
ABSTRACT
A principal function of endothelial cells is the formation of a barrier between the blood and tissues. This barrier arises from the physical connections at cell-cell junctions, which includes cytoskeletal tight junction and adherens junction proteins. Methods that alter barrier function must therefore affect these cell-cell connections. The blood brain barrier (BBB) represents perhaps the most selective endothelial barrier, which arises from endothelial cell interactions with astrocytes and pericytes. Even in non-central nervous system (CNS) endothelial cells, barrier properties can be enhanced, mimicking the BBB, through induction of intercellular junctions, by either direct co-culture with astrocytes, supplementation with astrocyte conditioned medium (ACM) and/or pharmacologic enhancement of cAMP. To understand how cell-cell junctions change during endothelial barrier enhancement, we examined the effects of ACM and/or cAMP donors added to standard media on human umbilical vein endothelial cells (HUVEC). HUVEC cultured with cAMP-elevating agents had the most enhanced barrier function as measured by Electric Cell-substrate Impedance Sensing (ECIS®), a real-time, label-free, impedance based method of studying cell barrier properties. However, subtle differences in actin and cell-cell junction proteins were seen across all four culture conditions. cAMP-elevating agents also triggered the redistribution of ZO-1 and VE-cadherin to cell-cell junctions, and intensified the actin microfilament network at the cell cortex. Using a VE-cadherin FRET-force sensor, we observed a decrease in VE-cadherin force in HUVEC cultured with ACM with cAMP donors. Our data indicate cAMP elevation induces both junctional strengthening and reduced VE-cadherin forces. Additionally, treatment with an inhibitor of formin, which reduced actin stress fibers, enhanced barrier function. These data suggest that barrier function is modulated both through the trafficking of proteins to cell-cell junctions, and through the modulation and a relaxation of mechanical force through adherens junctions as intercellular junctional complexes become established.
Introduction
A significant function of endothelial cells is to form a barrier between tissues and blood. Endothelial barriers arise through the formation of adherens and tight junctional complexes, which join endothelial cells together and restrict solute flux. During inflammation, cytokines alter adherens and tight junctions to allow transmigration of leukocytes and fluid leakage into the CNS.Citation1-5 Alterations in blood-brain barrier (BBB) permeability are observed in many disease states, including atherosclerosis, diabetes, hypertension, inflammation, sepsis, and HIV.Citation6,Citation7
Endothelial cells of the BBB are surrounded by other cell types, including astrocytic endfeet, pericytes, microglial and neuronal processes.Citation8 In particular, perivascular astrocytes are critical drivers of the induction of the BBB phenotype.Citation8-11 Astrocytes can lead to increases in expression of tight junctional proteins and decreases in barrier permeability to paracellular compounds—suggesting enhanced barrier function.Citation12 Astrocytes increase barrier tightening of endothelial cells through both contact- dependent and independent mechanisms suggesting the importance of both bound and soluble factors produced by astrocytes.Citation11,Citation13-16 Barrier properties can also be enhanced by activating key signaling events associated with the maintenance of junctional complexes such as through supplementation of the culture medium with cAMP analogues.Citation10,Citation14,Citation17
Myosin-dependent tension on cell-cell junctions is believed to play a key role in regulating junctional stability and hence vascular permeability.Citation18 Thus, mechanical forces may play an important role in endothelial barrier function. We have previously shown that VE-cadherin, the transmembrane protein in endothelial adherens junctions, is subject to myosin-dependent tension.Citation19 Barrier-disrupting agents, such as thrombin, have been reported to increase myosin phosphorylation and induce actin stress fibers. Additionally, blocking myosin inhibits the increase in endothelial permeability by these factors.Citation20 On the other hand, myosin also promotes assembly of adherens junctions and there is evidence that this process involves strengthening of junctions under applied force.Citation21-23 Thus, although forces play a role in the formation and destabilization of endothelial junctions, the relationship of the forces across cell-cell contacts to the strengthening of the endothelial barrier is not yet known.
To better understand how cell-cell junctions change during formation of the endothelial barrier, we sought to understand the differences between normal HUVEC endothelial monolayers cultured in one of four conditions; 1) regular HUVEC media, 2) astrocyte-conditioned media (ACM), 3) cAMP analogues and 4) ACM plus cAMP analogues. Endothelial cells stimulated with cAMP analogues had increased transendothelial electrical resistance, indicating a tighter barrier. These cells also exhibited dramatic increase in the localization of VE-cadherin and ZO-1 to cell-cell junctions. Additionally, cell-cell junctional morphology became more linear, with more cortical actin. Using a VE-cadherin FRET-force sensor, we observed that VE-cadherin force was decreased in these cells. Further supporting the role of mechanical force on barrier function, pharmacologic inhibition of formin proteins, which are known to regulate actin stress fiber formation, increased barrier function for cells grown in regular media. When taken together, our observations point to both junctional strengthening (more VE-cadherin at junctions) and reduced cell-cell junction forces as being associated with a stronger endothelial barrier.
Materials and reagents
Pooled primary human umbilical vein endothelial cells (HUVEC) were purchased from Thermo Fisher (C01510C) and Lonza Biosciences (CC-2519). Cells between passages 2 and 8 were maintained in Medium 200 from Thermo Fisher (M-200500) with supplements included via the low serum growth supplement (LSGS) kit from Thermo Fisher (S-003-K) at 37°C with 5% CO2. Final concentrations of supplements include 2% fetal bovine serum, hydrocortisone (1 μg/ml), human epidermal growth factor (10 ng/ml), basic fibroblast growth factor (3 ng/ml) and heparin (10 μg/ml).
HUVECs were treated with ACM (mixed 50:50 with HUVEC media) ± 8-CPT-cAMP (250 μM) and Ro-20-1724 (17.5 μM) from Sigma Aldrich (B8279 and C3912) to increase intracellular cAMP. In separate experiments, cells were also treated with the formin inhibitor, SMIFH2 (10 μM; Tocris). For immunostaining, immunocytochemistry, and FRET imaging, HUVEC were treated for 24 h. For the TEER measurements, experiments were allowed to continue for 36 h.
Transendothelial Resistance. Transendothelial resistance (TEER) of HUVEC monolayers was measured in real time using the ECIS system (1600R; Applied BioPhysics, Troy, NY). The Electric Cell-substrate Impedance Sensing (ECIS) system provides real-time monitoring of TEER changes. In brief, HUVECs (9000 cells/well) were seeded and grown to confluence on gelatin-coated arrays (96W10idf). After the cells reached confluence (typically 24-48 h), the medium was changed to include the treatments. To assess the effects of supplements on TEER, treatment groups were as follows; 1) standard HUVEC media, 2) standard media supplemented with cAMP-enhancing agents [8-CPT-cAMP (250 μM) and Ro-20-1724 (17.5 μM)], 3) ACM, or 4) ACM plus cAMP-enhancing agents. Measurements were recorded in real time at 4000 Hz and data were normalized to baseline measurements just prior to the onset of treatment (t = 0). The baseline TEER readings for confluent HUVEC cells were between 600 and 1200 Ω.
FRET image acquisition and analysis. Images were acquired and analyzed as previously described.Citation19,Citation24 Briefly, VE-cadherin tension sensor (TS) was expressed in HUVEC using adenovirus. VE-cadherin TS expressing cells were seeded at confluence on fibronectin coated glass-bottom dishes. 24 h later, cells were exposed to either regular HUVEC media or ACM (mixed 50:50 with HUVEC media) with cAMP enhancing agents for an additional 24 h. Cells were fixed in 4% PFA and imaged on an inverted Zeiss LSM 710 confocal (Oberkochen, Germany) using a 458 nm excitation with a plan-apochromat 63× oil objective lens. Live cells expressing either soluble teal (mTFP1) or venus fluorescent proteins were imaged in spectral mode using a 32-channel spectral META detector. Cells expressing the VE-cadherin tension sensor were imaged using online-unmixing mode in the Zeiss Zen Software. In each experiment, images were captured on the same day, with the gain and laser intensities fixed across all samples. All images were background subtracted on the teal and venus channels, respectively, to reduce noise, and all saturated pixels were removed. Ratio images were calculated by dividing the unmixed venus channel by the unmixed teal channel. To reduce FRET noise from edge artifacts, pixels with very large FRET ratios (>20) were removed from analysis. To examine FRET pixels of interest, ratio images were multiplied with binary image masks that outlined discrete cell-cell junctions. For each discrete group of junctions, a median FRET ratio was determined and considered as one independent observation.
Immunocytochemistry. Confluent HUVEC grown on glass bottom dishes were cultured for 24 h with 1) regular HUVEC medium, 2) regular HUVEC medium with cAMP agonists, 3) 50:50 mix of ACM and HUVEC medium, 4) 50:50 mix of ACM and HUVEC medium with cAMP agonists. Cells were rinsed in PBS with calcium and then fixed in 4% paraformaldehyde in phosphate buffer, pH 7.2. Cells were stained with rabbit ZO-1 (Thermo, #40-2200), mouse VE-cadherin (Santa Cruz, clone BV9), rabbit VE-cadherin (Cell Signaling, clone D87F2), mouse vinculin (Sigma, clone hVIN-1), and rabbit α-catenin (Sigma, # C2081) and Alexa Fluor® secondary antibodies (Life Technologies). Images were acquired using a Zeiss 710 LSM confocal.
Immunoblotting
Whole cell lysates were prepared using RIPA buffer, centrifuged to clear insoluble material, and then boiled in Laemmli sample buffer. Samples were resolved with SDS-PAGE, transferred to a PVDF membrane and analyzed using standard western blotting techniques. Proteins were detected using VE-cadherin (Cell signaling, clone D87F2) and α-tubulin (ThermoFisher, clone DM1A) antibodies using standard HRP-chemiluminescent techniques.
Statistical analysis
Barrier integrity, generated by ECIS analyses, were assessed. ECIS data was analyzed via repeated measures analysis of variance (ANOVA) with conditioned cell culture medium (control or ACM) and time (12 h prior to treatment (-12 h) up to 60 h post-treatment) as between- and within-subjects factors, respectively. Significant interactions were delineated via assessment of simple main effects and main effect contrasts (that were alpha corrected for family-wise error). Two-group analyses were assessed via Student's independent t-test. All effects were considered significant when p < 0.05.
Results
cAMP analogue treatment increased barrier integrity and function
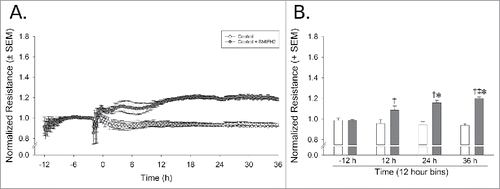
To assess the effects of medium supplements on an in vitro model of the BBB, HUVEC were exposed to 1) cAMP analogues, 2) ACM, or 3) ACM plus cAMP analogues and all conditions were compared to cells in normal growth medium. To confirm barrier function, barrier integrity was assessed using ECIS. Cells were plated on the electrode microarrays, allowed to grow until confluence, and then medium was replaced with one of the four treatment conditions. Electrical resistance was measured continuously throughout the experiment and the data was normalized to the baseline measurement just prior the application of the treatments.
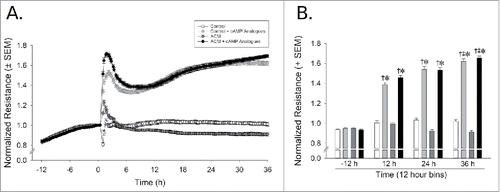
Normalized resistance (ohms) was influenced by the application of cAMP analogues which significantly interacted with time [F(3,24)=31.66, p < 0.05]. Compared to basal levels (−12 h from treatment), cAMP analogue supplementation significantly increased barrier resistance 12-36 h after treatment (p < 0.0001), and significantly differed from control cells at each time-point (p < 0.0001). This effect peaked at 36 h, when resistance was even greater than that observed 12 h post-treatment (p = 0.004) (). Pretreatment with ACM did not significantly influence resistance ()
Figure 1. cAMP enhances and maintains cell barrier function as compared to control media. Dynamic TEER monitoring reveals significant barrier enhancement with cAMP analogues by 12 h of exposure. (A) Normalized resistance versus time spanning from 12 h prior to treatment to 36 h post-treatment. (B) Data were binned in 12 h intervals. *Indicates cAMP analogue-treated cells significantly differ from control-treated cells, p < 0.05, †Indicates cAMP analogue-treated cells significantly differ from baseline measurements at −12 h, p < 0.05. ‡Indicates significant differences from the –12 h time-point, p < 0.05, n = 3 separate experiments/group, each performed in octuplicate.
cAMP analogue treatment alters tight junctions, adherens junctions, and the actin cytoskeleton
Next, we examined how the various treatments influenced both adherens and tight junctions. Confluent monolayers of endothelial cells grown on glass coverslips were exposed to 24 h of normal growth media, cAMP analogues, ACM, or ACM plus cAMP analogues. As compared to the cells grown without cAMP supplementation, treatment with cAMP analogues (in regular medium or ACM) increased ZO-1 accumulation at cell-cell junctions and resulted in a more linear morphology (), results which are consistent with increased barrier resistance (). Cells grown in the presence of the cAMP analogues had increased VE-cadherin recruitment to cell-cell contacts, along with a more continuous and linear VE-cadherin morphology at cell-cell junctions (). Interestingly, ACM with cAMP analogues resulted in the largest increase in cortical actin at cell-cell contacts ( and ).
Figure 2. ACM and cAMP treatments induce changes in the cytoskeleton and cell-cell junctions. (A) Cells treated with cAMP analogues had increased ZO-1 junctional recruitment and a more delineated, linear morphology. (B and C) Cells treated with ACM or cAMP enhancing agents had increased VE-cadherin and α-catenin junctional recruitment and more linear morphology. (D) Vinculin recruitment to cell-cell contacts was seen only with cAMP supplementation. Reduction of stress fibers and enhancement of cortical actin was most apparent in the ACM-treated cultures exposed to cAMP analogues (A through D).
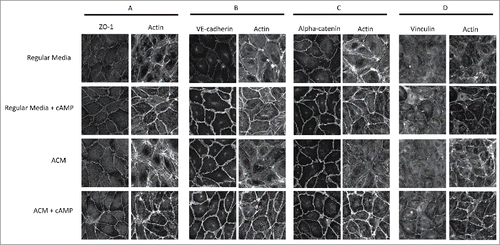
To investigate changes in the cytoskeletal connectivity of adherens junctions, vinculin and α-catenin were also stained in the four conditions. α-Catenin morphology () largely mirrored VE-cadherin morphology (), indicating that cAMP also recruits junctional proteins that link VE-cadherin to the cytoskeleton. Vinculin, a focal adhesion associated protein also known to be recruited to endothelial cell-cell junctions,Citation25 appeared to be enhanced by cAMP analogues (), which is consistent with a prior report showing that vinculin is important in barrier enhancement by oxidized phospholipids.Citation26
ACM plus cAMP treatment reduces the force across VE-cadherin
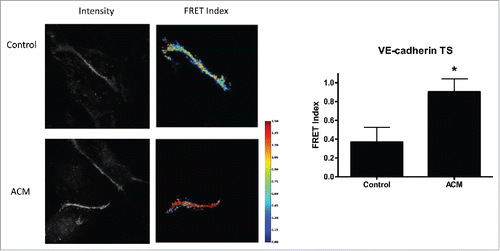
Because ACM with cAMP supplementation showed the most dramatic alterations of the actin cytoskeleton, we investigated how this treatment affected the mechanical force across VE-cadherin, using a previously developed and validated VE-cadherin tension biosensor which has an inverse FRET-force relationship.Citation19 Cells exposed to ACM plus cAMP analogues had reduced force (indicated by higher FRET) when compared to endothelial cells grown in normal cell culture medium (). These data indicate that enhanced barrier function is correlated to reduced mechanical force across VE-cadherin.
Figure 3. ACM reduces VE-cadherin force. Cells expressing VE-cadherin TS had higher FRET values, indicating reduced force, when cultured in ACM media as compared to cells cultured in standard medium. Example fluorescent images and corresponding FRET index images are shown; FRET index images were masked to segment individual junctions and remove intracellular signal. Median FRET was determined for each junction and plotted as mean ± SEM, with a minimum of 5 junctions per condition. Two additional experiments showed similar results.
ACM and cAMP analogues do not affect overall VE-cadherin expression
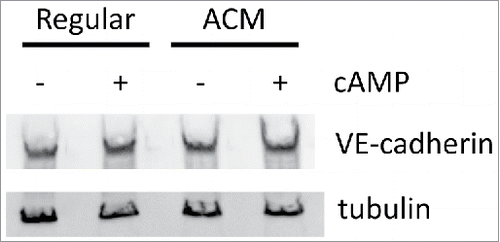
Because we observed increased VE-cadherin recruitment to cell-cell junctions (), we examined whether ACM or cAMP agonists affected VE-cadherin protein expression. Western blotting showed similar expression of VE-cadherin across all 4 culture conditions (), indicating that ACM and cAMP affect VE-cadherin localization and not protein expression.
Figure 4. ACM and cAMP do not affect protein expression of VE-cadherin. Total cell lysates were blotted for VE-cadherin and tubulin (as a loading control). No large differences were observed for VE-cadherin expression across the four treatment conditions.
Role of formin in mediating barrier function
In order to test the hypothesis that ACM and cAMP-induced changes in the actin cytoskeleton affect barrier function, we used the the formin inhibitor SMIFH2. We observed that SMIFH2 resulted in a loss of actin stress fibers within 4 h of treatment (Supplemental Figure 2). To confirm effects of formin inhibition on barrier function, barrier integrity was assessed using ECIS for SMIFH2-treated cells. HUVEC cells were plated on the electrode microarrays, allowed to grow until confluence, and then media was replaced with one of the four conditions; normal growth medium ± SMIFH2, and ACM plus cAMP analogues ± SMIFH2. TEER was measured continuously throughout the experiment and the data were normalized to the baseline measurement just prior to the application of the treatments. Data were binned in 12 h intervals, as described above. Inhibition of the formin FH2 domain, via application of SMIFH2, significantly interacted with time to increase normalized resistance [F(3,12)=17.79, p < 0.05]. SMIFH2-treated cells demonstrated significantly increased resistance at 12 (p = 0.02), 24 (p = 0.002), and 36 h (p = 0.0003) post-treatment, compared to baseline resistance (12 h prior to treatment) (). As with cAMP analogues, this effect peaked at 36 h when resistance was even greater than that observed 12 h post-treatment (p = 0.01). SMIFH2-treated cells significantly differed from control-treated cells at 24 (p = 0.006) and 36 h (p = 0.0003) post-treatment. No significant changes in barrier function were noted upon addition of SMIFH2 to ACM plus cAMP analogue medium, as compared to the same medium without SMIFH2 (data not shown).
Figure 5. Inhibition of stress fibers enhances barrier function. Dynamic TEER monitoring reveals significant barrier enhancement with the formin inhibitor SMIFH2 by 12 h of exposure. (A) Normalized resistance versus time spanning from 12 h prior to treatment to 36 h post-treatment. (B) Data were binned in 12 h intervals. *Indicates SMIFH2 treated cells significantly differ from control cells, p < 0.05. †Indicates SMIFH2 treated cells significantly differ from the −12 h time-point, p < 0.05. ‡Indicates significant differences from the –12 h time-point, p < 0.05, n = 3 separate experiments/group, each performed in octuplicate.
Discussion
Although cell-cell junctions have been shown to be necessary for barrier formation, the mechanisms by which individual junctional proteins participate in barrier strengthening are not fully understood. Mechanical force across the cell-cell junction, through the adherens junction, has been thought to be an important mediator of junctional stability and barrier function. In this study, we used cAMP analogues to induce stronger barrier function in HUVEC. We demonstrate that cAMP enhancement can tighten endothelial barrier properties as measured by increases in TEER (), beginning at 12 h of treatment and lasting at least until 36 h. Additionally at 24 h, it was observed that adherens junctions, tight junctions, and the actin cytoskeleton underwent dramatic changes in response to cAMP agonists (). These changes were also associated with reduced VE-cadherin force, measured using a VE-cadherin force biosensor. When taken together, our data indicate that increased localization of proteins to cell-cell junctions, cytoskeletal rearrangement, and reduced force across adherens junctions contribute to a stronger endothelial barrier.
Unstimulated HUVEC (grown in regular medium) form a uniform monolayer with cell-cell junctions when confluent; however, this monolayer does not have a strong or tight barrier characteristic of the BBB. Common approaches used to enhance barrier properties of endothelial cells include co-culturing endothelial cells with astrocytes in either a contact or non-contact formation, co-culturing endothelial cells with pericytes.Citation27,Citation28 and/or adding cAMP analogues to the culture medium.Citation10,Citation29 Astrocyte-endothelial cell co-cultures lead to increased tight junction and adherens junction protein expression, increased impedance across the barrier (TEER) and decreased permeability to paracellular compounds.Citation16,Citation30 The enhanced barrier seen when co-culturing endothelial cells with astroglia is observed in both the contact conformation (in which endothelial cells and the astrocytic cell body or endfeet are physically touching) and non-contact conformation (in which endothelial cells are plated on a transwell membrane and the astrocytes are plated on the bottom well compartment—so that direct contact is not possible). The enhancement in barrier function observed in the non-contact conformation suggests that a major contribution of astrocytes is the release of diffusible soluble factors to affect the endothelium.Citation31 Indeed, endothelial cells cultured in medium collected from astrocyte cultures (ACM) can also increase cellular junction expression and decrease permeability.Citation16,Citation17,Citation30,Citation32 Another method of tightening the barrier properties of endothelial cells exploits the second messenger cAMP effects on junctional proteins. cAMP modulates actin reorganization and distribution of tight junction and adherens junction proteins though its ability to modulate PKA.Citation29 The use of cAMP analogues can induce tight junction expression, increase TEER, and reduce paracellular permeability in HUVEC cells.Citation10,Citation14,Citation17,Citation29
In our studies, we observed that HUVEC cells cultured in medium supplemented with cAMP analogues resulted in a tighter barrier, as measured by an increased cellular TEER. On a similar timescale, these cells also displayed increases in ZO-1 and VE-cadherin expression, vinculin recruitment to cell-cell junctions, and decreased cell-cell junctional force. Furthermore, we observed that endothelial cells cultured with cAMP supplementation exhibited more cortical actin, whereas control cells had more stress fiber actin distribution. These data are consistent with stronger adhesion junctions in the cAMP supplemented groups. Others have demonstrated that actin tethering to adhesion complexes is necessary for the formation and stabilization of intercellular junctions.Citation33 Conversely, agents known to disrupt actin tethering (like thrombin and histamine) can result in actin reorganization from the cell cortex into stress fibers that stretch across the cell body. The stress fibers produce a tensile force to pull proteins away from the cell surface inward toward the center of the cell, creating a compromised barrier.Citation33 Our data is consistent with these findings. In these studies, cells were treated with SMIFH2, an inhibitor of formin. Formins act to promote the assembly of linear actin filaments and promote stress fiber formation.Citation34,Citation35 Inhibition of stress fiber formation by SMIFH2 resulted in an increased TEER, consistent with enhanced barrier function.
After demonstrating that cAMP supplementation resulted in a tighter barrier and that there was also an observable subcellular redistribution of actin consistent with an enhanced barrier, we then focused on measuring the mechanical tension across VE-cadherin in cells as they form an enhanced barrier in the presence of ACM. VE-cadherin is the principal component of the endothelial adherens junction. Interestingly, the redistribution of actin to the cell cortex and increases in VE-cadherin immunofluorescence observed with ACM treatment () coincided with a significant decrease in VE-cadherin mechanical tension (). Because the VE-cadherin force sensor measures average force per molecule, there are two possible explanations for the ACM + cAMP-induced decreased VE-cadherin force: 1) reduced actomyosin contractility or 2) recruitment of more VE-cadherin molecules at junctions (termed “junctional strengthening”), effectively reducing the average force across a single VE-cadherin molecule. It is difficult to parse contractility changes from junctional strengthening. Our observation of increased VE-cadherin and ZO-1 with cAMP agonists ( and ), coupled with increased recruitment of cytoskeletal connecting proteins α-catenin and vinculin ( and ), is suggestive of some junctional strengthening. However, the loss of actin stress fibers () and reduced VE-cadherin tension () with exposure to ACM and cAMP analogues () also suggests that there may be a simultaneous decrease in contractility. Liu et al.Citation23 were able to parse these two parameters using micropost arrays with pairs of cells. Using this technique with sphingosine-1-phosphate (S1P), a barrier enhancing agent, the authors demonstrated enhanced junctional strengthening with no effects on cellular contractility. While this technique is robust at distinguishing contractility from junctional strengthening, the use of shape-constrained cell pairs makes it difficult to directly compare it to our force data which came from confluent endothelial monolayers. Nevertheless, our data and the data of Liu et al., point to a model in which barrier-enhancing agents act to both strengthen junctions and possibly reduce contractile forces.Citation23
Furthermore, our data suggest that cortical actin may generate less force on VE-cadherin than actin stress fibers. We directly tested this hypothesis using the formin inhibitor SMIFH2, which decreases cell stress fibers. SMIFH2 increased barrier function in cells cultured in normal medium (). Although we hypothesized that the inhibitor SMIFH2 reduces forces at cell-cell contacts, high yellow fluorescence of the formin inhibitor made it impossible to use in conjunction with the VE-cadherin force sensor.
In conclusion, we characterized the response of HUVEC to astrocyte conditioned medium and to cAMP analogues with respect to changes in transendothelial electrical resistance, junction protein expression and through the measurement of cell-cell force changes. Barrier enhancement was observed within 24 h, and correlated with junctional strengthening, a shift from actin stress fibers to cortical actin, and reduced VE-cadherin force.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1405774_Supplemental_Figures.zip
Download Zip (949.9 KB)Funding
We acknowledge support from American Heart Association 16SDG27370007 (to DEC) and from the National Institutes of Health R00 DA039791 (JJP).
References
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/Monocyte Chemoattractant Protein-1 Mediates Enhanced Transmigration of Human Immunodeficiency Virus (HIV)-Infected Leukocytes across the Blood-Brain Barrier: A Potential Mechanism of HIV-CNS Invasion and NeuroAIDS. J Neurosci. 2006;26:1098–106. doi:10.1523/JNEUROSCI.3863-05.2006.
- Zlokovic B V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi:10.1016/j.neuron.2008.01.003.
- Labus J, Häckel S, Lucka L, Danker K. Interleukin-1 beta induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J Neurosci Methods. 2014;228:35–45. doi:10.1016/j.jneumeth.2014.03.002.
- Fletcher NF, Bexiga MG, Brayden DJ, Brankin B, Willett BJ, Hosie MJ, Jacque J-M, Callanan JJ. Lymphocyte migration through the blood-brain barrier (BBB) in feline immunodeficiency virus infection is significantly influenced by the pre-existence of virus and tumour necrosis factor (TNF)-α within the central nervous system (CNS): studies using an in. Neuropathol Appl Neurobiol. 2009;35:592–602. doi:10.1111/j.1365-2990.2009.01031.x.
- Abdullah Z, Rakkar K, Bath PMW, Bayraktutan U. Inhibition of TNF-alpha protects in vitro brain barrier from ischaemic damage. Mol Cell Neurosci. 2015;69:65–79. doi:10.1016/j.mcn.2015.11.003.
- Harris ES, Nelson WJ. VE-cadherin: at the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol 2010;22:651–8. doi:10.1016/j.ceb.2010.07.006.
- Leibrand CR, Paris JJ, Ghandour MS, Knapp PE, Kim W-K, Hauser KF, McRae M. HIV-1 Tat disrupts blood-brain barrier integrity and increases phagocytic perivascular macrophages and microglia in the dorsal striatum of transgenic mice. Neurosci Lett. 2017;640:136–43. doi:10.1016/j.neulet.2016.12.073.
- Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi:10.1038/nrn1824.
- Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi:10.1016/j.nbd.2009.07.030.
- Winger RC, Koblinski JE, Kanda T, Ransohoff RM, Muller WA. Rapid remodeling of tight junctions during paracellular diapedesis in a human model of the blood-brain barrier. J Immunol. 2014;193:2427–37. doi:10.4049/jimmunol.1400700.
- Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–7. doi:10.1038/325253a0.
- Sobue K, Yamamoto N, Yoneda K, Hodgson ME, Yamashiro K, Tsuruoka N, Tsuda T, Katsuya H, Miura Y, Asai K, et al. Induction of blood-brain barrier properties in immortalized bovine brain endothelial cells by astrocytic factors. Neurosci Res. 1999;35:155–64. doi:10.1016/S0168-0102(99)00079-6.
- Rist RJ, Romero IA, Chan MWK, Couraud PO, Roux F, Abbott NJ. F-actin cytoskeleton and sucrose permeability of immortalised rat brain microvascular endothelial cell monolayers: Effects of cyclic AMP and astrocytic factors. Brain Res. 1997;768:10–8. doi:10.1016/S0006-8993(97)00586-6.
- Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J. A cell culture model of the blood-brain barrier. J Cell Biol. 1991;115:1725–35. doi:10.1083/jcb.115.6.1725.
- Abbott NJ, Dolman DEM, Drndarski S, Fredriksson SM. An improved in vitro blood-brain barrier model: rat brain endothelial cells co-cultured with astrocytes. In: Milner R. (eds) Astrocytes. Methods in Molecular Biology (Methods and Protocols), vol 814. 2012. Humana Press.
- Hayashi Y, Nomura M, Yamagishi S, Harada S, Yamashita J, Yamamoto H. Induction of various blood-brain barrier properties in non-neural endothelial cells by close apposition to co-cultured astrocytes. Glia. 1997;19:13–26. doi:10.1002/(SICI)1098-1136(199701)19:1<13::AID-GLIA2>3.0.CO;2-B.
- Gaillard PJ, Voorwinden L, Nielsen J, Ivanov A, Atsumi R, Engman H, Ringbom C, de Boer AG, Breimer DD. Establishment and functional characterization of an in vitro model of the blood–brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur J Pharm Sci. 2001;12:215–22. doi:10.1016/S0928-0987(00)00123-8.
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi:10.1152/physrev.00012.2005.
- Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. 2013;23:1024–30. doi:10.1016/j.cub.2013.04.049.
- Gavard J. Breaking the VE-cadherin bonds. FEBS Lett. 2009;583:1–6. doi:10.1016/j.febslet.2008.11.032.
- Kris AS, Kamm RD, Sieminski AL. VASP involvement in force-mediated adherens junction strengthening. Biochem Biophys Res Commun. 2008;375:134–8. doi:10.1016/j.bbrc.2008.07.132.
- le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, de Rooij J. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol. 2010;189:1107–15. doi:10.1083/jcb.201001149.
- Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS. Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci U S A. 2010;107:9944–9. doi:10.1073/pnas.0914547107.
- Arsenovic PT, Ramachandran I, Bathula K, Zhu R, Narang JD, Noll NA, Lemmon CA, Gundersen GG, Conway DE. Nesprin-2G, a Component of the Nuclear LINC Complex, Is Subject to Myosin-Dependent Tension. Biophys J. 2016;110:34–43. doi:10.1016/j.bpj.2015.11.014.
- Huveneers S, Oldenburg J, Spanjaard E, van der Krogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol. 2012;196:641–52. doi:10.1083/jcb.201108120.
- Birukova AA, Shah AS, Tian Y, Moldobaeva N, Birukov KG. Dual role of vinculin in barrier-disruptive and barrier-enhancing endothelial cell responses. Cell Signal. 2016;28:541–51. doi:10.1016/j.cellsig.2016.02.015.
- Lai C-H, Kuo K-H. The critical component to establish in vitro BBB model: Pericyte. Brain Res Brain Res Rev. 2005;50:258–65. doi:10.1016/j.brainresrev.2005.07.004.
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–61. doi:10.1038/nature09522.
- Beese M, Wyss K, Haubitz M, Kirsch T. Effect of cAMP derivates on assembly and maintenance of tight junctions in human umbilical vein endothelial cells. BMC Cell Biol. 2010;11:68. doi:10.1186/1471-2121-11-68.
- Wuest DM, Wing AM, Lee KH. Membrane configuration optimization for a murine in vitro blood-brain barrier model. J Neurosci Methods. 2013;212:211–21. doi:10.1016/j.jneumeth.2012.10.016.
- Bicker J, Alves G, Fortuna A, Falcão A. Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: A review. Eur J Pharm Biopharm. 2014;87:1–24. doi:10.1016/j.ejpb.2014.03.012.
- Arthur FE, Shivers RR, Bowman PD. Astrocyte-mediated induction of tight junctions in brain capillary endothelium: an efficient in vitro model. Brain Res. 1987;433:155–9. doi:10.1016/0165-3806(87)90075-7.
- Abdullah Z, Bayraktutan U. NADPH oxidase mediates TNF-alpha-evoked in vitro brain barrier dysfunction: Roles of apoptosis and time. Mol Cell Neurosci. 2014;61:72–84. doi:10.1016/j.mcn.2014.06.002.
- Phng LK, Gebala V, Bentley K, Philippides A, Wacker A, Mathivet T, Sauteur L, Stanchi F, Belting HG, Affolter M, et al. Formin-mediated actin polymerization at endothelial junctions is required for vessel lumen formation and stabilization. Dev Cell. 2015;32:123–32. doi:10.1016/j.devcel.2014.11.017.
- Lampugnani MG. Endothelial adherens junctions and the actin cytoskeleton: An “infinity net”? J Biol. 2010;9:16. doi:10.1186/jbiol232.