Citation88 Our group has also shown that OSM was sufficient to induce epithelial barrier dysfunction and loss of tight junction structure in air-liquid interface cultures of differentiated nasal and bronchial epithelium. Additionally, we showed that levels of OSM correlated with levels of markers of epithelial leak in localized nasal secretions of CRS patients and in bronchoalveolar lavage fluids following allergen challenge in allergic asthmatic patients, suggesting that OSM may contribute to epithelial barrier dysfunction in vivo.Citation84
One potential mechanism for elevated OSM in type 2 inflammatory disease that has not been investigated is whether SNPs in the OSM or OSMR genes associate with the development of type 2 inflammatory disease. Gain of function SNPs in the OSM or OSMR gene could potentially result in elevated expression or increased receptor binding affinity or intrinsic activity that could amplify the effects of OSM and exacerbate type 2 inflammation, providing another potential mechanism of OSM mediated epithelial dysfunction. Alternatively, loss of function SNPs could be protective and confer reduced susceptibility to development of type 2 inflammation. SNPs in the OSMR gene have been found to associate with the severity of RA and systemic lupus erythematosus (SLE).Citation89 A SNP in the OSMR gene was shown to positively associate with tumor size in patients with papillary thyroid cancer.Citation90 Since SNPs in the OSMR gene have been shown to associate with disease, it would be worthwhile to seek SNPs in the OSM or OSMR gene that associate with type 2 inflammatory disease. If present, such SNPs could help to determine which patients are at risk and possibly provide insight about treatment modalities that patients might respond to.
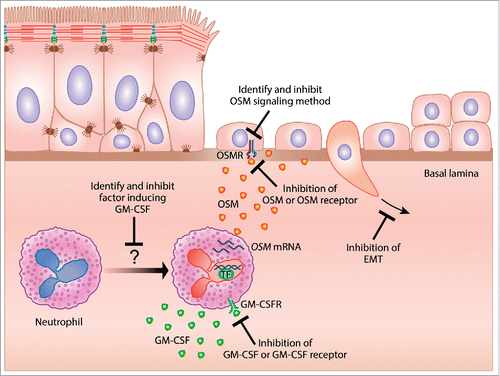
It is paradoxical that OSM can play both pathogenic and protective roles as well as be involved with both type 1 and type 2 inflammation. Although speculative, one factor that may determine whether OSM induces pathogenic or protective responses may simply be the concentration of OSM. Perhaps small quantities of OSM are important for the protective, pro-repair effects of OSM and higher levels of OSM are pathogenic. Another potential explanation for the paradoxical effects of OSM may be tissue specificity. This idea has been outlined in the danger hypothesis, and postulates that the “choice” of whether the immune system should mount a type 1 or type 2 response is driven by the tissue where this immune response will be deployed.Citation91 Type 1 immune responses have been shown to be very destructive in highly specialized tissues, such as the eye and brain.Citation92,93 These tissues may therefore skew the immune response toward a type 2 response in the event of an infection that would normally elicit a robust type 1 response to eradicate the pathogen and preserve tissue function. In support of this idea, fluid from the anterior chamber of the eye has been shown to be sufficient to both promote type 2 responses, and dampen type 1 responses.Citation94,95 Potentially, OSM may drive either a type 1 or type 2 response depending on which tissue is the site of inflammation. Another potential explanation for the paradoxical effects of OSM could relate to the signaling of OSM occurring through 2 distinct receptors, and activation by OSM of at least 5 different transcription factors. Activation of STAT3 may mediate the pro-repair effects of OSM, and OSM activation of other transcription factors may be responsible for OSM pathogenic effects. OSM roles in type 1 and type 2 inflammation may also be mediated through differing signaling mechanisms. Because OSM has so many varied effects throughout the body, therapeutically directly inhibiting OSM has the potential for having considerable side effects. Identifying the signaling mechanisms that mediate OSM induced barrier dysfunction could allow for a more targeted therapeutic approach while leaving the remaining OSM effects intact, thereby reducing the risk of side effects ().
Figure 2. Hypothetical mechanism of repair and potential therapeutic strategies for epithelial barrier restoration in the treatment of type 2 iflammatory disease. GM-CSF induces neutrophil derived OSM, which can mediate epithelial barrier dysfunction through the induction of EMT. Several potential mechanisms aimed at restoring epithelial barrier function for the treatment of type 2 inflammatory diseases are identified.
Neutrophil derived Oncostatin M in type 2 inflammation
Neutrophils are generally considered to be type 1 immune response effector cells and are not classically associated with type 2 immune responses. However, both CRS and asthma are known to have disease endotypes characterized by neutrophilia. While nasal polyps in Western countries tend to be highly eosinophilic, a significant proportion of nasal polyps from patients in Eastern Asian countries are neutrophilic.Citation96-98 Increased neutrophilia within nasal polyps has been linked to decreased responsiveness to corticosteroid treatment.Citation99 One endotype of severe asthma is often characterized by neutrophilia.Citation100,101 It is thought that neutrophilia in asthma associates with more severe disease, in part because neutrophils are not particularly responsive to corticosteroid treatment.Citation102 However, little is known about the role of neutrophils in type 2 inflammatory disease, outside of the existence of neutrophilic subtypes of CRS and asthma.
Neutrophil-derived OSM has been implicated in the pathogenesis of many conditions including acute lung injury, asthma, breast cancer, and rheumatoid arthritis.Citation81,103-105 Osteopontin, prostaglandin E2, follistatin-like 1 (FSTL1), complement factor 5a, and thrombin have all been shown to induce OSMCitation106-110 but, only GM-CSF has been shown to induce OSM in neutrophils.Citation105,111 Elbjeirami et al. showed that OSM was induced during neutrophil transendothelial migration in response to endothelial production of GM-CSF.Citation111 Queen et al. showed that co-culture of neutrophils with breast cancer cells induced OSM production, a response that was lost when GM-CSF was blocked.Citation105 Additionally, Miller et al. have shown that FSTL1 was necessary for OSM induction in a mouse model of chronic asthma.Citation109 Both GM-CSF and FSTL1 have been shown to be elevated in allergic airways disease.Citation112-114 Our studies showed that GM-CSF induced OSM in neutrophils isolated from peripheral blood, while FSTL1 did not. Interestingly, we also showed that the OSM-producing neutrophils in nasal polyps also expressed GM-CSF, which may indicate that neutrophils induce their own production of OSM through autocrine production of GM-CSF.Citation85 There is precedent for such a concept, as Wardlaw and colleagues showed that eosinophils prolong their own survival through the autocrine production of GM-CSF.Citation115 In addition, Durand et al, have shown that autoantibody ligation of FcγRIIIb (CD16) receptors on the surface of neutrophils protects against apoptosis through the induction of autocrine GM-CSF.Citation116 Our group has previously shown elevated levels of autoantibodies in nasal polyps, suggesting that elevated autoantibodies in nasal polyps may trigger GM-CSF expression in the infiltrating neutrophils by a similar mechanism.Citation117 Whether production of OSM by neutrophils via an autocrine GM-CSF dependent pathway occurs in type 2 inflammatory diseases is worthy of further investigation, and could potentially identify additional therapeutic targets ().
Neutrophils are classically thought to be a first line of defense in response to tissue damage,Citation118 and neutrophil-derived OSM has been shown to be important in the first stages of epithelial repair during which basal cells proliferate to cover the wound.Citation119 Neutrophils are known to be increased in CRS and severe asthma. Neutrophil-derived OSM could participate in both ongoing epithelial repair as well as promote the epithelial barrier dysfunction that is observed in these diseases. Several groups have shown that activated bronchial epithelium secretes GM-CSF into cell culture supernatants upon activation, e.g. by exposure to heat killed Staphylococcus aureus, which is a common pathogen that can cause epithelial damage.Citation85 One possible scenario is that epithelial damage induces GM-CSF, which in turn induces transient OSM production by the infiltrating neutrophils to promote the first stage of repair. If early OSM expression is transient, it would allow epithelial cells to redifferentiate back into functional epithelium during the later stages of repair. However, under pathological conditions, when neutrophils are more chronically recruited to the injury site, they may assume a phenotype that constitutively makes both OSM and GM-CSF.Citation85 The GM-CSF alone could be sufficient to induce chronic neutrophil-derived OSM in sufficient amounts to prevent late stage repair, causing a long-term state of barrier dysfunction. In addition, GM-CSF would also promote long-term survival of OSM-producing neutrophils. It will be important to understand the mechanism by which neutrophils “switch” from having a beneficial homeostatic role in epithelial repair to a pathogenic role in epithelial barrier dysfunction. Determining the molecular mechanism of this “switch” could uncover an important therapeutic target for the treatment of type 2 inflammatory disease.
Recent studies have described subtypes of polarized neutrophils, termed N1 and N2.Citation120 These N1 and N2 neutrophils have been shown to be very similar in function to their M1 and M2 macrophage counterparts.Citation120 N1 neutrophils are classical, proinflammatory effector cells, while N2 neutrophils specifically have been shown to promote tumor growth and metastasis, as well as play an important role in repair processes and the resolution of inflammation.Citation121-123 N1 and N2 neutrophil polarization has been best studied in mice, and N2 neutrophils have been shown to express the macrophage mannose receptor (MRC1), arginase 1 (ARG1), chitinase-like 3 (Ym1), IL-10 and TGFβ, which are also characteristic markers of M2 macrophages. Both M2 macrophages and N2 neutrophils have been associated with tissue repair mechanisms.Citation122 Compared to their N2 counterparts, N1 neutrophils have been shown to express more proinflammatory cytokines and chemokines such as TNF, IL-1β, CCL3, CCL5, IL-6 and IL-12.Citation122 We have shown that the majority of OSM+ neutrophils from nasal polyps expressed the N2 marker ARG1, indicating a phenotype resembling N2 neutrophils more than N1 neutrophils. We also showed that GM-CSF induced in vitro-stimulated neutrophils to express elevated MRC1 compared with unstimulated neutrophils and N1 polarized neutrophils, also indicating that OSM producing neutrophils could be skewed toward the N2 phenotype. However, since we did not see any induction of ARG1 in response to GM-CSF, another factor may be necessary for full induction of the N2 phenotype.Citation85 M2 macrophages have been shown to promote epithelial mesenchymal transition (EMT) in the context of cancer.Citation124 Given that M2 macrophages and N2 neutrophils have been shown to have similar functions, it is reasonable to speculate that N2 neutrophils could also induce EMT, potentially through the production of OSM, although this has not yet been studied. OSM has been shown to induce EMT,Citation125 which antagonizes epithelial differentiation, and expression of EMT biomarkers is well established in both CRS and asthma.Citation126
OSM has been shown to both promote and suppress cancer growth, and these effects should be considered in therapeutic targeting of OSM. OSM was originally discovered based on its ability to inhibit the proliferation of melanoma cell lines in vitro; however much of the data on OSM and cancer is conflicting and shows that OSM can both promote and prevent cancer cell growth. Some of this dichotomy is thought to reflect differential effects of OSM on tumor cells, where it may prevent growth, and on the immune cells within the tumor, where it may indirectly promote tumor growth. Whether OSM promotes or prevents cancer growth may depend on the relative abundance of immune cells within the tumor microenvironment. OSM has been shown to induce signatures of EMT in breast cancer cell lines, and the presence of OSM in breast carcinomas has been shown to be associated with increased evidence of EMT, decreased estrogen receptor expression, elevated risk of metastasis and poorer overall prognosis.Citation127-129 OSM has also been shown to associate with increased metastasis to the lung in a mouse model of cervical cancer.Citation130 Furthermore, OSM has been shown to promote the induction of M2 macrophages, and potentially could also promote N2 neutrophils through a similar mechanism.Citation131 M2 macrophages and N2 neutrophils have been referred to as tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs). Elevated TAMs have been linked with increased tumor growth, metastasis and poorer prognosis in many cancer types including breast, colon, gastric, lung, cervical and skin cancer.Citation132 Elevated TANs have been linked with poor survival in head and neck, liver, kidney and pancreatic cancers.Citation133 These observations suggest that OSM could potentially promote cancer progression either directly or indirectly through the induction of M2 macrophages and possibly N2 neutrophils. Additionally, other aspects of type 2 inflammation have been shown to promote cancer progression, although no role for OSM in these responses was evaluated.Citation134-136
Given the association of cancer risk with type 2 inflammation, it seems plausible that type 2 inflammatory diseases may confer an elevated risk of cancer development, particularly in patients with severe or unmanaged disease. A few studies support a potential link between type 2 inflammation and cancer risk. A Swedish study found that asthmatic patients had a generally increased risk of cancer compared with the general population, and asthmatic patients diagnosed with cancer had a worse prognosis compared with non-asthmatic cancer patients.Citation137 A meta-analysis of American populations showed an increased risk of lung cancer in asthmatic patients that were never smokers.Citation138 Another study found a higher risk of breast cancer and lymphoma in atopic patients that had a positive skin prick test.Citation139 Taken together these data seem to suggest the possibility that cytokines associated with type 2 inflammation may promote cancer development and growth. Because of this potential association, proper management and/or prevention of type 2 inflammatory disease may be important not only to treat the patient's disease, but also to potentially decrease their lifetime risk for cancer development.
The role of Oncostatin M mediated epithelial- mesenchymal transition on epithelial barrier function in type 2 inflammatory disease
Epithelial-mesenchymal transition (EMT) describes the process whereby polarized epithelial cells transition to become non-polarized mesenchymal cells that are capable of proliferation and migration, and are less susceptible to apoptosis.Citation140,141 EMT has been shown to be involved in many cellular processes including embryogenesis, tissue repair, and cancer metastasis.Citation142,143,144 Growth factors TGFβ, VEGF, FGF, and members of the EGF family are all able to induce EMT.Citation145 The induction of EMT can be mediated by many signaling cascades, including STAT3, the Notch pathway, the wnt pathway, SMAD pathways, gp130-YAP-Src, and various receptor tyrosine kinase pathways.Citation146,147
The transition from polarized epithelial cells to mesenchymal cells requires the downregulation of genes that are important for maintenance of the epithelial phenotype, namely junctional proteins, and the upregulation of genes important for maintenance of the mesenchymal phenotype. Downregulation of the adherens junction protein, E-cadherin destabilizes the intercellular junctions, and is a hallmark of EMT.Citation148 During EMT, decreased E-cadherin is balanced by elevated levels of the mesenchymal cadherin, N-cadherin, which further destabilizes the intercellular junctions.Citation149,150 Additionally, genes that are required for maintenance of the mesenchymal phenotype are upregulated during EMT. Fibroblast specific protein (FSP1), also known as S100A4, is an important mediator of the mesenchymal phenotype through the alteration of cell motility, survival and cytoskeletal organization.Citation151,152 Actin polymerization is important for motility of mesenchymal cells, and α-smooth muscle actin (ACTA) is elevated in mesenchymal cells.Citation153,154 Elevated levels of the intermediate filament protein vimentin (VIM) are a canonical indicator of EMT because mesenchyme-associated vimentin intermediate filaments replace the keratin intermediate filaments that are present in epithelium.Citation155,156 Heat shock protein 47 (SERPINH1), is also elevated during EMT, and plays a role in collagen synthesis.Citation157 Discoidin domain receptor 2 (DDR2) is a collagen receptor that is capable of inducing EMT when activated by type I collagens.Citation158 Integrin α5 (ITGA5) and fibronectin have been shown to be important markers of the mesenchymal phenotype.Citation142 Additionally, the extra domain A (EDA) splice variant has shown to be expressed in mesenchymal cells.Citation159 Expression of the genes necessary for the mesenchymal phenotype is mediated through transcription factors that drive EMT, including the Snail (SNAI) family proteins, twist-related protein (TWIST) family proteins, and zinc finger E-box binding homeobox (ZEB) family proteins.Citation160 As already mentioned, Oncostatin M (OSM) induces EMT, and we have implicated OSM in the barrier dysfunction that occurs in type 2 inflammatory disease.Citation84 In a cohort of CRS and control patients, we observed positive correlations of OSM with the mesenchymal markers, VIM, SERPINH1, ITGA5, and DDR2; with the EMT inducer TGFβ; and with the EMT transcription factors, TWIST1, TWIST2, ZEB1, and ZEB2 (unpublished). Additionally, when we treated bronchial epithelial ALI cultures with OSM we saw increased cell counts and elevated staining of the proliferation marker ki67 and the mesenchymal markers S100A4 and ACTA (unpublished). These data suggest that OSM is sufficient to induce EMT in airway epithelium, and that OSM induces epithelial barrier dysfunction through the induction of EMT.
The restoration of epithelial barrier function as a therapeutic strategy for the treatment of type 2 inflammatory disease
Our work has identified OSM as a potentially important inducer of barrier dysfunction in type 2 inflammatory diseases. Notably, OSM induced barrier disruption in nasal epithelial cells from CRS patients grown at ALI was reversible,Citation84 suggesting that therapeutic intervention targeting OSM has the potential to be beneficial in the treatment of CRS through the restoration of epithelial barrier function.
OSM has been therapeutically targeted in rheumatoid arthritis (RA) through the neutralization of OSM, and the inhibition of the JAK/STAT pathway.Citation161,162 JAK inhibitors CP-690, 550 and INCB028050, which inhibit JAK-1, JAK-2 and JAK-3, were shown to effectively block the activation of STAT1, STAT3 and STAT5 in OSM-treated fibroblasts in vitro.Citation162 In contrast, a phase II trial in rheumatoid arthritis (RA) using an anti-OSM monoclonal antibody, GSK315234, did not show any benefit, potentially due to low affinity OSM binding. However, the drug was well tolerated in patients and the study investigators concluded that a high-affinity anti-OSM antibody might be beneficial in the treatment of RA.Citation161 Another potential strategy for the neutralization of OSM is a receptor fusion protein (RFP) that consists of OSM ligand binding subunits of OSMR and gp130 covalently linked by a flexible domain, and such a construct has been shown to be a highly potent and specific inhibitor of OSM.Citation163
While the neutralization of OSM may result in either the prevention of barrier loss or the restoration of barrier function, it may not be optimal to target OSM to achieve these effects. As discussed earlier, OSM plays many diverse roles, including maintenance of liver homeostasis, bone metabolism, joint homeostasis and epithelial homeostasis. Broad systemic inhibition of OSM may thus interfere broadly with OSM functions and result in side effects associated with dysregulated OSM mediated homeostasis. OSM has also been shown to both inhibit growth of tumor cell lines in vitro, and promote tumor invasiveness and metastasis in vivo, as discussed earlier. Because OSM may play a role in the suppression of some types of cancer, further study is warranted to determine whether systemic inhibition of OSM could have the potential to elevate the risk of cancer development or progression. Given the considerable potential for unwanted side effects as a result broad systemic inhibition of OSM, such drugs should be approached carefully or alternatives should be explored. One potential alternative is local inhibition of OSM for the treatment of type 2 inflammatory disease, which could be achieved through medicated nasal lavages, nasal sprays, swallowed slurries or inhalers. Inhibition of OSM restricted locally in these diseases could potentially restore barrier function and allow chronic type 2 inflammation to resolve with a diminished risk of systemic side effects. As discussed earlier, the identification of the signaling pathway responsible for OSM induced barrier dysfunction could uncover alternative therapeutic targets that could help restore barrier function while leaving the remaining essential functions of OSM unchanged.
Another potential alternative to systemic inhibition of OSM is through inhibition of GM-CSF. GM-CSF is elevated in nasal polyps, expressed by neutrophils and sufficient to induce neutrophil derived OSM in vitro.Citation85 Given that GM-CSF may promote prolonged survival in neutrophils as well as induce OSM, therapeutic targeting of GM-CSF could potentially be beneficial in the treatment of type 2 inflammatory diseases through the restricted indirect inhibition of OSM. Inhibition of GM-CSF could block neutrophil-derived OSM production, allowing epithelium to enter the late phase of repair and differentiation, potentially resolving chronic inflammation resulting from barrier dysfunction. Two strategies have been used to inhibit GM-CSF in human trials, monoclonal antibodies against GM-CSF, and a monoclonal antibody against GM-CSFRα. Clinical trials have been conducted with monoclonal antibodies against GM-CSF (MOR103, KB003, and namilumab) in rheumatoid arthritis, plaque psoriasis, and severe asthma. A phase II trial using MOR103 in RA patients showed that patients given higher doses of the antibody had improved symptom scores compared with placebo control and low dose MOR103.Citation164 A phase II trial was conducted using KB003 in asthmatic patients that were poorly controlled with long-acting bronchodilators and either inhaled or oral corticosteroids. The investigators did not observe improved forced expiratory volume in 1 second (FEV1) in the entire patient cohort. However, when patients were separated based on markers of poorly controlled asthma, peripheral blood eosinophilia, low baseline FEV1, and high bronchodilator reversibility, improvement in FEV1 was detected in response to treatment with KB003.Citation165 Clinical trials have also been conducted to test a monoclonal antibody against GM-CSFRα, mavrilimumab, for the treatment of RA, and showed reduced symptom scores in RA patients. The investigators concluded that further study of mavrilimumab for the treatment of RA was warranted.Citation166 Additionally, mavrilimumab, a monoclonal antibody against GM-CSFRα, has been shown to be safe, and a phase 2 trial of this drug showed no difference in the frequency of adverse effects between mavrilimumab and placebo treatment.Citation167 Identification of the factor or factors that are necessary to induce neutrophil derived GM-CSF could also provide another exciting therapeutic target, although it is important to note that any effect of GM-CSF inhibition could be independent of OSM (). The success of this approach would require not only that OSM is the major inducer of barrier dysfunction but also that GM-CSF is the major inducer of OSM; neither of these conclusions is firmly established. Another potential alternative to broad systemic inhibition of OSM could be through blocking the downstream effects of OSM by blocking the induction of EMT, which could include the inhibition of transcription factors like Snail, Zeb or Twist that drive EMT, or the inhibition of the EMT inducers, TGFβ, EGF, VEGF and FGF ().
Conclusion
Epithelial barrier dysfunction associated with EMT is an important feature of the pathogenesis of many type 2 inflammatory diseases, including CRS, asthma and EoE. We have identified a novel pathogenic role for the neutrophil derived cytokine, OSM, in the disruption of epithelial barrier function through the induction of EMT. Therapeutic targeting of OSM, its signaling, mediators of EMT, neutrophils, or GM-CSF may be beneficial in the treatment of these diseases through the restoration of epithelial barrier function, and further work is warranted to determine the validity of these potential therapeutic strategies.
Disclosure of potential conflicts of interest
Disclosures for RPS are consultancies with: Intersect ENT, GlaxoSmithKline, Allakos, Aurasense, Merck, BioMarck, Sanofi, Astra/Zeneca/Medimmune, Genentech, Exicure Inc, Otsuka Inc.
Funding
The work was supported in part by Grants R37HL068546 and U19AI106683 (Chronic Rhinosinusitis Integrative Studies Program (CRISP)) from the NIH, and by the Ernest S. Bazley Charitable Fund.
References
- Podolsky DK. Mucosal immunity and inflammation. V. Innate mechanisms of mucosal defense and repair: the best offense is a good defense. Am J Physiol 1999; 277(3 Pt 1):G495-9; PMID:10484372
- Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem 1998; 273(45):29745-53; PMID:9792688; https://doi.org/10.1074/jbc.273.45.29745
- Oppenheim JJ, Tewary P, de la Rosa G, Yang D. Alarmins initiate host defense. Adv Exp Med Biol 2007; 601:185-94; PMID:17713005
- Porter RM, Lane EB. Phenotypes, genotypes and their contribution to understanding keratin function. Trends Genet 2003; 19(5):278-85; PMID:12711220; https://doi.org/10.1016/S0168-9525(03)00071-4
- Avila PC, Schleimer RP. Airway Epithelium. Allergy and Allergic Diseases. Wiley-Blackwell 2009. p. 366-97
- Casanova L, Bravo A, Were F, Ramirez A, Jorcano JJ, Vidal M. Tissue-specific and efficient expression of the human simple epithelial keratin 8 gene in transgenic mice. J Cell Sci 1995; 108(Pt 2):811-20; PMID:7539440
- Staskowski PA, McCaffrey TV. Effect of substance P on ciliary beat frequency in human adenoid explants. Otolaryngol Head Neck Surg 1992; 107(4):553-7; PMID:1279501; https://doi.org/10.1177/019459989210700407
- Albertine KH, Williams MC, Hyde DM. Anatomy and development of the repiratory tract. Murray JF, Nadel JA, Mason RJ, Boushey HA, editor. Philadelphia: W. B. Saunders; 2000
- Squier CA, Kremer MJ. Biology of oral mucosa and esophagus. J Natl Cancer Inst Monogr 2001; (29):7-15; PMID:11694559; https://doi.org/10.1093/oxfordjournals.jncimonographs.a003443
- Habif TP. Clinical Dermatology. 5th ed. Edinburgh: Mosby Elsevier; 2010
- Ross MH, Kaye GI, Pawlina W. Histology: A Text and Atlas with Cell and Molecular Biology: Lippincott Williams & Wilkins. 2002
- Neunlist M, Van Landeghem L, Mahe MM, Derkinderen P, des Varannes SB, Rolli-Derkinderen M. The digestive neuronal-glial-epithelial unit: a new actor in gut health and disease. Nat Rev Gastroenterol Hepatol 2013; 10(2):90-100; PMID:23165236; https://doi.org/10.1038/nrgastro.2012.221
- Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol 2006; 22:207-35; PMID:16771626; https://doi.org/10.1146/annurev.cellbio.22.010305.104219
- Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol 2006; 68:403-29; PMID:16460278; https://doi.org/10.1146/annurev.physiol.68.040104.131404
- Mattagajasingh SN, Huang SC, Hartenstein JS, Benz EJ, Jr. Characterization of the interaction between protein 4.1R and ZO-2. A possible link between the tight junction and the actin cytoskeleton. J Biol Chem 2000; 275(39):30573-85; PMID:10874042; https://doi.org/10.1074/jbc.M004578200
- Tsukita S, Yamazaki Y, Katsuno T, Tamura A, Tsukita S. Tight junction-based epithelial microenvironment and cell proliferation. Oncogene 2008; 27(55):6930-8; PMID:19029935; https://doi.org/10.1038/onc.2008.344
- Lilien J, Balsamo J, Arregui C, Xu G. Turn-off, drop-out: functional state switching of cadherins. Dev Dyn 2002; 224(1):18-29; PMID:11984870; https://doi.org/10.1002/dvdy.10087
- McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol 2003; 162(6):1831-43; PMID:12759241; https://doi.org/10.1016/S0002-9440(10)64318-0
- Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol 2005; 17(5):459-65; PMID:16099633; https://doi.org/10.1016/j.ceb.2005.08.009
- Nelson WJ, Dickinson DJ, Weis WI. Roles of cadherins and catenins in cell-cell adhesion and epithelial cell polarity. Prog Mol Biol Transl Sci 2013; 116:3-23; PMID:23481188
- Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell 2005; 123(5):889-901; PMID:16325582; https://doi.org/10.1016/j.cell.2005.09.020
- Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Eng J Med 2002; 346(22):1699-705; PMID:12037149; https://doi.org/10.1056/NEJMoa012705
- Wardlaw AJ, Brightling CE, Green R, Woltmann G, Bradding P, Pavord ID. New insights into the relationship between airway inflammation and asthma. Clin Sci 2002; 103(2):201-11; PMID:12149112; https://doi.org/10.1042/cs1030201
- Leckie MJ, ten Brinke A, Khan J, Diamant Z, O'Connor BJ, Walls CM, Mathur AK, Cowley HC, Chung KF, Djukanovic R, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 2000; 356(9248):2144-8; PMID:11191542; https://doi.org/10.1016/S0140-6736(00)03496-6
- Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet 2007; 370(9596):1422-31; PMID:17950857; https://doi.org/10.1016/S0140-6736(07)61600-6
- Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 2012; 18(5):716-25; PMID:22561835; https://doi.org/10.1038/nm.2678
- Hackett TL, de Bruin HG, Shaheen F, van den Berge M, van Oosterhout AJ, Postma DS, Heijink IH. Caveolin-1 controls airway epithelial barrier function. Implications for asthma. Am J Respir Cell Mol Biol 2013; 49(4):662-71; PMID:23742006; https://doi.org/10.1165/rcmb.2013-0124OC
- Hackett TL, Singhera GK, Shaheen F, Hayden P, Jackson GR, Hegele RG, Van Eeden S, Bai TR, Dorscheid DR, Knight DA. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am J Respir Cell Mol Biol 2011; 45(5):1090-100; PMID:21642587; https://doi.org/10.1165/rcmb.2011-0031OC
- Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol 2011; 128(3):549-56 e1-12; PMID:21752437; https://doi.org/10.1016/j.jaci.2011.05.038
- Grencis RK, Humphreys NE, Bancroft AJ. Immunity to gastrointestinal nematodes: mechanisms and myths. Immunol Rev 2014; 260(1):183-205; PMID:24942690; https://doi.org/10.1111/imr.12188
- Metz M, Piliponsky AM, Chen CC, Lammel V, Abrink M, Pejler G, Tsai M, Galli SJ. Mast cells can enhance resistance to snake and honeybee venoms. Science 2006; 313(5786):526-30; PMID:16873664; https://doi.org/10.1126/science.1128877
- Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, Palmiter RD, Chawla A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 2014; 157(6):1292-308; PMID:24906148; https://doi.org/10.1016/j.cell.2014.03.066
- Chu VT, Beller A, Rausch S, Strandmark J, Zanker M, Arbach O, Kruglov A, Berek C. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity 2014; 40(4):582-93; PMID:24745334; https://doi.org/10.1016/j.immuni.2014.02.014
- Isobe Y, Kato T, Arita M. Emerging roles of eosinophils and eosinophil-derived lipid mediators in the resolution of inflammation. Front Immunol 2012; 3:270; PMID:22973272; https://doi.org/10.3389/fimmu.2012.00270
- Goldie RG, Fernandes LB, Rigby PJ, Paterson JW. Epithelial dysfunction and airway hyperreactivity in asthma. Prog Clin Biol Res 1988; 263:317-29; PMID:2898153
- Heyman M, Desjeux JF. Cytokine-induced alteration of the epithelial barrier to food antigens in disease. Ann N Y Acad Sci 2000; 915:304-11; PMID:11193593; https://doi.org/10.1111/j.1749-6632.2000.tb05258.x
- Tieu DD, Kern RC, Schleimer RP. Alterations in epithelial barrier function and host defense responses in chronic rhinosinusitis. J Allergy Clin Immunol 2009; 124(1):37-42; PMID:19560577; https://doi.org/10.1016/j.jaci.2009.04.045
- Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Eng J Med 2011; 365(14):1315-27; PMID:21991953; https://doi.org/10.1056/NEJMra1011040
- De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, Berger AE, Zhang K, Vidyasagar S, Yoshida T, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 2011; 127(3):773-86 e1-7; PMID:21163515; https://doi.org/10.1016/j.jaci.2010.10.018
- Venkataraman D, Soto-Ramirez N, Kurukulaaratchy RJ, Holloway JW, Karmaus W, Ewart SL, Arshad SH, Erlewyn-Lajeunesse M. Filaggrin loss-of-function mutations are associated with food allergy in childhood and adolescence. J Allergy Clin Immunol 2014; 134(4):876-82 e4; https://doi.org/10.1016/j.jaci.2014.07.033
- Palmer CN, Ismail T, Lee SP, Terron-Kwiatkowski A, Zhao Y, Liao H, Smith FJ, McLean WH, Mukhopadhyay S. Filaggrin null mutations are associated with increased asthma severity in children and young adults. J Allergy Clin Immunol 2007; 120(1):64-8; PMID:17531295; https://doi.org/10.1016/j.jaci.2007.04.001
- Weidinger S, O'Sullivan M, Illig T, Baurecht H, Depner M, Rodriguez E, Ruether A, Klopp N, Vogelberg C, Weiland SK, et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J Allergy Clin Immunol 2008; 121(5):1203-9 e1; https://doi.org/10.1016/j.jaci.2008.02.014
- Dale BA, Holbrook KA, Steinert PM. Assembly of stratum corneum basic protein and keratin filaments in macrofibrils. Nature 1978; 276(5689):729-31; PMID:732879; https://doi.org/10.1038/276729a0
- Harding CR, Scott IR. Histidine-rich proteins (filaggrins): structural and functional heterogeneity during epidermal differentiation. J Mol Biol 1983; 170(3):651-73; PMID:6195345; https://doi.org/10.1016/S0022-2836(83)80126-0
- Mocsai G, Gaspar K, Nagy G, Irinyi B, Kapitany A, Biro T, Gyimesi E, Tóth B, Maródi L, Szegedi A. Severe skin inflammation and filaggrin mutation similarly alter the skin barrier in patients with atopic dermatitis. Br J Dermatol 2014; 170(3):617-24; PMID:24251354; https://doi.org/10.1111/bjd.12743
- Spergel JM, Paller AS. Atopic dermatitis and the atopic march. J Allergy Clin Immunol 2003; 112(6 Suppl):S118-27; PMID:14657842; https://doi.org/10.1016/j.jaci.2003.09.033
- Trejo Bittar HE, Yousem SA, Wenzel SE. Pathobiology of severe asthma. Annu Rev Pathol 2015; 10:511-45; PMID:25423350; https://doi.org/10.1146/annurev-pathol-012414-040343
- Shah A, Kagalwalla AF, Gonsalves N, Melin-Aldana H, Li BU, Hirano I. Histopathologic variability in children with eosinophilic esophagitis. Am J Gastroenterol 2009; 104(3):716-21; PMID:19209168; https://doi.org/10.1038/ajg.2008.117
- Rose TM, Bruce AG. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc Natl Acad Sci U S A 1991; 88(19):8641-5; PMID:1717982; https://doi.org/10.1073/pnas.88.19.8641
- Tamura S, Morikawa Y, Miyajima A, Senba E. Expression of oncostatin M in hematopoietic organs. Dev Dyn 2002; 225(3):327-31; PMID:12412016; https://doi.org/10.1002/dvdy.10156
- Brown TJ, Lioubin MN, Marquardt H. Purification and characterization of cytostatic lymphokines produced by activated human T lymphocytes. Synergistic antiproliferative activity of transforming growth factor beta 1, interferon-gamma, and oncostatin M for human melanoma cells. J Immunol 1987; 139(9):2977-83
- Salamon P, Shoham NG, Puxeddu I, Paitan Y, Levi-Schaffer F, Mekori YA. Human mast cells release oncostatin M on contact with activated T cells: possible biologic relevance. J Allergy Clin Immunol 2008; 121(2):448-55 e5; https://doi.org/10.1016/j.jaci.2007.08.054
- Grove RI, Mazzucco C, Allegretto N, Kiener PA, Spitalny G, Radka SF, Shoyab M, Antonaccio M, Warr GA. Macrophage-derived factors increase low density lipoprotein uptake and receptor number in cultured human liver cells. J Lipid Res 1991; 32(12):1889-97; PMID:1816320
- Scaffidi AK, Mutsaers SE, Moodley YP, McAnulty RJ, Laurent GJ, Thompson PJ, Knight DA. Oncostatin M stimulates proliferation, induces collagen production and inhibits apoptosis of human lung fibroblasts. Br J Pharmacol 2002; 136(5):793-801; PMID:12086989; https://doi.org/10.1038/sj.bjp.0704769
- Gearing DP, Comeau MR, Friend DJ, Gimpel SD, Thut CJ, McGourty J, Brasher KK, King JA, Gillis S, Mosley B, et al. The IL-6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science 1992; 255(5050):1434-7; PMID:1542794; https://doi.org/10.1126/science.1542794
- Mosley B, De Imus C, Friend D, Boiani N, Thoma B, Park LS, Cosman D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J Biol Chem 1996; 271(51):32635-43; PMID:8999038; https://doi.org/10.1074/jbc.271.51.32635
- Ichihara M, Hara T, Kim H, Murate T, Miyajima A. Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood 1997; 90(1):165-73; PMID:9207450
- Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 2009; 206(7):1465-72; PMID:19564350; https://doi.org/10.1084/jem.20082683
- Soldi R, Graziani A, Benelli R, Ghigo D, Bosia A, Albini A, Bussolino F. Oncostatin M activates phosphatidylinositol-3-kinase in Kaposi's sarcoma cells. Oncogene 1994; 9(8):2253-60; PMID:8036010
- Stancato LF, Sakatsume M, David M, Dent P, Dong F, Petricoin EF, Krolewski JJ, Silvennoinen O, Saharinen P, Pierce J, et al. Beta interferon and oncostatin M activate Raf-1 and mitogen-activated protein kinase through a JAK1-dependent pathway. Mol Cell Biol 1997; 17(7):3833-40; PMID:9199317; https://doi.org/10.1128/MCB.17.7.3833
- Lutticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, Harpur AG, Wilks AF, Yasukawa K, Taga T, et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 1994; 263(5143):89-92; PMID:8272872; https://doi.org/10.1126/science.8272872
- Ernst M, Oates A, Dunn AR. Gp130-mediated signal transduction in embryonic stem cells involves activation of Jak and Ras/mitogen-activated protein kinase pathways. J Biol Chem 1996; 271(47):30136-43; PMID:8939963; https://doi.org/10.1074/jbc.271.47.30136
- Lamb P, Seidel HM, Haslam J, Milocco L, Kessler LV, Stein RB, Rosen J. STAT protein complexes activated by interferon-gamma and gp130 signaling molecules differ in their sequence preferences and transcriptional induction properties. Nucleic Acids Res 1995; 23(16):3283-9; PMID:7667105; https://doi.org/10.1093/nar/23.16.3283
- Kuropatwinski KK, De Imus C, Gearing D, Baumann H, Mosley B. Influence of subunit combinations on signaling by receptors for oncostatin M, leukemia inhibitory factor, and interleukin-6. J Biol Chem 1997; 272(24):15135-44; PMID:9182534; https://doi.org/10.1074/jbc.272.24.15135
- Richards CD, Brown TJ, Shoyab M, Baumann H, Gauldie J. Recombinant oncostatin M stimulates the production of acute phase proteins in HepG2 cells and rat primary hepatocytes in vitro. J Immunol 1992; 148(6):1731-6
- Holzer RG, Ryan RE, Tommack M, Schlekeway E, Jorcyk CL. Oncostatin M stimulates the detachment of a reservoir of invasive mammary carcinoma cells: role of cyclooxygenase-2. Clin Exp Metastasis 2004; 21(2):167-76; PMID:15168734; https://doi.org/10.1023/B:CLIN.0000024760.02667.db
- Langdon C, Kerr C, Hassen M, Hara T, Arsenault AL, Richards CD. Murine oncostatin M stimulates mouse synovial fibroblasts in vitro and induces inflammation and destruction in mouse joints in vivo. Am J Pathol 2000; 157(4):1187-96; PMID:11021823; https://doi.org/10.1016/S0002-9440(10)64634-2
- Hintzen C, Quaiser S, Pap T, Heinrich PC, Hermanns HM. Induction of CCL13 expression in synovial fibroblasts highlights a significant role of oncostatin M in rheumatoid arthritis. Arthritis Rheum 2009; 60(7):1932-43; PMID:19565514; https://doi.org/10.1002/art.24602
- Malik N, Haugen HS, Modrell B, Shoyab M, Clegg CH. Developmental abnormalities in mice transgenic for bovine oncostatin M. Mol Cell Biol 1995; 15(5):2349-58; PMID:7739518; https://doi.org/10.1128/MCB.15.5.2349
- Zarling JM, Shoyab M, Marquardt H, Hanson MB, Lioubin MN, Todaro GJ. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc Natl Acad Sci U S A 1986; 83(24):9739-43; PMID:3540948; https://doi.org/10.1073/pnas.83.24.9739
- Beigel F, Friedrich M, Probst C, Sotlar K, Goke B, Diegelmann J, Brand S. Oncostatin M Mediates STAT3-Dependent Intestinal Epithelial Restitution via Increased Cell Proliferation, Decreased Apoptosis and Upregulation of SERPIN Family Members. PloS One 2014; 9(4):e93498; PMID:24710357; https://doi.org/10.1371/journal.pone.0093498
- Ganesh K, Das A, Dickerson R, Khanna S, Parinandi NL, Gordillo GM, Sen CK, Roy S. Prostaglandin E(2) induces oncostatin M expression in human chronic wound macrophages through Axl receptor tyrosine kinase pathway. J Immunol 2012; 189(5):2563-73; https://doi.org/10.4049/jimmunol.1102762
- Poling J, Gajawada P, Lorchner H, Polyakova V, Szibor M, Bottger T, Warnecke H, Kubin T, Braun T. The Janus face of OSM-mediated cardiomyocyte dedifferentiation during cardiac repair and disease. Cell Cycle 2012; 11(3):439-45; PMID:22262173; https://doi.org/10.4161/cc.11.3.19024
- Glezer I, Rivest S. Oncostatin M is a novel glucocorticoid-dependent neuroinflammatory factor that enhances oligodendrocyte precursor cell activity in demyelinated sites. Brain Behav Immun 2010; 24(5):695-704; PMID:20083191; https://doi.org/10.1016/j.bbi.2010.01.005
- Yamaguchi Y, Yoshikawa K. Cutaneous wound healing: an update. J Dermatol 2001; 28(10):521-34; PMID:11732719; https://doi.org/10.1111/j.1346-8138.2001.tb00025.x
- Jung ID, Noh KT, Lee CM, Chun SH, Jeong SK, Park JW, Park WS, Kim HW, Yun CH, Shin YK, et al. Oncostatin M induces dendritic cell maturation and Th1 polarization. Biochem Biophys Res Commun 2010; 394(2):272-8; PMID:20206608; https://doi.org/10.1016/j.bbrc.2010.02.153
- Broxmeyer HE, Bruns HA, Zhang S, Cooper S, Hangoc G, McKenzie AN, Dent AL, Schindler U, Naeger LK, Hoey T, et al. Th1 cells regulate hematopoietic progenitor cell homeostasis by production of oncostatin M. Immunity 2002; 16(6):815-25; PMID:12121663; https://doi.org/10.1016/S1074-7613(02)00319-9
- Koshy PJ, Lundy CJ, Rowan AD, Porter S, Edwards DR, Hogan A, Clark IM, Cawston TE. The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: a time-course study using real-time quantitative reverse transcription-polymerase chain reaction. Arthritis Rheum 2002; 46(4):961-7; PMID:11953973; https://doi.org/10.1002/art.10212
- Rioja I, Hughes FJ, Sharp CH, Warnock LC, Montgomery DS, Akil M, Wilson AG, Binks MH, Dickson MC. Potential novel biomarkers of disease activity in rheumatoid arthritis patients: CXCL13, CCL23, transforming growth factor alpha, tumor necrosis factor receptor superfamily member 9, and macrophage colony-stimulating factor. Arthritis Rheum 2008; 58(8):2257-67; PMID:18668547; https://doi.org/10.1002/art.23667
- Le Goff B, Singbrant S, Tonkin BA, Martin TJ, Romas E, Sims NA, Walsh NC. Oncostatin M acting via OSMR, augments the actions of IL-1 and TNF in synovial fibroblasts. Cytokine 2014; 68(2):101-9; PMID:24767864; https://doi.org/10.1016/j.cyto.2014.04.001
- Simpson JL, Baines KJ, Boyle MJ, Scott RJ, Gibson PG. Oncostatin M (OSM) is increased in asthma with incompletely reversible airflow obstruction. Exp Lung Res 2009; 35(9):781-94; PMID:19916861; https://doi.org/10.3109/01902140902906412
- Kang HJ, Kang JS, Lee SH, Hwang SJ, Chae SW, Woo JS, Lee HM. Upregulation of oncostatin m in allergic rhinitis. Laryngoscope 2005; 115(12):2213-6; PMID:16369169; https://doi.org/10.1097/01.mlg.0000187819.89889.4a
- Boniface K, Diveu C, Morel F, Pedretti N, Froger J, Ravon E, Garcia M, Venereau E, Preisser L, Guignouard E, et al. Oncostatin M secreted by skin infiltrating T lymphocytes is a potent keratinocyte activator involved in skin inflammation. J Immunol 2007; 178(7):4615-22; https://doi.org/10.4049/jimmunol.178.7.4615
- Pothoven KL, Norton JE, Hulse KE, Suh LA, Carter RG, Rocci E, Harris KE, Shintani-Smith S, Conley DB, Chandra RK, et al. Oncostatin M promotes mucosal epithelial barrier dysfunction, and its expression is increased in patients with eosinophilic mucosal disease. J Allergy Clin Immunol 2015; 136(3):737-46 e4; https://doi.org/10.1016/j.jaci.2015.01.043
- Pothoven KL, Norton JE, Suh LA, Carter RG, Harris KE, Biyasheva A, Welch K, Shintani-Smith S, Conley DB, Liu MC, et al. Neutrophils are a major source of the epithelial barrier disrupting cytokine Oncostatin M in patients with mucosal airways disease. J Allergy Clin Immunol, 2017; 139(6):p. 1966-1978 e9.
- Fritz DK, Kerr C, Fattouh R, Llop-Guevara A, Khan WI, Jordana M, Richards CD. A mouse model of airway disease: oncostatin M-induced pulmonary eosinophilia, goblet cell hyperplasia, and airway hyperresponsiveness are STAT6 dependent, and interstitial pulmonary fibrosis is STAT6 independent. J Immunol 2011; 186(2):1107-18; https://doi.org/10.4049/jimmunol.0903476
- Botelho FM, Rangel-Moreno J, Fritz D, Randall TD, Xing Z, Richards CD. Pulmonary expression of oncostatin M (OSM) promotes inducible BALT formation independently of IL-6, despite a role for IL-6 in OSM-driven pulmonary inflammation. J Immunol 2013; 191(3):1453-64; https://doi.org/10.4049/jimmunol.1203318
- Gazel A, Rosdy M, Bertino B, Tornier C, Sahuc F, Blumenberg M. A characteristic subset of psoriasis-associated genes is induced by oncostatin-M in reconstituted epidermis. J Invest Dermatol 2006; 126(12):2647-57; PMID:16917497; https://doi.org/10.1038/sj.jid.5700461
- Lin YZ, Li RN, Lin CH, Ou TT, Wu CC, Tsai WC, Liu HW, Yen JH. Association of OSMR gene polymorphisms with rheumatoid arthritis and systemic lupus erythematosus patients. Autoimmunity 2014; 47(1):23-6; PMID:24219225; https://doi.org/10.3109/08916934.2013.849701
- Hong IK, Eun YG, Chung DH, Kwon KH, Kim DY. Association of the oncostatin m receptor gene polymorphisms with papillary thyroid cancer in the korean population. Clin Exp Otorhinolaryngol 2011; 4(4):193-8; PMID:22232715; https://doi.org/10.3342/ceo.2011.4.4.193
- Matzinger P, Kamala T. Tissue-based class control: the other side of tolerance. Nat Rev Immunol 2011; 11(3):221-30; PMID:21350581; https://doi.org/10.1038/nri2940
- Knisely TL, Luckenbach MW, Fischer BJ, Niederkorn JY. Destructive and nondestructive patterns of immune rejection of syngeneic intraocular tumors. J Immunol 1987; 138(12):4515-23
- Linthicum DS, Mackay IR, Carnegie PR. Measurement of cell-mediated inflammation in experimental murine autoimmune encephalomyelitis by radioisotopic labeling. J Immunol 1979; 123(4):1799-805
- Wilbanks GA, Mammolenti M, Streilein JW. Studies on the induction of anterior chamber-associated immune deviation (ACAID). III. Induction of ACAID depends upon intraocular transforming growth factor-beta. Eur J Immunol 1992; 22(1):165-73; PMID:1530916; https://doi.org/10.1002/eji.1830220125
- Kosiewicz MM, Alard P, Streilein JW. Alterations in cytokine production following intraocular injection of soluble protein antigen: impairment in IFN-gamma and induction of TGF-beta and IL-4 production. J Immunol 1998; 161(10):5382-90
- Jankowski R, Bouchoua F, Coffinet L, Vignaud JM. Clinical factors influencing the eosinophil infiltration of nasal polyps. Rhinology 2002; 40(4):173-8; PMID:12526243
- Bachert C, Zhang N, Holtappels G, De Lobel L, van Cauwenberge P, Liu S, Lin P, Bousquet J, Van Steen K. Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J Allergy Clin Immunol 2010; 126(5):962-8, 8 e1-6; PMID:20810157; https://doi.org/10.1016/j.jaci.2010.07.007
- Cao PP, Li HB, Wang BF, Wang SB, You XJ, Cui YH, Wang DY, Desrosiers M, Liu Z. Distinct immunopathologic characteristics of various types of chronic rhinosinusitis in adult Chinese. J Allergy Clin Immunol 2009; 124(3):478-84, 84 e1-2; PMID:19541359; https://doi.org/10.1016/j.jaci.2009.05.017
- Wen W, Liu W, Zhang L, Bai J, Fan Y, Xia W, Luo Q, Zheng J, Wang H, Li Z, et al. Increased neutrophilia in nasal polyps reduces the response to oral corticosteroid therapy. J Allergy Clin Immunol 2012; 129(6):1522-8 e5; PMID:22460066; https://doi.org/10.1016/j.jaci.2012.01.079
- Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ. Neutrophilic inflammation in severe persistent asthma. Am J Respiratory Critical Care Med 1999; 160(5 Pt 1):1532-9; PMID:10556116; https://doi.org/10.1164/ajrccm.160.5.9806170
- Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest 2001; 119(5):1329-36; https://doi.org/10.1378/chest.119.5.1329
- Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet 2009; 373(9678):1905-17; https://doi.org/10.1016/S0140-6736(09)60326-3
- Grenier A, Combaux D, Chastre J, Gougerot-Pocidalo MA, Gibert C, Dehoux M, Chollet-Martin S. Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab Invest 2001; 81(2):133-41; PMID:11232634; https://doi.org/10.1038/labinvest.3780220
- Cross A, Edwards SW, Bucknall RC, Moots RJ. Secretion of oncostatin M by neutrophils in rheumatoid arthritis. Arthritis Rheum 2004; 50(5):1430-6; PMID:15146412; https://doi.org/10.1002/art.20166
- Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res 2005; 65(19):8896-904; PMID:16204061; https://doi.org/10.1158/0008-5472.CAN-05-1734
- Su CM, Chiang YC, Huang CY, Hsu CJ, Fong YC, Tang CH. Osteopontin Promotes Oncostatin M production in human osteoblasts: Implication of rheumatoid arthritis therapy. J Immunol 2015; 195(7):3355-64; https://doi.org/10.4049/jimmunol.1403191
- Kastl SP, Speidl WS, Kaun C, Katsaros KM, Rega G, Afonyushkin T, Bochkov VN, Valent P, Assadian A, Hagmueller GW, et al. In human macrophages the complement component C5a induces the expression of oncostatin M via AP-1 activation. Arterioscler Thromb Vasc Biol 2008; 28(3):498-503; PMID:18187666; https://doi.org/10.1161/ATVBAHA.107.160580
- Repovic P, Benveniste EN. Prostaglandin E2 is a novel inducer of oncostatin-M expression in macrophages and microglia. J Neurosci 2002; 22(13):5334-43; PMID:12097485
- Miller M, Beppu A, Rosenthal P, Pham A, Das S, Karta M, Song DJ, Vuong C, Doherty T, Croft M, et al. Fstl1 promotes asthmatic airway remodeling by inducing oncostatin M. J Immunol 2015; 195(8):3546-56; https://doi.org/10.4049/jimmunol.1501105
- Kastl SP, Speidl WS, Katsaros KM, Kaun C, Rega G, Assadian A, Hagmueller GW, Hoeth M, de Martin R, Ma Y, et al. Thrombin induces the expression of oncostatin M via AP-1 activation in human macrophages: a link between coagulation and inflammation. Blood 2009; 114(13):2812-8; PMID:19652200; https://doi.org/10.1182/blood-2009-01-200915
- Elbjeirami WM, Donnachie EM, Burns AR, Smith CW. Endothelium-derived GM-CSF influences expression of oncostatin M. Am J Physiol Cell Physiol 2011; 301(4):C947-53; PMID:21775705; https://doi.org/10.1152/ajpcell.00205.2011
- Ohno I, Lea R, Finotto S, Marshall J, Denburg J, Dolovich J, Gauldie J, Jordana M. Granulocyte/macrophage colony-stimulating factor (GM-CSF) gene expression by eosinophils in nasal polyposis. Am J Respiratory Cell Mol Biol 1991; 5(6):505-10; PMID:1958376; https://doi.org/10.1165/ajrcmb/5.6.505
- Kato M, Liu MC, Stealey BA, Friedman B, Lichtenstein LM, Permutt S, Schleimer RP. Production of granulocyte/macrophage colony-stimulating factor in human airways during allergen-induced late-phase reactions in atopic subjects. Lymphokine Cytokine Res 1992; 11(6):287-92; PMID:1477181
- Miller M, Esnault S, Kurten RC, Kelly EA, Beppu A, Das S, Rosenthal P, Ramsdell J, Croft M, Zuraw B, et al. Segmental allergen challenge increases levels of airway follistatin-like 1 in patients with asthma. J Allergy Clin Immunol 2016; 138(2):596-599. e4; https://doi.org/10.1016/j.jaci.2016.01.019
- Anwar AR, Moqbel R, Walsh GM, Kay AB, Wardlaw AJ. Adhesion to fibronectin prolongs eosinophil survival. J Exp Med 1993; 177(3):839-43; PMID:8436913; https://doi.org/10.1084/jem.177.3.839
- Durand V, Renaudineau Y, Pers JO, Youinou P, Jamin C. Cross-linking of human FcgammaRIIIb induces the production of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor by polymorphonuclear neutrophils. J Immunol 2001; 167(7):3996-4007; https://doi.org/10.4049/jimmunol.167.7.3996
- Tan BK, Li QZ, Suh L, Kato A, Conley DB, Chandra RK, Zhou J, Norton J, Carter R, Hinchcliff M, et al. Evidence for intranasal antinuclear autoantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 2011; 128(6):1198-206 e1; https://doi.org/10.1016/j.jaci.2011.08.037
- Engelhardt E, Toksoy A, Goebeler M, Debus S, Brocker EB, Gillitzer R. Chemokines IL-8, GROalpha, MCP-1, IP-10, and Mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am J Pathol 1998; 153(6):1849-60; PMID:9846975; https://doi.org/10.1016/S0002-9440(10)65699-4
- Goren I, Kampfer H, Muller E, Schiefelbein D, Pfeilschifter J, Frank S. Oncostatin M expression is functionally connected to neutrophils in the early inflammatory phase of skin repair: implications for normal and diabetes-impaired wounds. J Invest Dermatol 2006; 126(3):628-37; PMID:16410783; https://doi.org/10.1038/sj.jid.5700136
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009; 16(3):183-94; PMID:19732719; https://doi.org/10.1016/j.ccr.2009.06.017
- Mishalian I, Bayuh R, Levy L, Zolotarov L, Michaeli J, Fridlender ZG. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol Immunother 2013; 62(11):1745-56; PMID:24092389; https://doi.org/10.1007/s00262-013-1476-9
- Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res 2016; 110(1):51-61; PMID:26825554; https://doi.org/10.1093/cvr/cvw024
- Cuartero MI, Ballesteros I, Moraga A, Nombela F, Vivancos J, Hamilton JA, Corbí ÁL, Lizasoain I, Moro MA. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARgamma agonist rosiglitazone. Stroke 2013; 44(12):3498-508; PMID:24135932; https://doi.org/10.1161/STROKEAHA.113.002470
- Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P, Ding JL. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest 2013; 93(7):844-54; PMID:23752129; https://doi.org/10.1038/labinvest.2013.69
- Nightingale J, Patel S, Suzuki N, Buxton R, Takagi KI, Suzuki J, Sumi Y, Imaizumi A, Mason RM, Zhang Z. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J Am Society Nephrology 2004; 15(1):21-32; PMID:14694154; https://doi.org/10.1097/01.ASN.0000102479.92582.43
- Schleimer RP. Immunopathogenesis of Chronic Rhinosinusitis and Nasal Polyposis. Ann Rev Path 2017. 12: p. 331-357.
- West NR, Murphy LC, Watson PH. Oncostatin M suppresses oestrogen receptor-alpha expression and is associated with poor outcome in human breast cancer. Endocr Relat Cancer 2012; 19(2):181-95; PMID:22267707; https://doi.org/10.1530/ERC-11-0326
- West NR, Murray JI, Watson PH. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2014; 33(12):1485-94; PMID:23584474; https://doi.org/10.1038/onc.2013.105
- Bolin C, Tawara K, Sutherland C, Redshaw J, Aranda P, Moselhy J, Anderson R, Jorcyk CL. Oncostatin m promotes mammary tumor metastasis to bone and osteolytic bone degradation. Genes Cancer 2012; 3(2):117-30; PMID:23050044; https://doi.org/10.1177/1947601912458284
- Kucia-Tran JA, Tulkki V, Smith S, Scarpini CG, Hughes K, Araujo AM, Yan KY, Botthof J, Pérez-Gómez E, Quintanilla M, et al. Overexpression of the oncostatin-M receptor in cervical squamous cell carcinoma is associated with epithelial-mesenchymal transition and poor overall survival. Br J Cancer 2016; 115(2):212-22; PMID:27351213; https://doi.org/10.1038/bjc.2016.199
- Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, Kaur H, Bhatt ML, Bhadauria S. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget 2014; 5(14):5350-68; PMID:25051364; https://doi.org/10.18632/oncotarget.2110
- Heusinkveld M, van der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. J Transl Med 2011; 9:216; PMID:22176642; https://doi.org/10.1186/1479-5876-9-216
- Uribe-Querol E, Rosales C. Neutrophils in cancer: Two sides of the same coin. J Immunol Res 2015; 2015:983698; PMID:26819959; https://doi.org/10.1155/2015/983698
- Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D, Marches F, Banchereau J, Palucka AK. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med 2007; 204(5):1037-47; PMID:17438063; https://doi.org/10.1084/jem.20061120
- De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, Protti MP. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med 2011; 208(3):469-78; PMID:21339327; https://doi.org/10.1084/jem.20101876
- Lo Kuan E, Ziegler SF. Thymic stromal lymphopoietin and cancer. J Immunol 2014; 193(9):4283-8; https://doi.org/10.4049/jimmunol.1400864
- Liu X, Hemminki K, Forsti A, Sundquist J, Sundquist K, Ji J. Cancer risk and mortality in asthma patients: A Swedish national cohort study. Acta Oncol 2015; 54(8):1120-7; PMID:25608824; https://doi.org/10.3109/0284186X.2014.1001497
- Santillan AA, Camargo CA, Jr, Colditz GA. A meta-analysis of asthma and risk of lung cancer (United States). Cancer Causes Control 2003; 14(4):327-34; PMID:12846363; https://doi.org/10.1023/A:1023982402137
- Eriksson NE, Holmen A, Hogstedt B, Mikoczy Z, Hagmar L. A prospective study of cancer incidence in a cohort examined for allergy. Allergy 1995; 50(9):718-22; PMID:8546265; https://doi.org/10.1111/j.1398-9995.1995.tb01212.x
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119(6):1420-8; PMID:19487818; https://doi.org/10.1172/JCI39104
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest 2003; 112(12):1776-84; PMID:14679171; https://doi.org/10.1172/JCI200320530
- Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 2009; 119(6):1429-37; PMID:19487819; https://doi.org/10.1172/JCI36183
- Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 1997; 273(4 Pt 2):F563-74; PMID:9362334
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100(1):57-70; PMID:10647931; https://doi.org/10.1016/S0092-8674(00)81683-9
- Strutz F, Zeisberg M, Ziyadeh FN, Yang CQ, Kalluri R, Muller GA, Neilson EG. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int 2002; 61(5):1714-28; PMID:11967021; https://doi.org/10.1046/j.1523-1755.2002.00333.x
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14(6):818-29; PMID:18539112; https://doi.org/10.1016/j.devcel.2008.05.009
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015; 519(7541):57-62; PMID:25731159; https://doi.org/10.1038/nature14228
- Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci 2012; 125(Pt 19):4417-22; PMID:23165231; https://doi.org/10.1242/jcs.099697
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 2009; 28(1–2):15-33; PMID:19169796; https://doi.org/10.1007/s10555-008-9169-0
- Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR. Cadherin switching. J Cell Sci 2008; 121(Pt 6):727-35; PMID:18322269; https://doi.org/10.1242/jcs.000455
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E. Defining the epithelial stem cell niche in skin. Science 2004; 303(5656):359-63; PMID:14671312; https://doi.org/10.1126/science.1092436
- Stein U, Arlt F, Walther W, Smith J, Waldman T, Harris ED, Mertins SD, Heizmann CW, Allard D, Birchmeier W, et al. The metastasis-associated gene S100A4 is a novel target of beta-catenin/T-cell factor signaling in colon cancer. Gastroenterology 2006; 131(5):1486-500; PMID:17101323; https://doi.org/10.1053/j.gastro.2006.08.041
- Haynes J, Srivastava J, Madson N, Wittmann T, Barber DL. Dynamic actin remodeling during epithelial-mesenchymal transition depends on increased moesin expression. Mol Biol Cell 2011; 22(24):4750-64; PMID:22031288; https://doi.org/10.1091/mbc.E11-02-0119
- Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 2006; 5(15):1597-601; PMID:16880743; https://doi.org/10.4161/cc.5.15.3112
- Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn 2005; 233(3):706-20; PMID:15937929; https://doi.org/10.1002/dvdy.20345
- Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J 2010; 24(6):1838-51; PMID:20097873; https://doi.org/10.1096/fj.09-151639
- Vongwiwatana A, Tasanarong A, Rayner DC, Melk A, Halloran PF. Epithelial to mesenchymal transition during late deterioration of human kidney transplants: the role of tubular cells in fibrogenesis. Am J Transplant 2005; 5(6):1367-74; PMID:15888043; https://doi.org/10.1111/j.1600-6143.2005.00843.x
- Walsh LA, Nawshad A, Medici D. Discoidin domain receptor 2 is a critical regulator of epithelial-mesenchymal transition. Matrix Biol 2011; 30(4):243-7; PMID:21477649; https://doi.org/10.1016/j.matbio.2011.03.007
- Szell M, Bata-Csorgo Z, Koreck A, Pivarcsi A, Polyanka H, Szeg C, Gaál M, Dobozy A, Kemény L. Proliferating keratinocytes are putative sources of the psoriasis susceptibility-related EDA+ (extra domain A of fibronectin) oncofetal fibronectin. J Invest Dermatol 2004; 123(3):537-46; PMID:15304094; https://doi.org/10.1111/j.0022-202X.2004.23224.x
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15(3):178-96; PMID:24556840; https://doi.org/10.1038/nrm3758
- Choy EH, Bendit M, McAleer D, Liu F, Feeney M, Brett S, Zamuner S, Campanile A, Toso J. Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti- oncostatin M monoclonal antibody in rheumatoid arthritis: results from phase II randomized, placebo-controlled trials. Arthritis Res Therapy 2013; 15(5):R132; https://doi.org/10.1186/ar4312
- Migita K, Izumi Y, Torigoshi T, Satomura K, Izumi M, Nishino Y, Jiuchi Y, Nakamura M, Kozuru H, Nonaka F, et al. Inhibition of Janus kinase/signal transducer and activator of transcription (JAK/STAT) signalling pathway in rheumatoid synovial fibroblasts using small molecule compounds. Clin Exp Immunol 2013; 174(3):356-63; PMID:23968543; https://doi.org/10.1111/cei.12190
- Brolund L, Kuster A, Korr S, Vogt M, Muller-Newen G. A receptor fusion protein for the inhibition of murine oncostatin M. BMC Biotechnol 2011; 11:3; PMID:21223559; https://doi.org/10.1186/1472-6750-11-3
- Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, Berenfus VY, Vladeva S, Rech J, Rubbert-Roth A, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis 2015; 74(6):1058-64; PMID:24534756; https://doi.org/10.1136/annrheumdis-2013-204816
- Molfino NA, Kuna P, Leff JA, Oh CK, Singh D, Chernow M, Sutton B, Yarranton G. Phase 2, randomised placebo-controlled trial to evaluate the efficacy and safety of an anti-GM-CSF antibody (KB003) in patients with inadequately controlled asthma. BMJ Open 2016; 6(1):e007709; PMID:26739717; https://doi.org/10.1136/bmjopen-2015-007709
- Takeuchi T, Tanaka Y, Close D, Godwood A, Wu CY, Saurigny D. Efficacy and safety of mavrilimumab in Japanese subjects with rheumatoid arthritis: findings from a Phase IIa study. Mod Rheumatol 2015; 25(1):21-30; PMID:24720551; https://doi.org/10.3109/14397595.2014.896448
- Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, Szombati I, Esfandiari E, Sleeman MA, Kane CD, et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis 2013; 72(9):1445-52; PMID:23234647; https://doi.org/10.1136/annrheumdis-2012-202450