
ABSTRACT
Inflammatory bowel diseases broadly categorized into Crohn's disease (CD) and ulcerative colitis (UC), are chronic inflammatory disorders of the gastrointestinal tract with increasing prevalence worldwide. The etiology of the disease is complex and involves a combination of genetic, environmental, immunological and gut microbial factors. Recurring and bloody diarrhea is the most prevalent and debilitating symptom in IBD. The pathogenesis of IBD-associated diarrhea is multifactorial and is essentially an outcome of mucosal damage caused by persistent inflammation resulting in dysregulated intestinal ion transport, impaired epithelial barrier function and increased accessibility of the pathogens to the intestinal mucosa. Altered expression and/or function of epithelial ion transporters and channels is the principle cause of electrolyte retention and water accumulation in the intestinal lumen leading to diarrhea in IBD. Aberrant barrier function further contributes to diarrhea via leak-flux mechanism. Mucosal penetration of enteric pathogens promotes dysbiosis and exacerbates the underlying immune system further perpetuating IBD associated-tissue damage and diarrhea. Here, we review the mechanisms of impaired ion transport and loss of epithelial barrier function contributing to diarrhea associated with IBD.
Introduction
IBD is a chronic, inflammatory disorder of the gastrointestinal tract with immensely complex etiology. A combination of factors such as genetic predisposition, environmental stimuli, gut immune dysregulation and dysbiosis intersect resulting in persistent bowel inflammation either confined in the colon as seen in ulcerative colitis (UC) or involving complications in both small and the large intestine as observed in Crohn's disease (CD).Citation1-Citation3 Incidence of IBD has increased during the past decades affecting 3.1 million adults in the USACitation4 and 2.2 million people in Europe,Citation5 together with an increase observed in the low-risk Asian populations.Citation6 Both UC and CD manifest with alternating periods of relapse and remission with diarrhea, abdominal pain, gastrointestinal bleeding, and weight loss as key clinical symptoms of active disease. The health related quality of life (HRQOL) of IBD patients is severely impactedCitation7 and the fundamental management strategy is aimed at maintenance of remission, control of inflammation, restoration of nutritional deficits and treatment of symptoms like diarrhea.
Diarrhea is the hallmark symptom associated with IBD and is seen in almost 80% of the cases.Citation8 IBD-associated diarrhea is multifactorial and is the outcome of intricate pathophysiological events arising from widespread and sustained mucosal inflammation. Depending upon the site and magnitude of intestinal inflammation the severity of diarrhea varies in IBD patients, ranging from increased bowel frequency to chronic diarrhea requiring electrolyte supplementation and hospitalization.Citation8 The severity of diarrhea (stool frequency and consistency) is thus considered as an important determinant of the disease activity index. Therefore, the understanding of the molecular mechanisms leading to diarrhea in IBD is crucial for proper diagnosis and management.
Mechanistically, an imbalance in electrolyte absorption and/or secretion in the intestine disrupts the osmotic gradient resulting in water retention in the lumen and diarrhea. Broadly, excess production of inflammatory mediators in UC and CD generate a highly inflammatory milieu causing damage to the mucosal architecture, compromised epithelial barrier, increased antigen load into the intestinal lumen, altered immune responses, and loss of function and/or expression of various functional entities present on the luminal membrane such as ion transporters, channels and multiprotein junction complexes constituting the epithelial barrier (). This cascade of reactions contributes to inadequate water and solute transport leading to inflammatory diarrhea. In this review, we will summarize the impact of persistent or inadequately resolved chronic intestinal inflammation on electrolyte transport (ion transporters and channels) and epithelial barrier function (tight and adherens junctions) and the consequences of these alterations in pathogenesis of IBD-associated diarrhea.
Figure 1. Schematic of electrolyte transporters and junctional proteins in IECs: Ion transporters, channels, physical and chemical barriers are compromised in IBD leading to diarrhea. The transepithelial pathway of solutes and ions is mediated by ion/solute transporters and channels. As depicted in the figure, in steady state, apical transporters, NHE3 (Na+/H+ exchanger 3) and DRA (Down Regulated in Adenoma) work in conjunction to mediate electroneutral NaCl absorption. Electrogenic mode of Na+ absorption occurs in the distal part of colon via ENaC (Epithelial sodium channel). Intracellular Na+ gradient essential for sodium dependent transport processes is generated by the action of Na+/K+-ATPase present at the basolateral membrane. Cl− secretion across the membrane is facilitated by apical Cl− channel CFTR (cystic fibrosis transmembrane conductance regulator). Basolateral NKCC1 (Na+/K+/2Cl− cotransport system) is involved in uptake of Cl− from the serosal side. K+ taken up by NKCC1 and Na+/K+-ATPase is recycled back to the basolateral side by K+ channels localized to the basolateral membrane. Predominant mechanism of diarrhea in IBD involves impairment of electroneutral NaCl absorption accompanied with dysfunctional ENaC and Na+/K+-ATPase, with very little role if any played by anion secretion. Negative sign indicates the downregulation in function and/or expression of NHE3, DRA, ENaC and Na+/K+-ATPase in IBD. The mucus layers serves as a barrier and prevents the direct contact of gut microbes with the underlying epithelium. Adjacent intestinal epithelial cells are sealed together via an intricate network of junctional proteins including the tight junction (TJ) proteins (claudins, occludin, JAM and ZO-1), adherens junction (AJ, E-cadherin) and desmosomes. The optimal expression and function of tight and adherens junction proteins regulate the paracellular flux under physiological conditions. Negative sign indicates the downregulation in function and/or expression of TJ and AJ proteins in IBD. Breakdown of barrier function in IBD results in diarrhea via leak-flux mechanism. As summarized in the review, understanding of the mechanisms underlying loss of barrier function and defective ion absorption in IBD is essential for development of better treatment strategies for IBD and associated diarrhea.
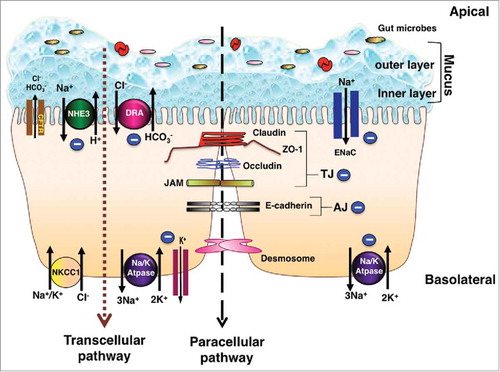
IBD and Altered Electrolyte Transport in the Intestine
Water and electrolyte transport mechanism
Epithelial cells lining the intestinal lumen not only serve as selectively permeable barriers but are also involved in bidirectional transport of nutrients, electrolytes and fluid from the intestinal lumen. The tremendous absorptive capacity of the intestinal epithelium is evident from the fact that on average 8–10 L/day of fluid enters the intestinal lumen and only <0.1-0.2 L/day is excreted in the feces.Citation9,Citation10 This process of water absorption is mainly passive and is governed by the movement of solutes and electrolytes (primarily Na+ and Cl−) across the membrane.Citation9-Citation11 Intestinal epithelial cells are highly polarized and express a variety of ion transporters and channels on the apical and the basolateral membrane that coordinate and maintain a dynamic balance between electrolyte absorption and secretion under normal physiological conditions (). Fluid loss in diarrhea occurs due to derangements in the equilibrium of electrolyte absorptive and secretory processes. During inflammatory insults in UC and CD the absorptive capacity of the colon is significantly compromised resulting in excessive loss of fluid in the stool. Accordingly, impairment in electrolyte absorption rather than secretion is now proposed as the major ion transport abnormality in diarrhea associated with IBD.Citation3,Citation9,Citation12-Citation17 Given the central role of electrolyte absorption in maintenance of fluid homeostasis in IBD, the current review focuses on ion transporters and channels involved in sodium and chloride absorption and alterations in their function and/or expression in IBD.
IBD and altered Na+ absorption in the intestine
Mechanisms for vectorial transport of Na+ from apical to the basolateral membrane of intestinal epithelial cells can be a) nutrient dependent or b) nutrient independent.Citation3,Citation9,Citation18,Citation19 The nutrient dependent transport of sodium is mediated via Na+-glucose/amino-acid symporters and is energized by the electrochemical gradient of Na+ established by basolaterally located Na+/K+ ATPase. The symport of these organic solutes (sugars and amino acids) and Na+ involves the cotransport of water, which is absorbed osmotically. This has formed the basis of Oral Rehydration Solution (ORS) used as the primary means for treating dehydration due to diarrhea.Citation9,Citation12,Citation20
Nutrient independent mode of sodium transport can be electroneutral where sodium is absorbed via Na+/H+ exchangers (NHEs) coupled to Cl− absorptionCitation3,Citation8,Citation12,Citation18,Citation19,Citation21,Citation22 or electrogenic which is Cl−-independent and is mediated by epithelial sodium channel (ENaC)Citation9,Citation14,Citation16,Citation18,Citation19 (). Electroneutral NaCl absorption is the predominant route for Na+, Cl− and water absorption in the ileum and colon of mammalian gastrointestinal tract whereas ENaC mediated electrogenic Na+ absorption is important for fluid homeostasis in the distal colon. Impaired Na+ absorption is a major mechanism resulting in diarrhea. Alterations in function and/or expression of epithelial sodium transporters such as Na+/H+ exchangers, ENaC and Na+/K+ATPase have been reported in experimental models of colitis and in biopsies from UC and CD patients.Citation3,Citation9,Citation13,Citation14,Citation16,Citation22-Citation25 The contribution of dysfunctional Na+/H+ exchangers, ENaC and Na+/K+ATPase in diarrhea associated with IBD is discussed in the upcoming sections of this review sections.
Sodium hydrogen exchangers (NHEs) in IBD associated diarrhea
Encoded by members of the SLC9 gene family, the Na+/H+ exchangers (NHEs) mediate the electroneutral exchange of an extracellular Na+ for a cytosolic H+. NHEs are of paramount importance in absorption of sodium and water, maintenance of intracellular pH and cell volume control. So far 11 NHE isoforms (NHE1-11) have been identified in mammals each having a different cellular localization and tissue distribution. In the intestinal epithelium, NHE2, NHE3 and NHE8 are present on the apical membrane and NHE1 is located on the basolateral membrane.Citation26-Citation30 Expression of other NHE isoforms in the intestine is debatable.Citation31
Among the four isoforms that are present in the intestine, the direct involvement of NHE1 (SLC9A1) and NHE2 (SLC9A2) in IBD associated diarrhea is not evident. Data obtained from experimental and clinical investigations are conflicting and indicate both an increaseCitation23,Citation32 as well as decreaseCitation33-Citation35 in NHE1 expression in response to inflammatory insults. Decrease in NHE2 function and expression has been reported in response to proinflammatory cytokines such as interferon γ (IFN-γ)Citation36 and Tumor necrosis factor α (TNFα).Citation37 A significant decrease in colonic NHE2 mRNA and protein expression is also reported in rat model of TNBS induced colitis.Citation38 Surprisingly, no change in the expression of NHE2 is reported in biopsies from UC patients and in mouse model of DSS induced colitis.Citation35 Also, NHE2 KO mice neither show alterations in intestinal Na+ absorption nor a diarrheal phenotype.Citation26,Citation28 Therefore, dysregulation of NHE2 function and/or expression is not of great significance for altered electrolyte absorption observed in IBD. However, contribution of impaired NHE1 expression in IBD associated diarrhea warrants further experimental validation.
NHE8 (SLC9A8) and NHE3 (SLC9A3) are ontogenetically regulated where NHE8 is predominantly expressed in the early life and its expression is later substituted by NHE3 in adulthood.Citation16,Citation26,Citation39 NHE8 KO mice do not have diarrhea probably because of compensatory increase in NHE2 and NHE3 in the small intestine.Citation29 However, it is interesting to note that expression of NHE8 is decreased by TNFα, LPS and in colitis models of DSS and TNBS.Citation40,Citation41 Furthermore, NHE8 KO mice are not only susceptible to DSS treatmentCitation42 but also have decreased levels of Muc2 (an important component of the mucus layer) and antimicrobial peptides with concomitant increase in bacterial adhesion in the colon.Citation29,Citation40,Citation42 This suggests that inhibition of NHE8 may not contribute to IBD associated diarrhea directly but perhaps plays a crucial role in maintenance of mucosal integrity during inflammation. Recent report confirming the expression of NHE8 in goblet cellsCitation43 further attests to this role of NHE8.
Of the isoforms present in the intestine, NHE3 is considered as the main player in intestinal Na+ absorptionCitation3,Citation16,Citation21,Citation26,Citation44 (). Genetic deletion of NHE3 in mice results in diarrhea, metabolic acidosis and impaired fluid homeostasis,Citation44,Citation45 a phenomenon not observed in knockout mouse models of other NHE isoforms. In fact, combined deficiency of both NHE3 and NHE2 in mice does not augment the severity of absorptive defects and diarrheal state of NHE3 KO mice.Citation46 Recent report by Janecke et al,Citation47 identified loss-of-function mutations in the SLC9A3 gene in 9 CSD (Congenital Sodium Diarrhea) patients. Interestingly, among this subset of 9 patients with mutated NHE3, 2 patients developed IBD implicating the deficiency of NHE3 in the pathophysiology of diarrhea in IBD. Another study reported the inhibitory impact of SNPs (single nucleotide polymorphisms) on NHE3 function.Citation48 Additionally, GWAS studies have established strong association between UC and SLC9A3 gene locus.Citation49,Citation50 Alterations in NHE3 function and/or expression observed in IBD patients have further substantiated that NHE3 dysfunction contributes to IBD associated diarrhea. However, the mechanism of NHE3 inhibition is controversial and is attributed to either i) decreased NHE3 activity as seen in human UC biopsies with unaltered protein, mRNA and surface expressionCitation23,Citation51-Citation53 or ii) decreased NHE3 protein levels observed in both UC and CD patient biopsiesCitation35,Citation54 with reduced NHE3 mRNA observed only in CD biopsiesCitation54 (). Consistent with the patient data, downregulation of NHE3 function has been reported in various mouse models of experimental colitis with varying pattern of changes seen in NHE3 expression. In DSS and TNBS induced colitis in mice a downregulation of NHE3 protein expression was found to occur in the mouse colon.Citation35 In TnfΔARE+/−, Rag2−/− CD4+ CD45RBhigh and 2% DSS-induced IL-10−/− models, a significant inhibition in NHE3 transport function was observed with no change in mRNA expression or membrane localization of NHE3.Citation53 Similarly, reduced NHE3 activity despite normal gene expression was observed in IL-10−/− mice.Citation55 In contrast, IL-2 deficient mice demonstrated a drastic reduction in NHE3 function (80%) with a concomitant decrease in mRNA and protein expression (41% and 24%, respectively). Collectively, the data obtained from IBD patients and murine models signify that compromised NHE3 function is a prime feature for pathogenesis of diarrhea in IBD. However, data on NHE3 expression in intestinal inflammation is inconsistent.
Table 1. Modulation of Ion transporters and channels involved in sodium and chloride absorption in IBD Patients.
It appears that the observed decrease in NHE3 function in IBD could be either transcriptional or post-translational. At the transcriptional level, proinflammatory cytokines such as IFN-γ and TNFα have been shown to decrease NHE3 mRNA expression both in vitroCitation36,Citation56 and in vivo.Citation36,Citation57 IFN-γ treatment decreased NHE3 mRNA and protein expression in C2/BBe cells and rat ileum and colon.Citation36 Along similar lines, our group demonstrated a decrease in NHE3 promoter activity by IFN-γ and TNFα via protein Kinase A (PKA) mediated phosphorylation of Sp1 and Sp3 transcription factors.Citation56
Clayburgh et alCitation57 demonstrated that in vivo, TNFα alters NHE3 surface expression post translationally. TNF-α promotes PKCα mediated redistribution of NHE3 from the brush border membrane resulting in NHE3 internalization and Na+ malabsorption in model of anti-CD3 antibody induced diarrhea. Furthermore, nitric oxide (NO), another inflammatory mediator has been shown to inhibit NHE3 function by activation of soluble guanylate cyclase/cGMP/protein kinase G (PKG) pathway in Caco-2 cells.Citation58 Interestingly, CSD patients also carry mutations in guanylate cyclase C (GUCY2C) where deficiency in Na+ absorption has been attributed to elevated cGMP levels resulting from gain-of-function mutations in GUCY2C. In these patients, NHE3 function is inhibited via its phosphorylation by cGMP kinase II.Citation59,Citation60 Many of the patients with germline GUCY2C mutations develop IBD with varying age of onset.Citation47,Citation49,Citation60 This further suggests that reduction in NHE3 function enhances the propensity for IBD pathogenesis.
The functional impairment in NHE3, despite normal expression and localization also suggests the potential involvement of other factors in UC associated diarrhea, such as NHE Regulatory Factors (NHERFs). The NHERFs (NHERF1, NHERF2, PDZK1 and IKEPP) are PDZ domain containing scaffolding proteins that interact directly with NHE3 and assist in the formation of NHE3 containing multiprotein complexes, endocytic recycling of NHE3 and its interaction with other proteins. Lenzen et alCitation55 showed diminished mRNA and protein of NHERF2 and PDZK1 (NHERF3) whereas, NHERF1 expression was unchanged in IL-10 deficient mice. On the other hand, Yeruva et alCitation53 found a significant downregulation of PDZK1 mRNA and protein expression in colon biopsies from UC patients, as well as in inflamed murine ileum and colon. As discussed above, both these studiesCitation53,Citation55 also reported a parallel decrease in NHE3 transporter activity with normal protein expression and membrane localization. This indicates that alterations in PDZK1 expression in presence of mucosal inflammation can be a potential cause for functional impairment of NHE3. Moreover, a decrease in NHE3 and NHERF1 protein expression is also reported in DSS and TNBS treated mice colon as well as mucosal biopsies from UC and CD patients.Citation35
It is evident that genetic deficiency of NHE3 in mice results in features of IBD. NHE3 knockout mice are presented not only with spontaneous colitis but also with enhanced vulnerability to experimental colitis.Citation61,Citation62 They exhibit mild diarrhea, occasional rectal prolapse and reduced body weight. Along with this, these mice also display histological damage characterized by crypt hyperplasia, diffuse neutrophilic infiltrate increase in matrix metalloproteinase 8 expression, marked decrease in PAS positive goblet cells and induction of inducible nitric oxide synthase (iNOS) and TNF-α. This indicates that NHE3 beyond its transporter role can participate in immunomodulation and maintenance of epithelial barrier integrity. This has been corroborated with studies where absence of NHE3 exacerbated spontaneous colitis in IL-10 KOCitation63 and Rag-2 KO mice.Citation64
Because NHE3 knockout mice develop spontaneous colitis,Citation61 they exhibit extremely reduced survival when challenged with even low dose of DSS. Due to fragile barrier, prevailing inflammation and tissue damage, DSS insult in these mice provokes intestinal bleeding, hypovolemic shock and sepsis.Citation62 This damage is not localized but spreads to small intestine where increased induction of pro-inflammatory genes has been evidenced. Though DSS exerts colon specific damage, small intestinal injury in NHE3 knockout mice is not surprising. A feeble barrier, dysbiosis and already existing inflammation, are sufficient to decrease the overall resilience of the system, where small intestine is but continuum of large intestine.
Another important feature of NHE3 knockout mice is dysbiosis that appears to be critical for inflammation. With intestinal epithelial barrier being tolerant to pathogenic insult, these mice show increased bacterial adhesion and translocation to distal colon which, interestingly, can be mitigated by orally administered antibiotics.Citation61 Increased cytokine expression in these mice in response to DSS (in small intestine) can also be restrained by antibiotic use. This indicates that gut microbiota plays a crucial role in pathophysiology of NHE3 knockout mice. In fact, spontaneous colitis in these mice is contingent on conventional housing. Conversely, NHE3 knockout mice are protected from colitis in ultraclean housing facility.Citation65 The exact mechanistic underpinnings of NHE3 function and gut microbiota are unclear. Apparently, NHE3 exerts important control on gut microbial remodeling.Citation64 Presence of NHE3 in mice deficient in T- and B- cell maturation (Rag2−/− mice) provides decisive advantage not only against T cell adoptive transfer colitis but also gut dysbiosis.Citation64 Probably, targeting NHE3 as therapy may prove beneficial not only in IBD but also in restoring normal gut flora. If interaction between gut microbiota and NHE3 function exists beyond the weak barrier needs further investigation.
Epithelial sodium channel (ENaC) and IBD associated diarrhea
ENaC is a Na+ selective channel expressed at the apical membrane of distal colon and rectum (). The channel is responsible for electrogenic Na+ absorption and plays an essential role in controlling salt and fluid homeostasis. Structurally, ENaC is a multimeric protein comprising of three homologous subunits (α, β and γ). α-subunit serves as a basic building block and is liable for the amiloride sensitivity of the ENaC. β and γ subunits are primarily engaged in sodium transportCitation66,Citation67 with a very little contribution from the α-subunit. However, the presence of all the three subunits is a prime requirement for efficient cell surface expression and Na+ transport.Citation66
In steady state, there are two components for sodium absorption in the colon, electroneutral Na+ absorption that predominates in the proximal part and electrogenic Na+ absorption via ENaC that occurs in the distal colon. Interestingly, in circumstances where Na+ absorption is compromised for example in NHE3 KO mice, there is a compensatory increase in ENaC function and expression to suffice for the electrolyte loss.Citation44,Citation61 However, with respect to the pathogenesis of diarrhea associated with IBD, a similar compensatory mechanism for ENaC induction is not observed. A substantial decrease in ENaC function is reported in the colonic mucosa of UC and CD patients.Citation23,Citation25,Citation35,Citation67-Citation71 Parallel to the reduced ENaC mediated Na+ transport, studies in IBD patient biopsies have also demonstrated downregulation of both mRNA and protein levels of β and γ subunits of ENaC.Citation25
Transcriptional regulation of β and γ subunits regulates ENaC expression in the intestine. In this regard, nanomolar concentrations of aldosterone have been shown to induce the mRNA expression of β and γ subunits.Citation72 However, in the inflamed colon of UC patients the mechanism for aldosterone-mediated increase in the β and γ subunits of ENaC was impaired despite unaltered expression of mineralocorticoid receptors.Citation25 This was attributed to the inhibitory effects of the proinflammatory cytokines such as IL-1β, TNF-α and IFN-γ in the colonic mucosa of UC patients and CD patients.Citation25,Citation44,Citation70 The data obtained from patient studies suggest that defective sodium absorption observed in IBD associated diarrhea involves coordinated down regulation of multiple sodium transporters including NHE3 and ENaC ().
Further, in vivo studies performed in experimental models of IBD also revealed a similar pattern of decrease in ENaC function and expression associated with impaired sodium absorption. Zeissig et alCitation70 showed that TNF-α activates MEK1/2 and ERK1/2 pathway and inhibit ENaC transcription in rat distal colon. Barmeyer et alCitation73 demonstrated that IL-2 deficient mice, which develop spontaneous colitis, exhibit severe defect in aldosterone-induced electrogenic Na+ absorptionCitation73 and concomitant decrease in mRNA and protein expression of β and γ subunits of ENaC. Defective Na+ absorption in these mice was attributed to downregulation of both NHE3 and ENaC.Citation73 Further evidence for reduced ENaC activity under inflammatory conditions was shown in JAK3 KO mice that develop spontaneous IBD like symptoms.Citation74
In a study by Sullivan et al,Citation35 diminished protein expression of β-ENaC and NHE3 protein in the colon of DSS and TNBS treated mice was found to occur. Interestingly, decreased α-ENaC expression as a result of inflammatory insult in DSS model of colitis was also shown in a separate study.Citation71 In contrast to DSS, TNBS and IL-2 KO models of colitis, K8 KO mice exhibited no change in ENaC protein expression. Interestingly, changes in the subcellular localization of γ-ENaC in K8 KO mice were correlated to the defects in electrogenic sodium transport.Citation75 The above observations in K8 KO mice imply that in addition to inflammation induced transcriptional suppression, alterations in ENaC trafficking can also contribute to defective electrogenic sodium absorption and associated diarrhea in IBD. Thus, there is strong evidence to suggest that decreased ENaC activity in the presence of mucosal inflammation also account for pathogenesis of diarrhea in IBD.
Sodium potassium ATPase (Na+/K+-ATPase) and IBD associated diarrhea
Na+/K+-ATPase is localized to the basolateral membrane (BLM) of () intestinal epithelial cells and is engaged in outward transport of 3 Na+ ions coupled to the movement 2 K+ ions into the cell. This process of active ion exchange establishes an inward Na+ gradient at the expense of ATP hydrolysis.Citation67,Citation69 Na+/K+-ATPase is a heterodimer comprised of α and β subunits, where the α subunit possesses the binding sites for ATP, Na+ and an extracellular binding site for K+.Citation76 The β subunit facilitates α/β heterodimerization and surface expression of the enzyme. Though optional, the presence of the γ subunit is also indicated and it is thought to possess a regulatory role.Citation19,Citation76 The α and the β subunits of Na+/K+-ATPase have several isoforms. However, α1 and β1 isoforms are more relevant in relation to ion transport processes.Citation69
Under normal physiological conditions, Na+/K+-ATPase plays a crucial role in nutrient coupled sodium absorption. The concentration gradient of sodium established by Na+/K+-ATPase facilitates the absorption of solutes (sugars and amino acids). This highlights the importance of optimal Na+/K+-ATPase activity in oral rehydration therapy and diarrheal diseases.Citation9,Citation19,Citation77 Additionally, inhibition of Na+/K+-ATPase has been shown to negatively regulate Na+ absorption and Cl− transport.Citation78 Apart from ion transport, its role in maintenance of tight junction integrity, cell polarity, stabilization of actin cytoskeleton and involvement in signaling pathways has also been elucidated.Citation19,Citation76
The impact of intestinal inflammation on the function and expression of Na+/K+-ATPase and its implications in associated diarrhea is evident from the studies conducted in biopsies from IBD patients. Several studies have shown that the activity of Na+/K+-ATPase was significantly down regulated in the inflamed mucosa of UC and CD patientsCitation68,Citation79-Citation81 (). The decrease in Na+/K+-ATPase activity in pediatric population was found to be complementary to the severity of inflammation.Citation80 Greig et alCitation69 reported a decreased expression of α1 and β1 isoforms of Na+/K+-ATPase in UC patients. Although the decrease in protein expression was consistent with reduced activity, concomitant reduction in mRNA levels was not observed in this study.Citation69 This observation suggested that a reduction in Na+/K+-ATPase protein expression during intestinal inflammation could be the outcome of either impaired trafficking of the enzyme to the BLM or defective translational processing. Along similar lines Sullivan et alCitation35 also showed decreased protein expression of α1 isoform of Na+/K+-ATPase in CD and UC patients. It is important to note that this studyCitation35 also reported a decline in expression of other ion transporters involved in sodium absorption such as NHE3 and ENaC. Therefore, diminished sodium absorption is a characteristic feature in IBD.
Interestingly, conflicting results have been obtained for Na+/K+-ATPase expression in most commonly used mouse models of DSS and TNBS induced colitis. Studies utilizing DSS model either showed no changeCitation35 or a decrease in α1-isoform of Na+/K+-ATPase with unaltered mRNA levels.Citation82 In TNBS model, Sullivan et alCitation35 noted a modest increase in α-subunit of Na+/K+-ATPase. However, none of these studies analyzed the transport activity. In another comprehensive study, Magro et alCitation83 studied function and expression of Na+/K+-ATPase along the length of TNBS treated mice intestine. Interestingly, a significant decrease in Na+/K+-ATPase function was observed in the proximal colon whereas in distal ileum a compensatory increase in Na+/K+-ATPase activity was noted. Surprisingly, the expression of Na+/K+-ATPase remained unchanged throughout the intestine. In contrast, in ileal enterocytes isolated from TNBS treated mice a decrease in Na+/K+-ATPase activity with a corresponding decrease in protein and mRNA expression of α1 and β1 subunits was observed.Citation84 Musch et al,Citation85 studied the mechanism of fluid loss induced by T cell activation in mice. The results showed that T-cell induced TNF-α production caused a marked decrease in Na+/K+-ATPase activity in juejunum resulting in diminished electroneutral and glucose dependent Na+ and water absorption.Citation85
A number of in vivo and in vitro studies have indicated that pro-inflammatory cytokines such as IFN-γ, TNFα and IL-1β are responsible for Na+/K+-ATPase dysfunction in IBD. For example, several studies demonstrated that IFN-γ exerts an inhibitory effect either on Na+/K+-ATPase activity or expression or both.Citation86-Citation89 Similarly, TNFα was shown to inhibit function and expression of Na+/K+-ATPase via prostaglandin E2 in rat colon.Citation90 Kreydiyyeh et al,Citation91 demonstrated a decrease in Na+/K+-ATPase activity with a corresponding reduction in protein expression following intraperitoneal administration of IL-1β in rat jejunum. Al Sadi et al,Citation92 provided the mechanistic link for inhibitory effect of IL-1β on Na+/K+-ATPase in Caco-2 cells. The results from this study indicated that IL-1β decreased Na+/K+-ATPase function and expression mainly via p38, COX2/PGE2 and JNK/AP1 pathways.Citation92
Taken together, these data suggest that diarrhea associated with IBD at least in part can be attributed to modulation in the Na+/K+-ATPase function. Importantly, absorption of Na+ across the intestine involves participation of several proteins including NHEs, ENaC and Na+/K+-ATPase. As discussed in previous sections of this review, defective sodium absorption arising from down regulation of various Na+ related transporter proteins in UC and CD serves as a critical factor leading to reduced electrolyte absorption and diarrhea.
As diarrhea in IBD mainly reflects impaired sodium/chloride and water absorption, the current review has mainly focused on the ion transporters and channels involved in sodium (discussed in previous section of this review) and chloride (discussed below) transport. However, it is important to note that potassium (K+) channels play an essential role in the recirculation of K+ ions taken up as a result of Na+/K+-ATPase activity, stabilization of membrane voltage and maintenance of the driving force for electrogenic transport as the exit of K+ ions results in generation of negative intracellular potential facilitating the absorption of sodium via ENaC. Based on the number of transmembrane (TM) helices, the K+ channels are categorized as Kv (voltage gated, 6 TMs), Kir (inwardly rectifying, 2 TMs), K2P (tandem pore domain, 4 TMs) and Kligand (ligand gated channels, 2–6 TMs).Citation16,Citation150 With respect to the pathogenesis of IBD associated diarrhea, marked reduction in the function and expression of basolateral intermediate conductance K+ channel (IK, ligand gated) has been reported.Citation16 Reduction in K+ channel activity results in epithelial cell depolarization and decreased electrical driving force further contributing to defective electrogenic Na+ absorption under inflammatory conditions. The role played by K+ channels is being increasingly recognized and warrants in-depth studies. Detailed structural classification and role of K+ channels in normal and pathophysiological situation has been reviewed elsewhere.Citation16,Citation150
IBD and altered Cl− absorption in the intestine
The connection between IBD-associated diarrhea and defective chloride transport has been debated over the mode of chloride transport-secretory versus absorptive pathway contributions. Initial conviction of increased net chloride secretion (electrogenic) in IBD-associated diarrhea driven by abundant pro-secretory immune mediators in the inflamed intestine was challenged by studies highlighting decreased sodium and chloride absorption as the major contributor.Citation9,Citation11,Citation13,Citation14,Citation22,Citation67,Citation93 Certainly, elevated colonic and fecal Cl− content in UC patientsCitation94-Citation96 could be explained by diminished Cl− uptake in UC colon.Citation94 Similarly, colonic mucosa and rectum of UC patients exhibited reduced net chloride absorption in in vitro flux studies.Citation97,Citation98 Colonic mucosa of UC patients is also characterized with extremely depressed or absent transmucosal electrical potential difference.Citation68,Citation98,Citation99 This also coincides with significantly reduced Cl−/HCO−3 exchange activity as has been observed in colonic crypts isolated from UC patients.Citation23 Decreased electrical potential difference and Cl−/HCO−3 exchange function is consistent with defective chloride absorption. Impaired Cl− absorption in IBD thus appears as one of the most critical events in IBD-associated diarrhea.
Chloride bicarbonate exchangers and diarrhea
Down Regulated in Adenoma (DRA or SLC26A3) and Putative Anion Transporter-1 (PAT-1 or SLC26A6) belong to SLC26 gene family and are the two main transporters involved in apical Cl−/HCO3− exchange in the mammalian intestine.Citation100,Citation101 DRA and PAT-1 exhibit an inverse pattern of expression along the length of the intestine. The expression of DRA increases from jejunum to colon whereas PAT-1 is mainly localized in the small intestine.Citation102 However, duodenum has a robust expression of both the anion exchangers. It is well established that diminished DRA expression and function results in diarrheal disorders. For instance, mutations in DRA cause congenital chloride diarrhea (CLD). CLD patients exhibit voluminous diarrhea, massive loss of Cl− in stool and metabolic alkalosis.Citation103,Citation104 DRA deficient mice reflect the CLD phenotype, exhibiting significantly reduced apical Cl−/HCO3− exchange activity, diarrhea, metabolic alkalosis and serum electrolyte imbalance.Citation105 Interestingly, PAT-1 KO mice exhibit defects in intestinal oxalate secretion hyperoxalemia, hyperoxaluria, increased incidence of CaOx (calcium oxalate) stones in kidney and do not show a diarrheal phenotype.Citation100,Citation106 Additionally, in hepatocyte nuclear factor (HNF) 1α and 1β double knockout miceCitation107 as well as Citrobacter rodentium-induced colitis modelCitation108 the complete loss of DRA has been correlated with severe diarrhea. Overall, these studies pinpoint that i) DRA serves as the major luminal intestinal Cl−/HCO3− exchanger responsible for bulk intestinal Cl− absorption and ii) DRA deficiency is one of the critical factors in intestinal diarrheal disorders. A conspicuous role of DRA in IBD associated diarrhea has been corroborated by both human and mouse studies, enumerated below.
DRA and IBD Associated Diarrhea
In IBD, loss of gene and/or protein expression of DRA occursCitation52,Citation109 though variability exists in the extent and type of this loss.Citation23,Citation110 One factor accounting for this variability can be the magnitude of intestinal inflammation.Citation52,Citation109 For instance, in moderately inflamed UC tissue complete absence of DRA protein expression coinciding with significantly reduced mRNA expression in surface epithelium has been reported by Yang et al.Citation109 Lohi et alCitation52 reported a similar reduction in DRA mRNA levels in pre-operative colonic samples of severely inflamed UC patients. Interestingly, a corresponding decline in DRA protein expression was not observed in this study.Citation52 In these two studies a comparison was drawn between healthy controls and UC patients. However, when examined in a wider spectrum of intestinal inflammation, UC, CD and ischemic colitis, an entirely different pattern emerged.Citation110 DRA mRNA expression and cytoplasmic (immunoreactive) DRA protein levels were intact among all the groups. But the characteristic apical membrane staining of DRA lacked exclusively in UC patientsCitation110 indicating inadequate membrane trafficking events. These studies with a limited sample size (≤10 UC tissues) were corroborated by a recent study conducted on a larger cohort (128 healthy individuals and 69 IBD patients with active UC and diarrheal symptoms).Citation23 Here, a strong reduction in Cl− absorption paralleled a decrease (∼50%) in DRA mRNA expression in UC colonic crypts.Citation23 Collectively, decreased DRA expression can lead to deficient Cl− absorption in UC. Evidence for a similar role of DRA in CD patients, however, is limited. These studies emphasize the conspicuity of DRA's role in IBD associated diarrhea ().
Murine inflammatory models being amenable to experimental toning have provided clearer insight into role of DRA in IBD. Similar to human studies described above, rodent models of spontaneous intestinal inflammation (HLA-B27/β2m transgenic rats and IL-10 knockout mice) exhibit significant reduction in colonic DRA mRNA expression.Citation109 Studies in IL-10 knockout model also revealed an important feature that loss of DRA expression is directly dependent on inflammation and presence of gut microbiota.Citation109 Germ free IL-10 knockout mice neither developed inflammation nor DRA deficiency in the colon. It could be that gut microbiota, a component overlooked in initial studies on UC patientsCitation23,Citation52,Citation109,Citation110 contributed to the contradictory observations on DRA expression noted in these studies.
The studies from our group reported a significant decline in DRA mRNA and protein expression in experimental model of DSS induced colitis.Citation111-Citation113 This loss of DRA was observed mainly in the distal colon. Notably, the loss of DRA expression observed in our studies is comparable to that observed by Yang et al.Citation109 in UC patients. Similar to moderately inflamed UC tissue, 3% DSS used in these studies caused mild colitis with no significant loss of surface epithelium. This reinstates the parallel association between loss of DRA expression and intestinal inflammation. Decreased intestinal DRA protein expression and coincidental diarrhea in DSS treated mice is also reported.Citation114 Other model of murine colitis induced by Citrobacter rodentium exhibit reduced DRA protein and mRNA levels with associated diarrhea.Citation108,Citation115 TNF+/ΔARE mice (that overexpress TNFα and display features of CD) exhibit a significant decrease in DRA mRNA and protein expression in both ileum and mid-distal colon.Citation116 These studies indicated that regardless of the origin of intestinal inflammation (genetic, chemically induced or pathogenic), DRA undergoes downregulation. Interestingly, these murine models collectively portray IBD pathogenesis, which involves a complex interplay of genetic, microbial and environmental components. These studies thus reaffirm that DRA plays crucial role in IBD associated diarrhea.
Whether downregulation of DRA is just the outcome or can instigate IBD pathogenesis is unclear. In a recent GWAS (Genome Wide Association Study), a single-nucleotide polymorphism in SLC26A3 gene associated with a decrease in DRA expression was identified as risk factor for UC development.Citation117 The use of knockout mice also showed that the lack of DRA, caused a significant increase in vulnerability to DSS induced colitis and lower survival rate subsequent to DSS insult.Citation118 These studies associate greater complexity to the role of DRA in IBD, definitely beyond its transporter function. Acute ionic imbalance (hence altered pH), expected gut microbial shift/increased propensity to pathogenic insult and consequent inflammation may underlie pathogenesis of IBD in the case of DRA deficiency. In fact, few of these presumptions have been validated. Colon of DRA knockout mice, unlike the wildtype mice, lack an adherent inner mucus layer despite sufficient number of goblet cells.Citation118 Low luminal pH owing to reduced bicarbonate secretion (DRA knockout) is thought to possibly inhibit the expansion and assembly of mucin granule sheets. Also, the absence of protective colonic mucus layer, as observed in DRA deficient mice, is a characteristic of both murine colitis and IBD patients that potentiates the development of colonic inflammation.Citation119 It also promotes breach of intestinal barrier by pathogenic bacteria and hence can possibly lead to dysbiosis.Citation93 Colon of DRA KO mice also exhibit a striking feature of “conjoined crypts” (crypts with merged crypt orifices) with a substantially elevated proliferation rate.Citation105 In comparison to the wild type mice where cell proliferation is zoned to crypt bottom, the proliferation zone occupied 30–50% of the crypt axis in DRA KO mice. It is compelling to note that a similar pattern is characterized in colitis patients.Citation120 Thus, DRA deficiency primarily causing decreased colonic absorptive efficiency eventually leads to altered proliferative homeostasis of the colonic crypts, flawed mucus layer formation, aggravated inflammation besides diarrhea associated with IBD.
So far, it is clear that DRA loss plays an integral part in IBD associated pathology and diarrhea. Mechanistically, it is highly probable that inflammatory mediators and events in IBD could directly affect DRA protein and mRNA expression. In vitro studies conducted utilizing human intestinal cell lines and mouse enteroids have yielded valuable information on regulation of DRA expression under inflammatory conditions such as those prevailing in IBD. Most pro-inflammatory cytokines abundant in IBD arrest DRA expression at transcriptional level.Citation109,Citation121,Citation122 For instance, treatment of Caco-2 cells with IL-1βCitation109 or IFN-γ 121 inhibit DRA expression transcriptionally. IFN-γ can downregulate DRA promoter activity via JAK/STAT1 pathway.Citation121 Specifically, IFN-γ responsive region located in the -933 to -925 bp region harbors a GAS (γ-activated site) cis element. This can be the mechanism operating in colonic mucosa of UC patients, where elevated STAT1 and diminished DRA function and expression have been reported. Similarly, we have shown a decrease in DRA mRNA and protein expression in response to TNFα treatment, in vitro and in cultured mouse enteroids.Citation122 TNF-α mediates its action through NF-κB pathway. Specifically, binding of p65 subunit of NF-κB at the regions of -935 to -629 and -375 to -84 of DRA promoter, downregulates its transcription.Citation122
Besides massive inflammation, oxidative stress is another potentially harmful event in IBD. In both human and animal models of IBD, elevated reactive oxygen and nitrogen species (ROS/RNS) contribute to IBD pathophysiology.Citation123-Citation125 As a matter of fact, production of ROS/RNS is part of inflammatory reactions and the booster as well. Characterizing the effects of these reactive species on DRA, we have found that both H2O2 126 and nitric oxide (NO)Citation127 suppress apical Cl−/OH− exchange activity. Surprisingly, the surface expression of DRA remained unaltered in presence of H2O2. This suggests involvement of other post-translational modifications such as phosphorylation, lipid-raft dependent pathways or of accessory proteins such as NHERFs as a possible cause in the reduction of Cl−/OH− exchange activity by H2O2. On the other hand, cGMP-PKG and PKC pathways mediated the inhibitory effect of NO on apical Cl−/OH− exchange activity.Citation127 Though the direct involvement of DRA in this studyCitation127 was not elucidated, it can be speculated that DRA being one of the key chloride bicarbonate exchangers could be affected.
Taken together, the above discussion highlights the correlation between loss of DRA function and/or expression in IBD, dampened chloride absorption and development of IBD associated diarrhea. Causality of this correlation is not completely known but can be attributed (a) primarily to pro-inflammatory mediators and oxidative stress prevalent in IBD that can affect DRA transcriptionally and post-translationally, respectively and (b) secondarily to lost or reduced DRA function resulting in aberrant mucus barrier.
DRA appears to be critical in the development of IBD associated diarrhea. Ongoing research in DRA knockout mice, its imperative translation to diseased and normal human intestinal enteroids and extrapolation to clinical studies should further highlight the role of DRA in IBD associated diarrhea.
IBD and Compromised Intestinal Epithelial Barriers
Components of intestinal barrier
Intestinal barrier serves as a protective layer and controls the unrestrained movement of pathogens and toxins from the lumen to the underlying tissue. However, it is selectively permeable and permits the absorption of solutes, ions and water. The term intestinal barrier represents a composite of variety of components that can be broadly categorized into a) the biochemical barriers comprising of the antimicrobial peptides and various elements of the mucosal immune system and b) the physical barriers constituted by the secreted mucus layer and the underlying single layer of intestinal epithelial cells.Citation128-Citation130 This epithelial layer is not impervious and has extensively developed mechanisms for both transcellular e.g. electrolyte and solute absorption, discussed in previous section of this review and paracellular transport. The paracellular transport is regulated by a complex network of intracellular junctions that stretch from the apical to the basolateral membrane of the neighboring intestinal epithelial cells. These include the apically located tight junctions (TJs) followed by sub-apically present adherens junctions (AJs) and the desmosomes present at the basolateral side.
TJs are the key components that serve as a seal between the adjacent cells and govern the passage of molecules; ions and water via ‘size and charge-selective’ pore (for passage of solutes ∼5-10 Å in diameter) and leak (for passage of solutes ∼125 Å in diameter) pathways.Citation131,Citation132 The TJs are composed of transmembrane-spanning proteins including claudins, tight junction associated MARVEL proteins (TAMPs-occludin, MARVEL and tricellulin), junctional adhesion molecule (JAM)-A and the coxsackie adenovirus receptor (CAR). The intracellular domains of the TJs are anchored to the actin cytoskeleton via scaffold of zonula occludens (ZO)-1, ZO-2, ZO-3 and cingulin.Citation133-Citation135 The paracellular trafficking and intestinal permeability are essentially controlled by the TJs.Citation130,Citation132,Citation133,Citation135-Citation137 On the other hand, AJs (consisting of cadherins and nectins) and desmosomes also play a role in regulating paracellular permeability but are more important for cell-cell adhesion, stabilization of TJ assembly, epithelial restitution and integrity.Citation129,Citation131,Citation132 Detailed anatomy, molecular composition, sub-types, location and function of TJs, AJs and desmosomes have been recently reviewed.Citation131,Citation134,Citation138 Here, we focus primarily on the modulation in function and expression of TJ components during intestinal inflammation, contributing to diarrhea in IBD. Additionally, inflammation induced alterations in AJs will also be discussed in brief.
Aberrant epithelial barrier and leak-flux diarrhea
Enhanced intestinal permeability associated with disrupted paracellular barrier is observed in both CD and UC patients. Two major consequences of compromised epithelial barrier in IBD are a) increased translocation of luminal antigens resulting in activation of mucosal immune system and perpetuation of inflammation and b) back flow of absorbed solutes and water via the paracellular pathway into the luminal compartment leading to “leak-flux” diarrhea.Citation128,Citation129,Citation139 A positive association between increased intestinal permeability and severity of diarrhea has been recently demonstrated in IBD patients.Citation140 However, it is interesting to note that deficiency of apical junctional complexes alone does not lead to diarrhea. Diminished electrolyte absorption in presence of compromised barrier integrity appears to aggravate diarrhea observed in IBD.Citation14,Citation130
Data obtained from IBD patients, genetically modified mouse models, mouse models of IBD and in vitro models of inflammation support the critical role of TJs and AJs in regulating paracellular barrier integrity. The mechanisms involved in their regulation during intestinal inflammation are reviewed in the following section.
Mechanisms of TJs and AJs regulation in intestinal inflammation
Emerging evidence suggests that intestinal barrier defects can either predispose or enhance IBD progression.Citation9,Citation128,Citation133-Citation137,Citation141 For example, it has been reported that CD patients in remission with increased intestinal permeability are at a higher risk of disease reactivation.Citation132 Parallel to this, a significant decrease in the epithelial resistance (indicative of leakiness of the paracellular barrier) in the colonic biopsies from UC (85%) and CD (60%) patients as compared to the controls is also observed (utilizing one-path impedance spectroscopy).Citation134,Citation136 However, whether altered gut barrier is the primary cause of the disease or is the consequence of prevailing inflammation during the course of CD and UC is yet unsettled. It has been noticed that the first-degree relatives of CD patients demonstrate increased intestinal permeability and are at increased risk for developing IBD.Citation129,Citation132,Citation142 This suggests that genetic and environmental factors can lead to IBD associated barrier loss, which in turn serves as a contributing factor for enhanced mucosal permeability and disease pathogenesis. Taken together, these observations emphasize the clinical relevance of the permeability defects and altered paracellular junctions (especially TJs) in IBD.
Intestinal permeability primarily reflects the state of the epithelial TJs.Citation129,Citation143 Disturbances in the ultrastructure and protein composition of TJs are identified as morphological correlate for increased gut permeability and decreased epithelial resistance in IBD patients. Reduction in depth of TJ meshwork, number of TJ strands together with strand breaks and discontinuous strand architecture are identified as possible causes of barrier dysfunction in both CD and UC.Citation133,Citation136,Citation144 These perturbations in the gross morphology of the TJs are mainly due to a) alterations in protein expression, b) altered membrane trafficking and localization and c) changes in actomyosin cytoskeleton.Citation134 In this regard, investigations in patients with CD revealed unaltered expression for claudins-1 and 4, decrease in protein expression of occludin, claudins- 3, 5, 7, 8 and JAM-A.Citation129,Citation131,Citation133,Citation134,Citation136 Claudin 5 and 8 also exhibited a shift from the TJ domain towards the lateral plasma membrane in CD biopsies as compared to the control. Of note, protein level of claudin-2 was found to be significantly elevated in CD (). Comparable changes in TJs were also seen in UC patients with contradictory reports for claudins-1, 4 and elevation in claudin-2 protein levelsCitation129,Citation132-Citation134,Citation136,Citation144-Citation147 (). It is worth noting that claudin-2 expression in active UC patients is significantly higher to that observed in CD patients.Citation144 Interestingly, reduced claudin-3 protein expression in colonic biopsies of UC patients was found to be associated with a concomitant increase in urinary concentration of claudin-3. Therefore, measurement of urinary claudin-3 was proposed as a non-invasive marker to assess tight junction impairment.Citation129,Citation133 The above discussion implies that in IBD there is a decrease and/or redistribution in the expression of a subset of claudins that seal the tight junctions (e.g. claudins-1, 3, 4, 5, 7, 8) with a concomitant increase in pore forming claudin-2. This phenomenon of “claudin switching” together with the alterations in other TJ proteins result in an increase in the paracellular permeability and may underlie leak-flux associated diarrhea observed in IBD.Citation12,Citation131-Citation134,Citation144
Table 2. Alterations in Tight junction proteins in IBD Patients.
The focus of the research on barrier dysfunction in IBD centers mainly on TJ impairment and the studies focusing on dysregulation in AJs are quite limited. A dampened cell-cell adhesion observed in UC and CD patients is attributed to downregulation of E-cadherin protein expression measured in tissue biopsies of the IBD patients. Additionally, inflammation induced reduction in the levels of p120-catenin and β-catenin, binding partners for E-cadherin, have also been shown to further decrease the junctional integrity.Citation134 Notably, E-cadherin polymorphism has been shown to be associated with IBD.Citation131,Citation134
Similar to the evidence obtained from IBD patients, the data from mouse models of experimental colitis support the hypothesis that decrease/redistribution of TJ complexes results in enhanced intestinal permeability and promote IBD pathogenesis. A substantial decrease in protein expression of claudins-3, 4, 5, 7, 8, occludin, JAM-A and ZO-1 has been reported in DSS treated mice with a parallel increase in intestinal permeability.Citation142 Similar pattern of change in expression for claudins-3 and 8, occludin, JAM-A and ZO-1 were reported in TNBS colitis model.Citation142 Contrary to the patient data, expression of claudin-2 in both these models of colitis was inconsistent and showed a varying pattern in different studies.Citation142 However, this could be attributed to differences in mouse strain, experimental conditions at the facility and the influence of the host microbiome. These factors play a definitive role in the development of colitis in experimental models.
Furthermore, studies using genetically modified mice with mucosal barrier dysfunction recapitulated IBD phenotype. For example, intestine specific knockdown of pore-sealing claudin-7 in mice demonstrated increased intestinal permeability and developed spontaneous inflammation.Citation131,Citation134,Citation142 On the other hand, JAM-A KO mice do not develop spontaneous colitis but exhibit impaired intestinal permeability accompanied by elevated inflammatory responses.Citation134 Furthermore, overexpression of dominant-negative N-cadherin mutant in mice resulted in functional disruption of E-cadherin associated with development of spontaneous inflammation, crypt hyperproliferation and development of adenomas.Citation131,Citation134,Citation142 Along similar lines, intestine specific knockdown of E-cadherin in mice resulted in increased paracellular permeability, bloody diarrhea, deranged mucosal architecture, compromised paneth and goblet cell maturation.Citation131,Citation134,Citation142 These findings provided evidence that apart from junctional integrity, E-cadherin plays an essential role in intestinal homeostasis and differentiation of secretory cell lineages. Interestingly, intestine specific knockdown of p-120 catenin (cytoplasmic binding partner of E-cadherin) has also been reported to have loss of cell-cell adhesion, significant inflammatory response, intestinal bleeding and mucosal damage.Citation142 Antigen leakage across the defective barrier in these KO mice results in spontaneous inflammation and exacerbated immune response, characteristic of what is observed in IBD patients. Taken together, barrier defects exacerbate inflammation in IBD (triggered by antigen leakage), which together with defective ion transport processes contribute to the pathogenesis of IBD associated diarrhea.
It is well accepted that expression of claudin-2 is highly upregulated in both UC and CD. As it is a pore forming claudin, increase in its expression is correlated with enhanced paracellular flux, diarrhea and augmented inflammatory responses. Interestingly, villin-claudin-2 transgenic mice (with intestine specific overexpression of claudin 2) show increased barrier permeability but are resistant to experimentally induced colitis and develop compensatory adaptive immune responses. In stark contrast, claudin-2 knockdown in mice was not protective. Instead, claudin-2 KO mice are more susceptible to DSS induced colitis.Citation133,Citation134,Citation142 This hints towards the possibility that increase in claudin-2 expression in IBD could be an adaptive mechanism for maintaining mucosal homeostasis. However, the exact explanation for these claudin-2 related changes remains to be established. Although the expression of occludin was downregulated in IBD, occludin KO mice had normal gut permeability and TJ architecture. In fact, rather than the paracellular pathway, occludin deficiency in mice resulted in dysregulated epithelial and immune homeostasis.Citation134,Citation142 It is important to appreciate that junctional proteins like occludin and E-cadherin impact not only the permeability related issues in the gut but are also relevant in maintenance of overall homeostasis in the intestine. This is further attested by the fact that ZO-1 KO mice are embryonically lethal.Citation131
Apart from the targeted deletion of TJ or AJ protein in mice, the gut permeability is also affected by the alterations in cytoskeletal proteins. One such example is regulation of myosin II phosphorylation by MLCK (myosin light chain kinase). The active enzyme phosphorylates myosin II leading to contraction of actomyosin ring thereby upregulating TJ permeability.Citation142 Studies have shown that MLCK is upregulated in IBD and in models of experimental colitis.Citation143 The transgenic model of mice with constitutively active MLCK, show increased gut permeability and susceptibility to experimentally induced colitis regardless of preserved occludin, ZO-1, claudin1 and JAM expression and absence of spontaneous inflammation. Taken together, studies using genetically modified mice with targeted disruption of TJ or AJ proteins highlighted the facts that deficiency of barrier associated proteins i) impairs paracellular permeability ii) perturbs immune homeostasis and intensifies mucosal inflammatory responses and iii) affects epithelial restitution, proliferation and apoptosis and thus can contribute to the pathogenesis of IBD associated diarrhea. These models also elucidated the non-barrier roles of these junctional proteins in the intestine.
Several lines of evidence implicate pro-inflammatory cytokines as key contributors for TJ dysregulation in IBD. Detailed discussion of molecular pathways regulating tight and adherens junction during inflammation is beyond the scope of the present review. In general, pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-1β, IL-6, IL-13 and IL-23 impact barrier integrity and gut permeability via a) expressional regulation of the junctional complex (including upregulation of endocytosis, decreased exocytosis and altered transcription), b) redistribution of TJs causing junctional destabilization, c) activation of MLCK and other signaling pathways such as PI3Kinase/Akt, NF-κB, JNK etc. and d) apoptosis dependent mechanism which lead to erosion and ulceration of the epithelial surface and increased mucosal penetration of the antigens.Citation128,Citation129,Citation131,Citation134,Citation135,Citation143,Citation148 Additionally, the role of microRNAs (miR) in regulating the expression of tight junction proteins has also surfaced. Down regulation of claudins-1, 3 and 8 by miR 29, 93 and 223 respectively and that of occludin by TNF-α induced miR 122a has been demonstrated.Citation18,Citation134 Interestingly, aberrant microRNA profiles observed in UC and CD patientsCitation149 raise the possibility of utilizing microRNAs as marker for permeability defects.
From above, it is clear that sustained inflammation and breach of barrier integrity promotes “leakiness” in the gut, which together with defective ion transport processes contribute to pathogenesis of IBD associated diarrhea.
Conclusion
The incidence for chronic inflammatory disorders of the intestine such as ulcerative colitis and Crohn's disease has significantly increased in the past years. The etiology for both UC and CD is complex with diarrhea as the most debilitating symptom. As summarized in the review, ample evidence indicates that defects in absorptive ion transport accompanied with barrier dysregulation in the intestine are major contributors for diarrhea. Inflammation induced decrease in function and/or expression of major sodium and chloride transporters and channels such as NHE3, ENaC, Na+/K+ ATPase and DRA severely perturbs the hydroelectrolytic balance leading to diarrhea. Of note, chloride secretion does not appear to be affected in IBD and hence its role in diarrheal pathogenesis is negligible. While the drift from normal physiological functions of these transporter proteins is well described, it is interesting to note that studies in the knockout mouse models (especially NHE3 and DRA KO) have highlighted their other important and novel roles such as effects on gut barrier, immunomodulation and dysbiosis. These data are fascinating, as targeting NHE3 and possibly DRA could restore normal microbiota under conditions other than IBD as well. However, further validation using animal models and translation to humans will be important. Also, fecal microbiota transfer studies from IBD (UC and CD) patients and healthy individuals to knockout mice will be crucial to define the role of microbiota to altered expression profiles of NHE3 and DRA in IBD. Furthermore, utilizing cultured enteroids/colonoids prepared from diseased and normal individual's tissue biopsies should provide mechanistic insights into changes in intestinal ion transporters in response to inflammation. Human data (from UC and CD patients) corroborated with several murine models of IBD combined with observations made using knockout or transgenic mice have revealed crucial but complex role of TJ and AJ in IBD associated diarrhea via leak-flux mechanism. Aberrant expression and/or localization of tight and adherens junction during inflammation leads to enhanced intestinal permeability, ease of penetration for the pathogens and perpetuation of inflammation. However, whether the loss of barrier integrity is the “cause or the consequence” in IBD has to be delineated. Investigating the mechanisms responsible for changes in intestinal ion transporters and tight/adherens junctions offer potential therapeutic targets for more efficient treatment of IBD and associated diarrhea.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Zhang M, Sun K, Wu Y, Yang Y, Tso P, Wu Z. Interactions between Intestinal Microbiota and Host Immune Response in Inflammatory Bowel Disease. Front Immunol. 2017;8:942. doi:10.3389/fimmu.2017.00942.
- Shouval DS, Rufo PA. The Role of Environmental Factors in the Pathogenesis of Inflammatory Bowel Diseases: A Review. JAMA pediatrics. 2017;171(10):999–1005. doi:10.1001/jamapediatrics.2017.2571. PMID: 28846760.
- Priyamvada S, Gomes R, Gill RK, Saksena S, Alrefai WA, Dudeja PK. Mechanisms Underlying Dysregulation of Electrolyte Absorption in Inflammatory Bowel Disease-Associated Diarrhea. Inflamm Bowel Dis. 2015;21(12):2926–35. doi:10.1097/MIB.0000000000000504. PMID: 26595422.
- Inflammatory Bowel Disease Is More Common Than Earlier Studies Showed. Jama. 2016;316(24):2590. doi:10.1001/jama.2016.18375..
- Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46–54 e42; quiz e30. doi:10.1053/j.gastro.2011.10.001. PMID: 22001864.
- Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12(4):205–17. doi:10.1038/nrgastro.2015.34.
- Umanskiy K, Fichera A. Health related quality of life in inflammatory bowel disease: the impact of surgical therapy. World J Gastroenterol. 2010;16(40):5024–34. PMID: 20976838.
- Wenzl HH. Diarrhea in chronic inflammatory bowel diseases. Gastroenterol Clin North Am. 2012;41(3):651–75. doi:10.1016/j.gtc.2012.06.006. PMID: 22917170.
- Binder HJ. Mechanisms of diarrhea in inflammatory bowel diseases. Ann N Y Acad Sci. 2009;1165:285–93. doi:10.1111/j.1749-6632.2009.04039.x.
- Ward JB, Keely SJ, Keely SJ. Oxygen in the regulation of intestinal epithelial transport. J Physiol. 2014;592(12):2473–89. doi:10.1113/jphysiol.2013.270249. PMID: 24710059.
- Seidler U, Lenzen H, Cinar A, Tessema T, Bleich A, Riederer B. Molecular mechanisms of disturbed electrolyte transport in intestinal inflammation. Ann N Y Acad Sci. 2006;1072:262–75. doi:10.1196/annals.1326.024..
- Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111(7):931–43. doi:10.1172/JCI18326. PMID: 12671039.
- Barkas F, Liberopoulos E, Kei A, Elisaf M. Electrolyte and acid-base disorders in inflammatory bowel disease. Ann Gastroenterol. 2013;26(1):23–8. PMID: 24714322.
- Sandle GI. Pathogenesis of diarrhea in ulcerative colitis: new views on an old problem. J Clin Gastroenterol. 2005;39(4 Suppl 2):S49–52. PMID: 15758659.
- Surawicz CM. Mechanisms of diarrhea. Current gastroenterology reports. 2010;12(4):236–41. PMID: 20532705.
- Magalhaes D, Cabral JM, Soares-da-Silva P, Magro F. Role of epithelial ion transports in inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2016;310(7):G460–76. doi:10.1152/ajpgi.00369.2015. PMID: 26744474.
- McCole DF, Barrett KE. Decoding epithelial signals: critical role for the epidermal growth factor receptor in controlling intestinal transport function. Acta Physiol. 2009;195(1):149–59. doi:10.1111/j.1748-1716.2008.01929.x. PMID: 18983445.
- Tang Y, Forsyth CB, Keshavarzian A. New molecular insights into inflammatory bowel disease-induced diarrhea. Expert Rev Gastroenterology Hepatol. 2011;5(5):615–25. doi:10.1586/egh.11.64.
- Ghishan FK, Kiela PR. Epithelial transport in inflammatory bowel diseases. Inflamm Bowel Dis. 2014;20(6):1099–109. doi:10.1097/MIB.0000000000000029. PMID: 24691115.
- Suh JS, Hahn WH, Cho BS. Recent Advances of Oral Rehydration Therapy (ORT). Electrolyte Blood Press: E & BP. 2010;8(2):82–6. doi:10.5049/EBP.2010.8.2.82.
- Gill RK, Alrefai W, Ramaswamy A, Dudeja PK. Mechanisms and regulation of NaCl absorption in the human intestine. Recent Res Devel Physiol. 2003;1:643–77.
- Martinez-Augustin O, Romero-Calvo I, Suarez MD, Zarzuelo A, de Medina FS. Molecular bases of impaired water and ion movements in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15(1):114–27. doi:10.1002/ibd.20579. PMID: 18626965.
- Farkas K, Yeruva S, Rakonczay Z, Jr., Ludolph L, Molnar T, Nagy F, Szepes Z, Schnur A, Wittmann T, Hubricht J, et al. New therapeutic targets in ulcerative colitis: the importance of ion transporters in the human colon. Inflamm Bowel Dis. 2011;17(4):884–98. PMID: 20722063.
- Barmeyer C, Erko I, Fromm A, Bojarski C, Allers K, Moos V, Zeitz M, Fromm M, Schulzke JD. Ion transport and barrier function are disturbed in microscopic colitis. Ann N Y Acad Sci. 2012;1258:143–8. doi:10.1111/j.1749-6632.2012.06631.x.
- Amasheh S, Barmeyer C, Koch CS, Tavalali S, Mankertz J, Epple HJ, Gehring MM, Florian P, Kroesen AJ, Zeitz M, et al. Cytokine-dependent transcriptional down-regulation of epithelial sodium channel in ulcerative colitis. Gastroenterology. 2004;126(7):1711–20. PMID: 15188166.
- Donowitz M, Ming Tse C, Fuster D. SLC9/NHE gene family, a plasma membrane and organellar family of Na(+)/H(+) exchangers. Mol Aspects Med. 2013;34(2-3):236–51. doi:10.1016/j.mam.2012.05.001. PMID: 23506868.
- Kiela PR, Xu H, Ghishan FK. Apical NA+/H+ exchangers in the mammalian gastrointestinal tract. J Physiol Pharmacol. 2006;57 Suppl 7:51–59.
- Kiela PR, Ghishan FK. Na+-H+Exchange in Mammalian Digestive Tract. Physiology of the Gastrointestinal Tract (Fourth Edition). 2006;2:1847–1879.
- Xu H, Zhang B, Li J, Wang C, Chen H, Ghishan FK. Impaired mucin synthesis and bicarbonate secretion in the colon of NHE8 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2012;303(3):G335–43. doi:10.1152/ajpgi.00146.2012. PMID: 22575219.
- Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol. 2005;67:411–443. doi:10.1146/annurev.physiol.67.031103.153004.
- Gurney MA, Laubitz D, Ghishan FK, Kiela PR. Pathophysiology of Intestinal Na(+)/H(+) exchange. Cell Mol Gastroenterol Hepatol. 2017;3(1):27–40. doi:10.1016/j.jcmgh.2016.09.010. PMID: 28090568.
- Khan I, al-Awadi FM, Abul H. Colitis-induced changes in the expression of the Na+/H+ exchanger isoform NHE-1. J Pharmacol Exp Ther. 1998;285(2):869–75. PMID: 9580638.
- Khan I, Siddique I, Al-Awadi FM, Mohan K. Role of Na+/H+ exchanger isoform-1 in human inflammatory bowel disease. Canadian journal of gastroenterology = Journal canadien de gastroenterologie. 2003;17(1):31–6. PMID: 12560852.
- Siddique I, Khan I. Mechanism of regulation of Na-H exchanger in inflammatory bowel disease: role of TLR-4 signaling mechanism. Dig Dis Sci. 2011;56(6):1656–62. doi:10.1007/s10620-010-1524-7. PMID: 21221801.
- Sullivan S, Alex P, Dassopoulos T, Zachos NC, Iacobuzio-Donahue C, Donowitz M, Brant SR, Cuffari C, Harris ML, Datta LW, et al. Downregulation of sodium transporters and NHERF proteins in IBD patients and mouse colitis models: potential contributors to IBD-associated diarrhea. Inflamm Bowel Dis. 2009;15(2):261–74. doi:10.1002/ibd.20743. PMID: 18942765.
- Rocha F, Musch MW, Lishanskiy L, Bookstein C, Sugi K, Xie Y, Chang EB. IFN-gamma downregulates expression of Na(+)/H(+) exchangers NHE2 and NHE3 in rat intestine and human Caco-2/bbe cells. Am J Physiol Cell Physiol. 2001;280(5):C1224–32. doi:10.1152/ajpcell.2001.280.5.C1224. PMID: 11287336.
- Amin MR, Orenuga T, Tyagi S, Dudeja PK, Ramaswamy K, Malakooti J. Tumor necrosis factor-alpha represses the expression of NHE2 through NF-kappaB activation in intestinal epithelial cell model, C2BBe1. Inflamm Bowel Dis. 2011;17(3):720–31. doi:10.1002/ibd.21419. PMID: 20722069.
- Soleiman AA, Thameem F, Khan I. Mechanism of down regulation of Na-H exchanger-2 in experimental colitis. PloS One. 2017;12(5):e0176767. doi:10.1371/journal.pone.0176767. PMID: 28493993.
- Xu H, Chen H, Dong J, Lynch R, Ghishan FK. Gastrointestinal distribution and kinetic characterization of the sodium-hydrogen exchanger isoform 8 (NHE8). Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2008;21(1-3):109–16. doi:10.1159/000113752. PMID: 18209477.
- Liu C, Xu H, Zhang B, Johansson ME, Li J, Hansson GC, Ghishan FK. NHE8 plays an important role in mucosal protection via its effect on bacterial adhesion. Am J Physiol Cell Physiol. 2013;305(1):C121–8. doi:10.1152/ajpcell.00101.2013. PMID: 23657568.
- Xu H, Chen H, Dong J, Li J, Chen R, Uno JK, Ghishan FK. Tumor necrosis factor-{alpha} downregulates intestinal NHE8 expression by reducing basal promoter activity. Am J Physiol Cell Physiol. 2009;296(3):C489–97. doi:10.1152/ajpcell.00482.2008. PMID: 19109523.
- Wang A, Li J, Zhao Y, Johansson ME, Xu H, Ghishan FK. Loss of NHE8 expression impairs intestinal mucosal integrity. Am J Physiol Gastrointest Liver Physiol. 2015;309(11):G855–64. doi:10.1152/ajpgi.00278.2015. PMID: 26505975.
- Xu H, Li Q, Zhao Y, Li J, Ghishan FK. Intestinal NHE8 is highly expressed in goblet cells and its expression is subject to TNF-alpha regulation. Am J Physiol Gastrointest Liver Physiol. 2016;310(2):G64–9. doi:10.1152/ajpgi.00367.2015. PMID: 26564720.
- Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19(3):282–5. doi:10.1038/969. PMID: 9662405.
- Gawenis LR, Stien X, Shull GE, Schultheis PJ, Woo AL, Walker NM, Clarke LL. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2002;282(5):G776–84. doi:10.1152/ajpgi.00297.2001. PMID: 11960774.
- Ledoussal C, Woo AL, Miller ML, Shull GE. Loss of the NHE2 Na(+)/H(+) exchanger has no apparent effect on diarrheal state of NHE3-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2001;281(6):G1385–96. doi:10.1152/ajpgi.2001.281.6.G1385. PMID: 11705743.
- Janecke AR, Heinz-Erian P, Yin J, Petersen BS, Franke A, Lechner S, Fuchs I, Melancon S, Uhlig HH, Travis S, et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum Mol Genet. 2015;24(23):6614–23. doi:10.1093/hmg/ddv367. PMID: 26358773.
- Zhu XC, Sarker R, Horton JR, Chakraborty M, Chen TE, Tse CM, Cha B, Donowitz M. Nonsynonymous single nucleotide polymorphisms of NHE3 differentially decrease NHE3 transporter activity. Am J Physiol Cell Physiol. 2015;308(9):C758–66. doi:10.1152/ajpcell.00421.2014. PMID: 25715704.
- Janecke AR, Heinz-Erian P, Muller T. Congenital Sodium Diarrhea: A Form of Intractable Diarrhea, With a Link to Inflammatory Bowel Disease. J Pediatr Gastroenterol Nutr. 2016;63(2):170–6. doi:10.1097/MPG.0000000000001139. PMID: 26835907.
- Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–52. doi:10.1038/ng.764. PMID: 21297633.
- Yeruva S, Farkas K, Hubricht J, Rode K, Riederer B, Bachmann O, Cinar A, Rakonczay Z, Molnar T, Nagy F, et al. Preserved Na(+)/H(+) exchanger isoform 3 expression and localization, but decreased NHE3 function indicate regulatory sodium transport defect in ulcerative colitis. Inflamm Bowel Dis. 2010;16(7):1149–61. doi:10.1002/ibd.21183. PMID: 20027604.
- Lohi H, Makela S, Pulkkinen K, Hoglund P, Karjalainen-Lindsberg ML, Puolakkainen P, Kere J. Upregulation of CFTR expression but not SLC26A3 and SLC9A3 in ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. 2002;283(3):G567–75. doi:10.1152/ajpgi.00356.2001. PMID: 12181169.
- Yeruva S, Chodisetti G, Luo M, Chen M, Cinar A, Ludolph L, Lunnemann M, Goldstein J, Singh AK, Riederer B, et al. Evidence for a causal link between adaptor protein PDZK1 downregulation and Na(+)/H(+) exchanger NHE3 dysfunction in human and murine colitis. Pflugers Archiv: European journal of physiology. 2015;467(8):1795–807. doi:10.1007/s00424-014-1608-x. PMID: 25271043.
- Siddique I, Hasan F, Khan I. Suppression of Na+/H+ exchanger isoform-3 in human inflammatory bowel disease: lack of reversal by 5'-aminosalicylate treatment. Scand J Gastroenterol. 2009;44(1):56–64. doi:10.1080/00365520802321253. PMID: 18785066.
- Lenzen H, Lunnemann M, Bleich A, Manns MP, Seidler U, Jorns A. Downregulation of the NHE3-binding PDZ-adaptor protein PDZK1 expression during cytokine-induced inflammation in interleukin-10-deficient mice. PloS one. 2012;7(7):e40657. doi:10.1371/journal.pone.0040657. PMID: 22848392.
- Amin MR, Malakooti J, Sandoval R, Dudeja PK, Ramaswamy K. IFN-gamma and TNF-alpha regulate human NHE3 gene expression by modulating the Sp family transcription factors in human intestinal epithelial cell line C2BBe1. Am J Physiol Cell Physiol. 2006;291(5):C887–96. doi:10.1152/ajpcell.00630.2005. PMID: 16760259.
- Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116(10):2682–94. doi:10.1172/JCI29218. PMID: 17016558.
- Gill RK, Saksena S, Syed IA, Tyagi S, Alrefai WA, Malakooti J, Ramaswamy K, Dudeja PK. Regulation of NHE3 by nitric oxide in Caco-2 cells. Am J Physiol Gastrointest Liver Physiol. 2002;283(3):G747–56. doi:10.1152/ajpgi.00294.2001. PMID: 12181191.
- Chen T, Kocinsky HS, Cha B, Murtazina R, Yang J, Tse CM, Singh V, Cole R, Aronson PS, de Jonge H, et al. Cyclic GMP kinase II (cGKII) inhibits NHE3 by altering its trafficking and phosphorylating NHE3 at three required sites: identification of a multifunctional phosphorylation site. J Biol Chem. 2015;290(4):1952–65. doi:10.1074/jbc.M114.590174. PMID: 25480791.
- Muller T, Rasool I, Heinz-Erian P, Mildenberger E, Hulstrunk C, Muller A, Michaud L, Koot BG, Ballauff A, Vodopiutz J, et al. Congenital secretory diarrhoea caused by activating germline mutations in GUCY2C. Gut. 2016;65(8):1306–13. doi:10.1136/gutjnl-2015-309441. PMID: 25994218.
- Laubitz D, Larmonier CB, Bai A, Midura-Kiela MT, Lipko MA, Thurston RD, Kiela PR, Ghishan FK. Colonic gene expression profile in NHE3-deficient mice: evidence for spontaneous distal colitis. Am J Physiol Gastrointest Liver Physiol. 2008;295(1):G63–77. doi:10.1152/ajpgi.90207.2008. PMID: 18467500.
- Kiela PR, Laubitz D, Larmonier CB, Midura-Kiela MT, Lipko MA, Janikashvili N, Bai A, Thurston R, Ghishan FK. Changes in mucosal homeostasis predispose NHE3 knockout mice to increased susceptibility to DSS-induced epithelial injury. Gastroenterology. 2009;137(3):965–75, 75 e1-10. doi:10.1053/j.gastro.2009.05.043. PMID: 19450596.
- Larmonier CB, Laubitz D, Thurston RD, Bucknam AL, Hill FM, Midura-Kiela M, Ramalingam R, Kiela PR, Ghishan FK. NHE3 modulates the severity of colitis in IL-10-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2011;300(6):G998–G1009. doi:10.1152/ajpgi.00073.2011. PMID: 21415416.
- Laubitz D, Harrison CA, Midura-Kiela MT, Ramalingam R, Larmonier CB, Chase JH, Caporaso JG, Besselsen DG, Ghishan FK, Kiela PR. Reduced Epithelial Na+/H+ Exchange Drives Gut Microbial Dysbiosis and Promotes Inflammatory Response in T Cell-Mediated Murine Colitis. PloS one. 2016;11(4):e0152044. doi:10.1371/journal.pone.0152044. PMID: 27050757.
- Larmonier CB, Laubitz D, Hill FM, Shehab KW, Lipinski L, Midura-Kiela MT, McFadden RM, Ramalingam R, Hassan KA, Golebiewski M, et al. Reduced colonic microbial diversity is associated with colitis in NHE3-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2013;305(10):G667–77. doi:10.1152/ajpgi.00189.2013. PMID: 24029465.
- Firsov D, Gautschi I, Merillat AM, Rossier BC, Schild L. The heterotetrameric architecture of the epithelial sodium channel (ENaC). EMBO J. 1998;17(2):344–52. doi:10.1093/emboj/17.2.344. PMID: 9430626.
- Greig E, Sandle GI. Diarrhea in ulcerative colitis. The role of altered colonic sodium transport. Ann N Y Acad Sci. 2000;915:327–32. PMID: 11193595.
- Sandle GI, Higgs N, Crowe P, Marsh MN, Venkatesan S, Peters TJ. Cellular basis for defective electrolyte transport in inflamed human colon. Gastroenterology. 1990;99(1):97–105. PMID: 2344946.
- Greig ER, Boot-Handford RP, Mani V, Sandle GI. Decreased expression of apical Na+ channels and basolateral Na+, K+-ATPase in ulcerative colitis. J Pathol. 2004;204(1):84–92. PMID: 15307141.
- Zeissig S, Bergann T, Fromm A, Bojarski C, Heller F, Guenther U, Zeitz M, Fromm M, Schulzke JD. Altered ENaC expression leads to impaired sodium absorption in the noninflamed intestine in Crohn's disease. Gastroenterology. 2008;134(5):1436–47. PMID: 18355814.
- McCole DF, Rogler G, Varki N, Barrett KE. Epidermal growth factor partially restores colonic ion transport responses in mouse models of chronic colitis. Gastroenterology. 2005;129(2):591–608. PMID: 16083715.
- Bergann T, Zeissig S, Fromm A, Richter JF, Fromm M, Schulzke JD. Glucocorticoids and tumor necrosis factor-alpha synergize to induce absorption by the epithelial sodium channel in the colon. Gastroenterology. 2009;136(3):933–42. PMID: 19185581.
- Barmeyer C, Harren M, Schmitz H, Heinzel-Pleines U, Mankertz J, Seidler U, Horak I, Wiedenmann B, Fromm M, Schulzke JD. Mechanisms of diarrhea in the interleukin-2-deficient mouse model of colonic inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;286(2):G244–52. PMID: 14715519.
- Umbach AT, Luo D, Bhavsar SK, Hosseinzadeh Z, Lang F. Intestinal Na+ loss and volume depletion in JAK3-deficient mice. Kidney & blood pressure research. 2013;37(4-5):514–20.
- Toivola DM, Krishnan S, Binder HJ, Singh SK, Omary MB. Keratins modulate colonocyte electrolyte transport via protein mistargeting. J Cell Biol. 2004;164(6):911–21. PMID: 15007064.
- Rajasekaran SA, Barwe SP, Rajasekaran AK. Multiple functions of Na,K-ATPase in epithelial cells. Semin Nephrol. 2005;25(5):328–34. doi:10.1016/j.semnephrol.2005.03.008. PMID: 16139688.
- Hamilton KL, Robert K. Crane-Na(+)-glucose cotransporter to cure? Front Physiol. 2013;4:53. doi:10.3389/fphys.2013.00053.
- Ewe K. Intestinal transport in constipation and diarrhoea. Pharmacology. 1988;36 Suppl 173–84.
- Allgayer H, Kruis W, Paumgartner G, Wiebecke B, Brown L, Erdmann E. Inverse relationship between colonic (Na+ + K+)-ATPase activity and degree of mucosal inflammation in inflammatory bowel disease. Dig Dis Sci. 1988;33(4):417–22. PMID: 2832138.
- Ejderhamn J, Finkel Y, Strandvik B. Na,K-ATPase activity in rectal mucosa of children with ulcerative colitis and Crohn's disease. Scand J Gastroenterol. 1989;24(9):1121–5. PMID: 2556782.
- Rachmilewitz D, Karmeli F, Sharon P. Decreased colonic Na-K-ATPase activity in active ulcerative colitis. Isr J Med Sci. 1984;20(8):681–4. PMID: 6088428.
- Hirota CL, McKay DM. Loss of Ca-mediated ion transport during colitis correlates with reduced ion transport responses to a Ca-activated K channel opener. Br J Pharmacol. 2009;156(7):1085–97. doi:10.1111/j.1476-5381.2009.00122.x. PMID: 19298254.
- Magro F, Fraga S, Ribeiro T, Soares-da-Silva P. Regional intestinal adaptations in Na+,K+-ATPase in experimental colitis and the contrasting effects of interferon-gamma. Acta physiologica Scandinavica. 2005;183(2):191–9. doi:10.1111/j.1365-201X.2004.01388.x. PMID: 15676060.
- Wild GE, Thomson AB. Na(+)-K(+)-ATPase alpha 1- and beta 1-mRNA and protein levels in rat small intestine in experimental ileitis. Am J Physiol. 1995;269(5 Pt 1):G666–75. doi:10.1152/ajpgi.1995.269.5.G666. PMID: 7491957.
- Musch MW, Clarke LL, Mamah D, Gawenis LR, Zhang Z, Ellsworth W, Shalowitz D, Mittal N, Efthimiou P, Alnadjim Z, et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J Clin Invest. 2002;110(11):1739–47. doi:10.1172/JCI15695. PMID: 12464679.
- Bertelsen LS, Eckmann L, Barrett KE. Prolonged interferon-gamma exposure decreases ion transport, NKCC1, and Na+-K+-ATPase expression in human intestinal xenografts in vivo. Am J Physiol Gastrointest Liver Physiol. 2004;286(1):G157–65. doi:10.1152/ajpgi.00227.2003. PMID: 12958023.
- Yoo D, Lo W, Goodman S, Ali W, Semrad C, Field M. Interferon-gamma downregulates ion transport in murine small intestine cultured in vitro. Am J Physiol Gastrointest Liver Physiol. 2000;279(6):G1323–32. doi:10.1152/ajpgi.2000.279.6.G1323. PMID: 11093956.
- Magro F, Fraga S, Ribeiro T, Soares-da-Silva P. Intestinal Na+-K+-ATPase activity and molecular events downstream of interferon-gamma receptor stimulation. Br J Pharmacol. 2004;142(8):1281–92. doi:10.1038/sj.bjp.0705895. PMID: 15277314.
- Sugi K, Musch MW, Field M, Chang EB. Inhibition of Na+,K+-ATPase by interferon gamma down-regulates intestinal epithelial transport and barrier function. Gastroenterology. 2001;120(6):1393–403. PMID: 11313309.
- Markossian S, Kreydiyyeh SI. TNF-alpha down-regulates the Na+-K+ ATPase and the Na+-K+-2Cl-cotransporter in the rat colon via PGE2. Cytokine. 2005;30(6):319–27. doi:10.1016/j.cyto.2004.11.009. PMID: 15935952.
- Kreydiyyeh SI, Al-Sadi R. Interleukin-1beta increases urine flow rate and inhibits protein expression of Na(+)/K(+)-ATPase in the rat jejunum and kidney. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2002;22(10):1041–8. doi:10.1089/107999002760624279..
- Al-Sadi RM, Kreydiyyeh SI. Mediators of interleukin-1 beta action Na(+)-K(+)ATPase in Caco-2 cells. Eur Cytokine Netw. 2003;14(2):83–90. PMID: 12957788.
- Bachmann O, Seidler U. News from the end of the gut–how the highly segmental pattern of colonic HCO(3)(-) transport relates to absorptive function and mucosal integrity. Biological & pharmaceutical bulletin. 2011;34(6):794–802.
- Roediger WE, Lawson MJ, Kwok V, Grant AK, Pannall PR. Colonic bicarbonate output as a test of disease activity in ulcerative colitis. J Clin Pathol. 1984;37(6):704–7. PMID: 6327778.
- Caprilli R, Frieri G, Latella G, Vernia P, Santoro ML. Faecal excretion of bicarbonate in ulcerative colitis. Digestion. 1986;35(3):136–42. PMID: 3781109.
- Schilli R, Breuer RI, Klein F, Dunn K, Gnaedinger A, Bernstein J, Paige M, Kaufman M. Comparison of the composition of faecal fluid in Crohn's disease and ulcerative colitis. Gut. 1982;23(4):326–32. PMID: 7076010.
- Hawker PC, McKay JS, Turnberg LA. Electrolyte transport across colonic mucosa from patients with inflammatory bowel disease. Gastroenterology. 1980;79(3):508–11. PMID: 7429111.
- Sandle GI, Hayslett JP, Binder HJ. Effect of glucocorticoids on rectal transport in normal subjects and patients with ulcerative colitis. Gut. 1986;27(3):309–16. PMID: 3699552.
- Edmonds CJ, Pilcher D. Electrical potential difference and sodium and potassium fluxes across rectal mucosa in ulcerative colitis. Gut. 1973;14(10):784–9. PMID: 4758659.
- Kato A, Romero MF. Regulation of electroneutral NaCl absorption by the small intestine. Annu Rev Physiol. 2011;73:261–81. doi:10.1146/annurev-physiol-012110-142244.
- Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Archiv: European journal of physiology. 2004;447(5):710–21. PMID: 12759755.
- Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med. 2013;34(2-3):494–515. PMID: 23506885.
- Makela S, Kere J, Holmberg C, Hoglund P. SLC26A3 mutations in congenital chloride diarrhea. Human mutation. 2002;20(6):425–38. PMID: 12442266.
- Wedenoja S, Pekansaari E, Hoglund P, Makela S, Holmberg C, Kere J. Update on SLC26A3 mutations in congenital chloride diarrhea. Human mutation. 2011;32(7):715–22. PMID: 21394828.
- Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, Soleimani M. slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J Biol Chem. 2006;281(49):37962–71. PMID: 17001077.
- Jiang Z, Asplin JR, Evan AP, Rajendran VM, Velazquez H, Nottoli TP, Binder HJ, Aronson PS. Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nat Genet. 2006;38(4):474–8. doi:10.1038/ng1762. PMID: 16532010.
- D'Angelo A, Bluteau O, Garcia-Gonzalez MA, Gresh L, Doyen A, Garbay S, Robine S, Pontoglio M. Hepatocyte nuclear factor 1alpha and beta control terminal differentiation and cell fate commitment in the gut epithelium. Development. 2010;137(9):1573–82. doi:10.1242/dev.044420. PMID: 20388655.
- Borenshtein D, Schlieper KA, Rickman BH, Chapman JM, Schweinfest CW, Fox JG, Schauer DB. Decreased expression of colonic Slc26a3 and carbonic anhydrase iv as a cause of fatal infectious diarrhea in mice. Infect Immun. 2009;77(9):3639–50. PMID: 19546193.
- Yang H, Jiang W, Furth EE, Wen X, Katz JP, Sellon RK, Silberg DG, Antalis TM, Schweinfest CW, Wu GD. Intestinal inflammation reduces expression of DRA, a transporter responsible for congenital chloride diarrhea. Am J Physiol. 1998;275(6 Pt 1):G1445–53. PMID: 9843783.
- Haila S, Saarialho-Kere U, Karjalainen-Lindsberg ML, Lohi H, Airola K, Holmberg C, Hastbacka J, Kere J, Hoglund P. The congenital chloride diarrhea gene is expressed in seminal vesicle, sweat gland, inflammatory colon epithelium, and in some dysplastic colon cells. Histochem Cell Biol. 2000;113(4):279–86. PMID: 10857479.
- Singh V, Kumar A, Raheja G, Anbazhagan AN, Priyamvada S, Saksena S, Jhandier MN, Gill RK, Alrefai WA, Borthakur A, et al. Lactobacillus acidophilus attenuates downregulation of DRA function and expression in inflammatory models. Am J Physiol Gastrointest Liver Physiol. 2014;307(6):G623–31. doi:10.1152/ajpgi.00104.2014. PMID: 25059823.
- Jayawardena D, Anbazhagan AN, Guzman G, Dudeja PK, Onyuksel H. Vasoactive Intestinal Peptide Nanomedicine for the Management of Inflammatory Bowel Disease. Molecular pharmaceutics. 2017;14(11):3698–708. doi:10.1021/acs.molpharmaceut.7b00452. PMID: 28991483.
- Anbazhagan AN, Thaqi M, Priyamvada S, Jayawardena D, Kumar A, Gujral T, Chatterjee I, Mugarza E, Saksena S, Onyuksel H, et al. GLP-1 nanomedicine alleviates gut inflammation. Nanomedicine: nanotechnology, biology, and medicine. 2017;13(2):659–65. doi:10.1016/j.nano.2016.08.004. PMID: 27553076.
- Xu L, Xiao F, He J, Lan X, Ding Q, Li J, Seidler U, Zheng Y, Tian D. Lysophosphatidic acid increases SLC26A3 expression in inflamed intestine and reduces diarrheal severity in C57BL/6 mice with dextran-sodium-sulfate-induced colitis. Chin Med J. 2014;127(9):1737–43. PMID: 24791884.
- Kumar A, Anbazhagan AN, Coffing H, Chatterjee I, Priyamvada S, Gujral T, Saksena S, Gill RK, Alrefai WA, Borthakur A, et al. Lactobacillus acidophilus counteracts inhibition of NHE3 and DRA expression and alleviates diarrheal phenotype in mice infected with Citrobacter rodentium. Am J Physiol Gastrointest Liver Physiol. 2016;311(5):G817–G26. doi:10.1152/ajpgi.00173.2016. PMID: 27634011.
- Xiao F, Juric M, Li J, Riederer B, Yeruva S, Singh AK, Zheng L, Glage S, Kollias G, Dudeja P, et al. Loss of downregulated in adenoma (DRA) impairs mucosal HCO3(-) secretion in murine ileocolonic inflammation. Inflamm Bowel Dis. 2012;18(1):101–11. doi:10.1002/ibd.21744. PMID: 21557395.
- Asano K, Matsushita T, Umeno J, Hosono N, Takahashi A, Kawaguchi T, Matsumoto T, Matsui T, Kakuta Y, Kinouchi Y, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009;41(12):1325–9. doi:10.1038/ng.482. PMID: 19915573.
- Xiao F, Yu Q, Li J, Johansson ME, Singh AK, Xia W, Riederer B, Engelhardt R, Montrose M, Soleimani M, et al. Slc26a3 deficiency is associated with loss of colonic HCO3 (-) secretion, absence of a firm mucus layer and barrier impairment in mice. Acta Physiol. 211(1):161–75. PMID: 24373192.
- Johansson ME, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, Ghishan FK, Carvalho FA, Gewirtz AT, Sjovall H, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63(2):281–91. doi:10.1136/gutjnl-2012-303207. PMID: 23426893.
- Serafini EP, Kirk AP, Chambers TJ. Rate and pattern of epithelial cell proliferation in ulcerative colitis. Gut. 1981;22(8):648–52. PMID: 7286781.
- Saksena S, Singla A, Goyal S, Katyal S, Bansal N, Gill RK, Alrefai WA, Ramaswamy K, Dudeja PK. Mechanisms of transcriptional modulation of the human anion exchanger SLC26A3 gene expression by IFN-{gamma}. Am J Physiol Gastrointest Liver Physiol. 2010;298(2):G159–66. doi:10.1152/ajpgi.00374.2009. PMID: 19940027.
- Kumar A, Chatterjee I, Gujral T, Alakkam A, Coffing H, Anbazhagan AN, Borthakur A, Saksena S, Gill RK, Alrefai WA, et al. Activation of Nuclear Factor-kappaB by Tumor Necrosis Factor in Intestinal Epithelial Cells and Mouse Intestinal Epithelia Reduces Expression of the Chloride Transporter SLC26A3. Gastroenterology. 2017;153(5):1338–50 e3. doi:10.1053/j.gastro.2017.08.024. PMID: 28823863.
- Cross RK, Wilson KT. Nitric oxide in inflammatory bowel disease. Inflammatory bowel diseases. 2003;9(3):179–89. PMID: 12792224.
- Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med. 2012;237(5):474–80. doi:10.1258/ebm.2011.011358. PMID: 22442342.
- Kolios G, Valatas V, Ward SG. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113(4):427–37. doi:10.1111/j.1365-2567.2004.01984.x. PMID: 15554920.
- Saksena S, Gill RK, Tyagi S, Alrefai WA, Ramaswamy K, Dudeja PK. Role of Fyn and PI3K in H2O2-induced inhibition of apical Cl-/OH- exchange activity in human intestinal epithelial cells. Biochem J. 2008;416(1):99–108. PMID: 18564062.
- Saksena S, Gill RK, Syed IA, Tyagi S, Alrefai WA, Ramaswamy K, Dudeja PK. Modulation of Cl-/OH- exchange activity in Caco-2 cells by nitric oxide. Am J Physiol Gastrointest Liver Physiol. 2002;283(3):G626–33. PMID: 12181176.
- Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nature reviews Gastroenterology & hepatology. 2017;14(1):9–21. doi:10.1038/nrgastro.2016.169.
- Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, Wells JM. Intestinal permeability–a new target for disease prevention and therapy. BMC Gastroenterol. 2014;14:189. doi:10.1186/s12876-014-0189-7.
- Camilleri M, Sellin JH, Barrett KE. Pathophysiology, Evaluation, and Management of Chronic Watery Diarrhea. Gastroenterology. 2017;152(3):515–32 e2. doi:10.1053/j.gastro.2016.10.014. PMID: 27773805.
- France MM, Turner JR. The mucosal barrier at a glance. J Cell Sci. 2017;130(2):307–14. doi:10.1242/jcs.193482. PMID: 28062847.
- Choi W, Yeruva S, Turner JR. Contributions of intestinal epithelial barriers to health and disease. Exp Cell Res. 2017;358(1):71–7. doi:10.1016/j.yexcr.2017.03.036. PMID: 28342899.
- Barmeyer C, Fromm M, Schulzke JD. Active and passive involvement of claudins in the pathophysiology of intestinal inflammatory diseases. Pflugers Archiv: European journal of physiology. 2017;469(1):15–26. doi:10.1007/s00424-016-1914-6. PMID: 27904960.
- Lechuga S, Ivanov AI. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim Biophys Acta. 2017;1864(7):1183–94. doi:10.1016/j.bbamcr.2017.03.007. PMID: 28322932.
- Lee SH. Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intestinal research. 2015;13(1):11–8. doi:10.5217/ir.2015.13.1.11. PMID: 25691839.
- Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H, Richter J, Bojarski C, Schumann M, Fromm M. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci. 2009;1165:294–300. doi:10.1111/j.1749-6632.2009.04062.x.
- Salim SY, Soderholm JD. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17(1):362–81. doi:10.1002/ibd.21403. PMID: 20725949.
- Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol. 2014;36:157–65. doi:10.1016/j.semcdb.2014.08.011.
- Mankertz J, Schulzke JD. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol. 2007;23(4):379–83. doi:10.1097/MOG.0b013e32816aa392. PMID: 17545772.
- Chang J, Leong RW, Wasinger VC, Ip M, Yang M, Phan TG. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients With Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology. 2017;153(3):723–31 e1. doi:10.1053/j.gastro.2017.05.056. PMID: 28601482.
- Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. 2013;4:280. doi:10.3389/fimmu.2013.00280.
- Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal immunology. 2017;10(2):307–17. doi:10.1038/mi.2016.128. PMID: 28120842.
- Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend Your Fences: The Epithelial Barrier and its Relationship With Mucosal Immunity in Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol. 2017;4(1):33–46. doi:10.1016/j.jcmgh.2017.03.007. PMID: 28560287.
- Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. 2007;56(1):61–72. doi:10.1136/gut.2006.094375. PMID: 16822808.
- Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159(6):2001–9. doi:10.1016/S0002-9440(10)63051-9. PMID: 11733350.
- Hering NA, Fromm M, Schulzke JD. Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J Physiol. 2012;590(5):1035–44. doi:10.1113/jphysiol.2011.224568. PMID: 22219336.
- Vetrano S, Rescigno M, Cera MR, Correale C, Rumio C, Doni A, Fantini M, Sturm A, Borroni E, Repici A, et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135(1):173–84. doi:10.1053/j.gastro.2008.04.002. PMID: 18514073.
- Boivin MA, Roy PK, Bradley A, Kennedy JC, Rihani T, Ma TY. Mechanism of interferon-gamma-induced increase in T84 intestinal epithelial tight junction. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2009;29(1):45–54. doi:10.1089/jir.2008.0128.
- Chapman CG, Pekow J. The emerging role of miRNAs in inflammatory bowel disease: a review. Ther Adv Gastroenterol. 2015;8(1):4–22. doi:10.1177/1756283X14547360. PMID: 25553076.
- Kuang Q, Purhonen P, Hebert H. Structure of potassium channels. Cell Mol Life Sci. 2015;72(19): 3677–93. doi:10.1007/s00018-015-1948-5. Epub 2015 Jun 13. PMID: 26070303.