
ABSTRACT
Chronic inflammatory skin disorders are frequently associated with impaired skin barrier function. Selective phosphodiesterase-4 (PDE4) inhibition constitutes an effective therapeutic strategy for the treatment of inflammatory skin diseases. We now report the pharmacological anti-inflammatory profile of DRM02, a novel pyrazolylbenzothiazole derivative with selective in vitro inhibitory activity toward PDE4 isoforms A, B and D. DRM02 treatment of cultured primary human and mouse epidermal keratinocytes interfered with pro-inflammatory cytokine production elicited by interleukin-1α and tumor necrosis factor-α. Similarly, DRM02 inhibited the production of pro-inflammatory cytokines by human peripheral blood mononuclear cells ex vivo and cultured THP-1 monocyte-like cells, with IC50 values of 0.6–14 µM. These anti-inflammatory properties of DRM02 were associated with dose-dependent repression of nuclear factor-κB (NF-κB) transcriptional activity. In skin inflammation in vivo mouse models, topically applied DRM02 inhibited the acute response to phorbol ester and induced Th2-type contact hypersensitivity reactivity. Further, DRM02 also decreased cutaneous clinical changes and expression of Th17 immune pathway cytokines in a mouse model of psoriasis evoked by repeated topical imiquimod application. Thus, the overall pharmacological profiling of the PDE4 inhibitor DRM02 has revealed its potential as a topical therapy for inflammatory skin disorders and restoration of skin homeostasis.
Introduction
The skin plays indispensable roles as an immune and physical barrier, and is a first line of defense against pathogens and environmental challenges. The etiology of a number of common dermatological diseases includes prominent inflammatory and immune components. Citation1 Indeed, inflammatory skin diseases are major health issues, as they constitute the most common problem in dermatology. Citation2 Although acute inflammation contributes to the homeostatic protective and regenerative properties of the skin, inflammatory memory is retained in epidermal stem cells and may contribute to recurrent skin inflammation and development of maladaptive responses. Citation3
Therapies for chronic inflammatory cutaneous diseases, such as atopic dermatitis and psoriasis, often include the use of corticosteroids or calcineurin inhibitors. Topical corticosteroids are highly effective, but adverse consequences, including skin atrophy, systemic absorption, and suppression of the hypothalamic-pituitary-adrenal axis, limit their long-term use. Citation4 Topical calcineurin inhibitors interfere with T-lymphocyte and dendritic cell activation, and have been used as a second-line therapy for short-term or intermittent treatment. Limitations of calcineurin inhibitors include contraindication in immunocompromised individuals, and the relatively common occurrence of burning or stinging with application, which negatively affects patient compliance. Citation4 Over the past two decades, phosphodiesterases (PDE) have also emerged as druggable targets in the treatment of inflammatory skin disorders. Citation5
Within the superfamily of PDE enzymes, the type-4 (PDE4) family is a major player in the regulation of cellular pro-inflammatory responses. PDE4 exerts key roles in inflammatory disorders, and is relatively abundant in myeloid and lymphoid cells. Citation6,Citation7 Several PDE4 isoforms are also expressed in epidermal keratinocytes. Citation8 PDE4 catalyzes the hydrolysis of cyclic adenosine monophosphate (cAMP), thereby inactivating this second messenger. Citation6 Inhibition of PDE4 increases cAMP levels, which, in turn, interferes with pro-inflammatory processes in epidermal cells in vitro, and in preclinical in vivo models of cutaneous inflammatory disorders. Citation9–Citation11 Several PDE4 inhibitors, including apremilast and cilomilast, are now clinically used to decrease the expression of key pro-inflammatory cytokines, including tumor necrosis factor (TNF) α, interleukin (IL)-6, IL-17 and IL-23. Citation7,Citation9,Citation11,Citation12 However, there remains an ongoing need for the introduction of effective novel anti-inflammatory drugs, as some clinically approved PDE4 inhibitors can also exert dose-limiting systemic toxicities. Citation13
In the present study, we describe the pharmacological effects of DRM02, a novel and selective PDE4 inhibitor. DRM02 interferes with pro-inflammatory cytokine production in response to a wide variety of stimuli both in epidermal and in immune cells. Furthermore, we also demonstrate the effectiveness of DRM02 in several mouse models of skin inflammation elicited by a variety of agents.
Results
Activity profiling of DRM02
DRM02 is a pyrazolylbenzothiazole derivative (), which we developed as a potential topical anti-inflammatory reagent. To identify the principal targets of DRM02 modulation, this compound was first profiled against >600 proteins in cell-free activity assay panels containing various human cellular enzymes, including PDE subtypes, kinases, phosphatases, and receptor kinases. In these assays, DRM02 decreased U937 cell-derived PDE4 activity with a half-maximal inhibitory concentration (IC50) of 0.4 μM (). DRM02 also decreased kinase activity present in immunoprecipitates containing exogenously expressed human integrin-linked kinase (ILK) isolated from Hi-5 insect cells, with a mean IC50 of 20.6 nM (). DRM02 had no significant activity at concentrations lower than 1.0 μM against any of the other proteins evaluated.
Figure 1. Pharmacological activity of DRM02 in vitro.
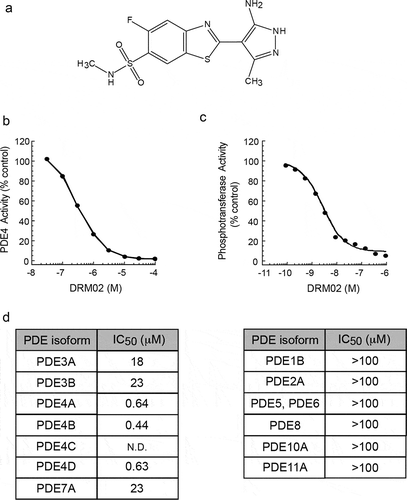
To further characterize the modulatory activity of DRM02 toward phosphodiesterases, we next interrogated a panel of purified and recombinant human PDE enzymes. Using similar in vitro assays, we found that DRM02 effectively inhibited PDE4A, PDE4B and PDE4D isoforms with IC50 values of 0.64 μM, 0.44 μM and 0.63 μM, respectively (). Notably, DRM02 was substantially less potent against PDE3A, PDE3B and PDE7A, and did not significantly affect the activity of PDE1B, PDE2A, PDE5, PDE6, PDE8A, PDE10A or PDE11A (). Collectively, these results identify DRM02 as a selective inhibitor of the PDE4 isoforms A, B and D.
DRM02 is a potent inhibitor of pro-inflammatory cytokine expression in epidermal keratinocytes
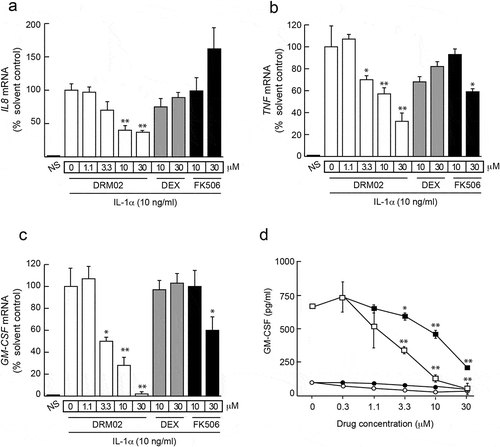
In the epidermis, keratinocytes are both key cellular targets for and producers of pro-inflammatory mediators, contributing to the activation of self-sustaining inflammatory loops. Citation14 We examined the effect of DRM02 on inflammatory cytokine expression in primary cultured human epidermal keratinocytes stimulated with IL-1α, which induces expression of multiple pro-inflammatory factors in these cells. Four hours after IL-1α addition to the culture medium, we observed increases in cellular IL8, TNF and GM-CSF mRNA levels of approximately 10-, 25- and 18-fold, respectively (). The presence of DRM02 attenuated the stimulatory effects of IL-1α on IL8, TNF and GM-CSF transcript levels in a dose-dependent fashion, with IC50 values in the 3–20 μM range for IL8 and GM-CSF, and 10–30 μM for TNF. The glucocorticoid dexamethasone and the calcineurin inhibitor FK506, two potent anti-inflammatory drugs, had lesser or no effects on the levels of these mRNA species (). IL8, TNF and GM-CSF mRNA levels remained 10- to 20-fold higher after 24 h of IL-1α treatment, relative to those in cultures without IL-1α stimulation, and significant reductions in those levels persisted in the presence of DRM02 (data not shown). In the culture medium of non-stimulated keratinocytes, low granulocyte-macrophage colony-stimulating factor (GM-CSF) concentrations were detected (~ 100 pg/ml), but these levels increased substantially by stimulation with IL-1α. Significantly, DRM02 was a more potent inhibitor of IL-1α-induced increases in GM-CSF in culture medium (IC50 ~ 3 μM) than FK506 (IC50 ~ 20 μM, ). DRM02 also effectively dampened increases in GM-CSF secretion into culture medium by keratinocytes stimulated with TNFα, epidermal growth factor (EGF), transforming growth factor-α (TGFα), or with IL-1α/EGF, IL-1α/TGFα, TNFα/EGF or TNFα/TGFα combinations (Supplemental Figure 1).
Figure 2. DRM02 inhibition of inflammatory cytokine expression in human epidermal keratinocytes.
ILK is dispensable for the anti-inflammatory effects of DRM02
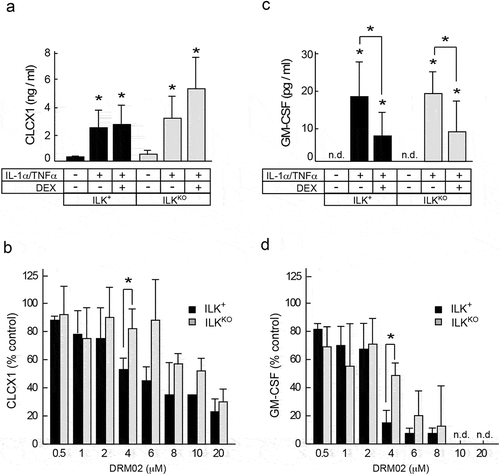
We next determined if ILK is involved in the observed anti-inflammatory properties of DRM02 in keratinocytes. To this end, we compared the responses to IL-1α/TNFα of primary keratinocytes isolated from mice with epidermis-specific inactivation of the Ilk gene (hereafter termed ILKKO) and ILK-expressing littermates (hereafter termed ILK+). Citation15 Stimulation with IL-1α and TNFα increased the levels of secreted CXCL1, the mouse orthologue of human IL-8, by ~23- and ~8-fold, respectively, in ILK+ and ILKKO cells, and these increases were insensitive to the presence of dexamethasone (). In contrast, DRM02 impaired stimulation of CXCL1 in a dose-dependent manner in all cultures tested, irrespective of whether or not the cells expressed ILK (). IL-1α/TNFα treatment also increased levels of secreted GM-CSF in both cell types, and these increases were sensitive to inhibition by dexamethasone and by DRM02 (). No reduction in viability was observed in either cell type with any of these treatments (data not shown). These data indicate that ILK is not necessary for keratinocyte inflammatory responses to IL-1α/TNFα or for the pharmacological action of DRM02 in this model.
Figure 3. ILK-independent inhibition of pro-inflammatory cytokine expression by DRM02.
DRM02 inhibits expression of inflammation-regulating factors in THP-1 and in peripheral blood mononuclear cells (PBMC)
Antigen-presenting cells, including monocytes and macrophages, can enter the skin from the peripheral circulation, playing key roles in initiating and modulating cutaneous inflammation. Citation16 Hence, we further characterized the effects of DRM02 on responses that model inflammatory conditions, using THP-1 monocyte-like cells and human PBMC from healthy donors.
THP-1 cells pre-treated with vehicle or with increasing concentrations of DRM02 were then stimulated with lipopolysaccharide (LPS) and interferon (IFN)-γ for 48 h, to increase cytokine production that reflects pro-inflammatory M1 macrophage polarization in vitro. Citation17 Under these conditions, DRM02 decreased levels of secreted TNFα, IL-1β, IL-6 and IL-8 in a dose-dependent manner, with IC50 values in the 1.4–9.0 μM range (). DRM02 concentrations as high as 30 μM were without effect on THP-1 cell viability or proliferation ( and data not shown).
Table 1. DRM02 inhibits proliferation and cytokine expression of stimulated PBMC and THP-1 cells. PBMC were stimulated with either phytohemagglutinin (PHA; 1 μg/ml) or the combination of LPS (10 ng/ml) plus IFNγ (33 ng/ml). THP-1 monocyte-like cells were treated with LPS (10 ng/ml) plus IFNγ (100 ng/ml). Conditioned cell culture media were collected after 48 hours, and were analyzed to determine cytokine levels using bead arrays and flow cytometry. DRM02 IC50 determinations are expressed as the mean ± SD for each test parameter. The number or experiments is indicated in parentheses. ND, not determined.
To complement these studies using primary cells, we next examined the effect of DRM02 on pro-inflammatory responses of normal human PBMC ex vivo, stimulated with either an LPS/IFN-γ combination or the mitogen phytohemagglutinin. Both LPS/IFNγ and phytohemagglutinin promoted cell proliferation, which was sensitive to inhibition by DRM02, as evidenced by the ability of this drug to reduce [3 H] thymidine ([3 H]dThd) incorporation into DNA, with IC50 values of 2.90 ± 1.4 μM and 2.2 ± 1.2 μM, respectively (). In addition to functioning as potent mitogens in PBMC, these agents also induce secretion of pro-inflammatory cytokines. Citation18 DRM02 decreased LPS/IFNγ-induced secretion of TNFα, IL-1β, IL-6, IL-8 and IL-12p70 (mean IC50 0.6 μM–14.5 μM; ). Similarly, stimulation of PBMC with phytohemagglutinin resulted in DRM02-sensitive secretion of TNFα, IFNγ and IL-10, with IC50 values for DRM02 inhibition in the low micromolar range (). Thus, DRM02 efficiently lowered pro-inflammatory signatures of activated PBMC and THP-1 cells.
Inhibition of NF-κB activity by DRM02
NF-κB is a central component of pro-inflammatory signaling pathways activated by cytokines such as TNFα and IL-1. Citation19 Cellular NF-κB activation by TNFα and IL-1 occurs through a canonical pathway that involves activation of IκB kinase (IKK) and IκB dissociation from NF-κB. Free NF-κB dimers then translocate from the cytoplasm to the nucleus to activate transcription of pro-inflammatory and other target genes (reviewed in Citation20 ). Interactions between the cAMP and NF-κB pathways have been reported for many cell types, including keratinocytes, monocytes, THP-1 and HEK293 cells. Citation20 To determine the effects of DRM02 on NF-κB transcriptional activity, we used a HEK293-derived line that stably maintains a chromosomal integration of a luciferase reporter construct regulated by multiple copies of the NF-κB response element, hereafter termed 293-Luc cells. Treatment with TNFα increased luciferase activity by approximately 100-fold, relative to unstimulated cells (Supplemental Figure 2). The presence of DRM02 interfered with this induction in a dose-dependent manner, with IC50 and IC90 values of ~7 μM and ~30 μM, respectively, without measurable effects on cell viability (Supplemental Figure 2). For comparison, treatment with the well-established NF-κB inhibitor quinazoline Citation21 at the highest test concentration that did not induce cytotoxicity (10 μM) decreased TNFα-induced, NF-κB-dependent luciferase output by ~43% (Supplemental Figure 2).
Inhibition of intercellular adhesion molecule-1 (ICAM-1) expression by DRM02
In the skin, certain pro-inflammatory stimuli induce expression of adhesion molecules, which are important contributors to the entry and persistence of immune cells in affected sites. ICAM-1 is a cell surface glycoprotein present in epithelial, endothelial and immune cells. In epidermal keratinocytes, ICAM-1 is an adhesion molecule that serves as a ligand for the leukocyte adhesion protein LFA-1, facilitating their interactions with infiltrating leukocytes. Citation22 Significantly, ICAM-1 expression can be up-regulated through the canonical NF-κB signaling pathway. Citation20 Because DRM02 interfered with NF-κB transcriptional activity, and given that changes in ICAM-1 expression substantially influence the course of inflammatory and immune responses in the skin, Citation22 we next examined the effects of DRM02 on inflammation-induced ICAM-1 expression in human epidermal HaCaT keratinocytes.
Stimulation of HaCaT cells with TNFα (10 ng/ml), IFNγ (2.5 ng/ml) or both cytokines increased cell surface levels of ICAM-1 by approximately 20-, 10- and >100-fold above those in non-stimulated cells, respectively (). The presence of DRM02 attenuated in a dose-dependent manner the increases in ICAM-1 surface levels induced by TNFα alone or in combination with IFNγ, achieving a 75% reduction at a concentration of 20 μM (,c). Under these conditions, the effect of DRM02 was distinct from that of dexamethasone, which did not alter increases in surface ICAM-1 levels induced by TNFα or TNFα/IFNγ combinations (,c). Similarly, DRM02 induced concentration-dependent decreases in surface ICAM-1 levels in HaCaT cells treated with IL-1β/TNFα or IL-1β/IFNγ combinations, although it was relatively ineffective against surface ICAM-1 increases produced by treatment with IFNγ alone (Supplemental Figure 3).
Figure 4. DRM02 inhibition of ICAM-1 expression.
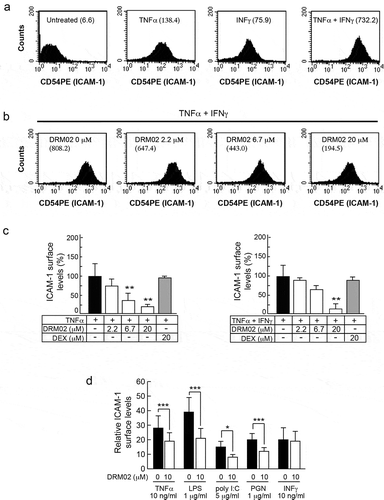
Under inflammatory conditions, monocyte expression of ICAM-1 and other adhesion molecules is also activated. Citation23,Citation24 Thus, we next explored the ability of DRM02 to modulate ICAM-1 expression in stimulated THP-1 cells. To this end, we incubated the cells with TNFα, LPS, polyinosinic:polycytidylic acid (poly I:C), or peptidoglycan (PGN) and observed 14- to 39-fold increases in surface ICAM-1 levels, relative to non-stimulated cells (). DRM02 (10 μM) suppressed these increases in ICAM-1 levels by 35%-45%. Similar to HaCaT cells, DRM02 was without effect on enhanced surface ICAM-1 levels induced by IFNγ (). Collectively, these data indicate that DRM02 effectively interferes with ICAM-1 induction by a broad range of pro-inflammatory stimuli, both in epithelial and in immune cell types.
DRM02 inhibition of LPS-induced inflammation in vivo
Activated macrophages release pro-inflammatory mediators through activation of NF-κB and other pathways. Citation25 Aberrant macrophage responses contribute to various inflammatory disorders, and, therefore, their pharmacological modulation has untapped therapeutic potential. Macrophages activated by bacterial LPS exhibit multiple responses, including increased secretion of inflammatory mediators and expression of adhesion molecules representing a characteristic “inflammation signature”. Citation26 Given the range of anti-inflammatory properties of DRM02 observed in cultured cells, we next investigated whether DRM02 might exert protective effects on systemic inflammatory responses in vivo. To this end, we administered LPS intravenously (i.v. 1 mg/kg body weight) to mice pretreated with control vehicle or with DRM02, and determined serum concentrations of various cytokines and other factors at timed intervals thereafter.
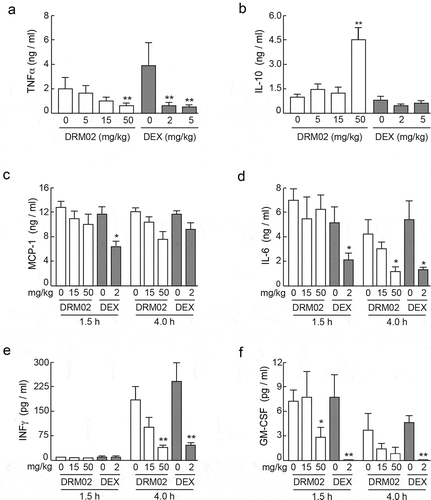
TNFα, IFNγ, GM-CSF, macrophage chemo-attractant protein-1 (MCP-1), IL-6 and IL-10 were undetectable in serum samples prepared from naive mice or from animals given PBS. In contrast, TNFα, IL-6 and GM-CSF were readily detected in serum as early as 1.5 h post-LPS injection (). DRM02 pretreatment (50 mg/kg body weight) significantly mitigated LPS-mediated increases in serum levels of these three cytokines by 60%-80% (). Increases in IFNγ levels were observed 4 h following LPS administration, and they were similarly reduced by DRM02 (). Notably, the magnitude of DRM02 reduction in cytokine serum levels was comparable to that observed in mice treated with dexamethasone, albeit a lower dose of dexamethasone was used due to the combination of dexamethasone solubility and maximum volume restrictions for s.c. administration in mice (). However, unlike dexamethasone, DRM02 also increased serum levels of the anti-inflammatory cytokine IL-10 within the time frame of these experiments (). Neither DRM02 nor dexamethasone modulated LPS-mediated increases in MCP-1 serum levels (). These studies indicate that that DRM02 effectively attenuates systemic expression of a variety of pro-inflammatory factors following exposure to intravenous LPS.
Figure 5. Systemic administration of DRM02 modulates serum levels of inflammatory mediators in LPS-challenged mice.
Topical DRM02 inhibits acute irritant dermatitis and T-helper type 2 (Th2) contact hypersensitivity in mice
Skin inflammation can be induced in mouse models by a single topical dose of phorbol myristate acetate (PMA). This acute inflammatory reaction, termed irritant contact dermatitis (ICD), is characterized by epidermal erythema, edema, and inflammatory cell infiltration. Citation1 To explore the anti-inflammatory activity of DRM02 in the skin, we next assessed its effects on ear edema associated with PMA-induced ICD in mice. Auricles of mice were treated with PMA, followed 3 h later by DRM02 or matched vehicle, and ear thickness was measured 24 h after the initial PMA treatment. The average increase in auricular thickness caused by PMA with no additional treatment was 0.6 mm (). In four independent experiments using groups of 6 PMA-treated mice each, topical application of a 20 mM DRM02 solution reduced ear swelling by 55.3 ± 10.1%. In comparison, auricular thickness in animals treated with dexamethasone (6.6 mM) was reduced by 38.7 ± 7.0% ().
Figure 6. Topical DRM02 inhibits PMA-induced skin inflammation and contact hypersensitivity responses to FITC.
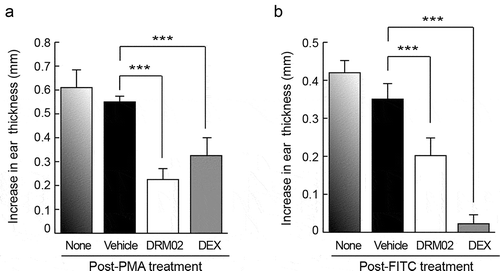
Contact hypersensitivity (CHS) is an inflammatory skin reaction mediated by T cells, which can be elicited in mice by cutaneous exposure to fluorescein isothiocyanate (FITC). Citation27 Similar to human allergic contact dermatitis, FITC-induced CHS consists of a sensitization and an elicitation phase, characterized by local infiltration of Th2 helper type lymphocytes. Citation27 To investigate the activity of DRM02 in FITC-induced CHS, we first sensitized mice by exposure to topical FITC on abdominal skin four times over a period of two weeks. The animals were then challenged by topically applying FITC on the ears, followed 2 h later by DRM02 treatment. We measured ear thickness 24 h after FITC challenge, and determined that auricular thickness on average had increased by about 0.42 mm, relative to the mean pre-treatment thickness of 0.25 mm. FITC-induced changes in ear thickness were 42.6% ± 6.6% lower upon treatment with DRM02 (). For comparison, we also treated FITC-challenged mice with dexamethasone, and observed a 90.3 ± 3.8% reduction in ear swelling (). Collectively, these observations indicate that DRM02 interferes with both acute and hypersensitivity-associated pro-inflammatory processes in the skin.
DRM02 suppresses imiquimod-induced skin inflammation in mice
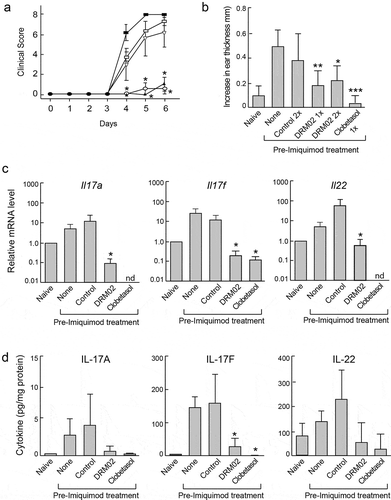
Repeated topical application of imiquimod to mouse skin induces psoriasiform dermatitis, characterized by TNFα-dependent development of inflamed scaly lesions driven by the IL-23/IL-17 immune axis, which resemble human plaque-type psoriasis. Citation28–Citation30 To examine the ability of DRM02 to modulate psoriasiform inflammation, we topically administered 5% imiquimod thrice to mouse dorsal skin, at 24- to 26-h intervals. The animals were also pretreated with control vehicle, DRM02 or the corticosteroid clobetasol 2 h prior to each imiquimod administration. Clobetasol was included in these experiments as a clinically used, reference anti-psoriasis treatment for comparison with DRM02. In these studies, we first investigated if DRM02 activity could affect clinical scores in imiquimod-induced psoriasiform lesions on mouse dorsal skin. To this end, we employed an established scoring system to assess skin inflammation, based on the clinically used Psoriasis Area and Severity Index (PASI). Citation31 Skin plaques (scored 0–4) and erythema (scored 0–4) were evaluated independently, with higher scores indicating increased severity. DRM02 gel applied twice daily for 7 days led to significantly lower imiquimod-induced psoriasiform scores (), without observable deleterious systemic effects, as evidenced by unaltered body, draining lymph node, and spleen weights (Supplemental Figure 4). Clobetasol similarly prevented the clinical skin changes produced by imiquimod. However, clobetasol treatment was associated with significant systemic outcomes, including a 6% decrease in body weight, as well as significant reductions in relative lymph node and spleen weights (Supplemental Figure 4). As an additional test of the effects of DRM02 on inflammation, we measured changes in ear thickness associated with exposure to imiquimod. Topical application of DRM02 either once or twice daily onto ears just prior to imiquimod treatment significantly reduced ear swelling to a similar extent to that observed with clobetasol (). We also harvested tissues from treated animals, to assess changes in inflammatory gene expression 6 h after the last imiquimod dose. We analyzed a panel of cytokines and chemokines associated with inflammation and/or psoriasis, and observed significant increases in mRNA and protein levels of the pro-psoriatic cytokines IL-17a, IL-17 f and IL-22 in imiquimod-treated skin relative to those in skin of naive mice. These imiquimod-induced increases were mitigated by treatment with DRM02 or clobetasol (,d; Supplemental Tables S1 and S2). Together, these observations indicate that DRM02 significantly improves psoriasiform clinical scores in the imiquimod mouse model, likely through mechanisms that involve interference with production of pro-inflammatory mediators.
Figure 7. DRM02 ameliorates imiquimod-induced inflammation.
Discussion
In these studies, we have demonstrated a wide range of anti-inflammatory properties exhibited by DRM02, a novel and selective phosphodiesterase PDE4 inhibitor. During in vitro profiling, we observed that DRM02 also reduced phosphotransferase activity present in ILK immunoprecipitates. The role of ILK in inflammation appears to be complex. For example, ILK is necessary for neutrophil trafficking to sites of inflammation and activation of epithelial inflammatory signaling in experimental colitis mouse models. Citation32 ILK is also important for leukocyte chemotaxis and immune cell trafficking, as ILK-deficient thymic T lymphocytes exhibit reduced chemotaxis to the chemokines CXCL12 and CCL19. Citation33 In contrast, cytokine production and proliferation of B-lymphocytes following LPS challenge does not require ILK. Citation34 Similarly, our studies found that ILK is neither required in epidermal keratinocytes for responses to IL-1α and TNFα, nor for the anti-inflammatory effects of DRM02 in these cells, suggesting that DRM02 may exert its anti-inflammatory effects predominantly through PDE4 modulation.
Cyclic AMP plays pivotal roles in all the cell types that contribute to the pathophysiology of inflammatory diseases in the skin and other organs. Citation35 For example, leukocytes from individuals with atopic dermatitis exhibit increased phosphodiesterase activity and cAMP degradation, resulting in activation of pro-inflammatory cascades. Citation36 Thus, the pharmacological modulation of phosphodiesterases has been identified as an important therapeutic strategy in the management of mild-to-moderate skin inflammatory conditions.
In humans, over one hundred forms of cyclic nucleotide phosphodiesterases have been identified. Amongst individual phosphodiesterase families, the C-terminal catalytic domains are conserved and determine substrate recognition and binding selectivity for inhibitors. Citation37 Within the PDE4 family, the Q2 domain contains a unique pocket that can accommodate a water molecule, further contributing to the ability of specific small molecules to selectively interact with a given PDE4 isoform(s). Citation36 Whether DRM02 binds PDE4 isoforms through this water-containing pocket remains an important area for future research, which may also aid in the development of additional analogues with improved potency and activity.
PDE4 inhibitors increase intracellular cAMP levels, leading to impaired transcription of pro-inflammatory NF-κB target genes, through mechanisms that involve protein kinase A activation, up-regulation of cAMP responsive element binding protein (CREB) and CRE-driven gene transcription. Citation12,Citation38 Our experiments using 293Luc reporter cells showed that DRM02 decreased NF-κB transcriptional activity in cells stimulated with TNFα. PDE4 inhibitors exhibit cell type- and stimulus-specific effects on NF-κB pathways. For example, apremilast reportedly interferes with NF-κB transcriptional activity in THP-1 and Jurkat cells without altering the ability of this transcription factor to translocate to the nucleus. Citation12 In contrast, PDE4 inhibition by rolipram in human leukemia cells activated by TLR7/8 and TLR9- agonists decreased IκBα phosphorylation and NF-κB p65 nuclear translocation. Citation39 Our studies showed that DRM02 exhibits anti-inflammatory activity in multiple cultured cell types exposed to a variety of stimuli without detectable cytotoxicity. DRM02 interfered with activation of keratinocyte and immune cell responses to IL-1 and TNFα, two key pro-inflammatory cytokines, and also reduced immune cell responses to TLR ligands.
Inflammatory cues stimulate keratinocytes to express ICAM-1, which promotes leukocyte recruitment to the skin, thereby playing a pivotal role in promoting and amplifying inflammatory responses. Citation40 DRM02 decreased up-regulation of ICAM-1 induced by TNFα or TNFα/IFNγ combinations in human HaCaT keratinocytes and THP-1 monocyte-like cells. DRM02 also inhibited ICAM-1 expression in response to TLR2, TLR3 and TLR4 stimulation in THP-1 cells. In contrast, DRM02 had no detectable effect on ICAM-1 upregulation in response to IFNγ alone. Whereas TNFα and TLR ligands exert many of their biological effects through NF-κB activation, Citation41 IFNγ preferentially stimulates signaling through JAK/STAT pathways. These observations further underline the possibility that the anti-inflammatory activity of DRM02 may be predominantly associated with NF-κB modulation. An important area for future research will be to determine the mechanisms involved in DRM02 regulation of NF-κB activity in epidermal keratinocytes and other cell types associated with skin inflammation.
DRM02 attenuated IL-1α-induced increases in GM-CSF expression in both mouse and human keratinocytes. GM-CSF is highly expressed in skin regions affected by atopic dermatitis Citation42 or psoriasis. Citation43 Importantly, GM-CSF may foster eosinophil, monocyte-macrophage and Langerhans cell viability and activity in chronic inflammatory skin lesions, thus contributing to persistence of disease. Citation44 The prominent inhibitory effects of DRM02 on the expression of GM-CSF and other inflammatory mediators in epidermal keratinocytes may be a key mechanism involved in the observed efficacy of DRM02 against inflammatory skin diseases.
In vivo, the TLR4 ligand LPS elicits responses that reflect activation of the innate immune system, such as release into the bloodstream of a wide variety of cytokines and chemokines, mainly produced by monocytes. Citation26 Significantly, DRM02 mitigated LPS-induced increases in TNFα, GM-CSF and IFN-γ serum levels, and increased those of the anti-inflammatory cytokine IL-10. These effects are consistent with the known mode of action of other PDE4 inhibitors, which induce PKA activation via cAMP, leading to IL-10 expression and inhibition of NF-κB, Citation9 further suggesting that DRM02 likely reduces inflammatory responses in vivo through pathways that involve PDE4 inhibition. In these experiments, there were no discernible adverse side effects of DRM02 administration. However, although DRM02 has shown marked selectivity for PDE4 enzymes in pharmacological profiling assays, detailed pharmacodynamic characterization using a range of DRM02 doses in mice will be an important area for future studies, to rule out possible off-target effects under the conditions of our experiments.
Topical DRM02 treatment in mice reduced the acute, edematous reaction to PMA, the antigen-specific Th2 type immune contact hypersensitivity response to FITC, and the development of Th17 immune pathway-related psoriasiform lesions elicited by the TLR7/8 agonist IMQ. DRM02 also reduced mRNA and protein levels of IL-17A, IL-17 F, and IL-22 in the skin. These cytokines, released by T lymphocytes and natural killer cells, trigger innate immunity responses in epithelial cell types, but with distinct response patterns. IL-17 induces inflammatory-type responses, whereas IL-22 predominantly influences epithelial cell differentiation. Citation45,Citation46 Importantly, DRM02 exerted anti-inflammatory effects in a variety of mouse models of inflammation, without detectable negative systemic effects. In contrast, we found that adverse systemic effects accompanied clobetasol treatment in IMQ-induced psoriasiform inflammation, including weight loss and immune-system organ size alterations.
Chronic inflammatory skin disorders, such as psoriasis and atopic dermatitis, are associated with impaired skin barrier function. For example, imiquimod-induced psoriasis in mouse models is characterized by increased transepidermal water loss, indicative of impairment in the barrier function of the skin. Citation47 In this and other preclinical models, interference with inflammatory pathway activation alleviates dermatitis symptoms and skin barrier dysfunction, Citation47 and a key question to address in the future is whether the anti-inflammatory properties of DRM02 contribute to the restoration of skin barrier capacity. Overall, however, our studies have shown that DRM02 is a novel anti-inflammatory drug that exhibits activity in a wide array of in vitro and in vivo models of skin inflammation elicited by distinct insults and stimuli. Importantly, the anti-inflammatory activity that DRM02 exhibited in these models was comparable or superior to that observed with commonly used therapeutic corticosteroids, such as dexamethasone and clobetasol.
Chronic inflammatory skin diseases, such as psoriasis and atopic dermatitis, affect many individuals. Major therapies for these disorders consist of topical administration of costicosteroids, vitamin D analogues, retinoids, and calcineurin inhibitors. Citation48 However, these therapies are not equally effective for all patients, and can be associated with severe undesired side effects. For example, chronic topical corticosteroid use produces skin atrophy, delayed wound healing, and increased susceptibility to infections. Citation49,Citation50 Further, systemic corticosteroid distribution following topical administration can result in hypothalamic-pituitary axis suppression. Citation49,Citation50 These limitations have spurred the development of novel therapies, such as those that selectively target PDE4.
PDE4 inhibition reduces cellular cAMP hydrolysis, which in turn interferes with synthesis and release of pro-inflammatory cytokines, including TNFα, IFNγ and various interleukins. Citation51 Clinically used oral PDE4 inhibitors, such as apremilast, are very effective, but are still associated with gastrointestinal and neurological adverse effects. Citation48,Citation52,Citation53 Several small molecule PDE4 inhibitors for topical use are currently at various stages of development. For example, CHF6001 is a potent PDE4 inhibitor administered by inhalation, and used for inflammatory pulmonary diseases. Citation54 Preclinical studies have shown that CHF6001 interferes with NF-kB nuclear translocation and reduces oxidative stress in cultured epidermal keratinocytes, although its cutaneous anti-inflammatory effects in vivo have yet to be reported. Citation53 E6005, another novel PDE4 inhibitor developed for topical use, suppressed pro-inflammatory cytokine production in human lymphocytes in vitro, exhibited anti-inflammatory effects in mouse models of atopic dermatitis, and showed acceptable tolerability in randomized controlled trials. Citation55 Crisaborole is a potent boron-based PDE4 and TNFα release inhibitor in PBMC (IC50 = 0.49–5.0 μM). Citation56,Citation57 Topically administered 2% crisaborole ointment is well tolerated and has been approved for the treatment of mild or moderate atopic dermatitis. Citation56,Citation57 Important features of crisaborole in these trials were its low systemic exposure and mild treatment-related adverse events that spontaneously resolved in 16%-57% of patients. Citation58,Citation59 Further, crisaborole reduced expression of inflammatory mediators and improved skin barrier function in atopic dermatitis patients, Citation60 indicating that PDE4 inhibition may be an effective approach to restore the epidermal barrier, at least under some circumstances.
Our studies demonstrate that topical DRM02 is well tolerated and exhibits effective anti-inflammatory activity in a variety of cutaneous inflammatory mouse disease models. An exploratory, double-blinded, randomized, within-subject control Phase-II clinical study (NCT01993420) evaluated 0.25% DRM02 topical gel given twice daily for six weeks to 21 adult individuals with atopic dermatitis. DRM02 treatment was well tolerated locally, and without any discernible systemic effects. Both DRM02 and the control gel produced marked reductive effects on target lesion scores, potentially masking any DRM02-invoked pharmacological activity. Given that the preclinical pharmacological profile of DRM02 is consistent with pronounced selectivity for PDE4, the development of DRM02 formulations optimized for topical delivery in humans may thus constitute an effective alternative therapeutic approach to the use of corticosteroids and other current treatments for inflammatory skin conditions.
Materials and methods
Ethics approvals
All studies using human PBMC were approved by the ethics committee of QLT Inc. Experiments using human PBMC were conducted with blood drawn from healthy adult human male and female volunteers under written informed consent, and in accordance with the Helsinki Declaration of 1975. All animal experiments received ethics approvals in accordance with guidelines and regulations of the Canadian Council on Animal Care. Experiments using ILK-expressing and ILK-deficient primary keratinocytes were approved by the University of Western Ontario Animal Care Committee (Protocol No. 2015–21). Study protocols for imiquimod-induced inflammation in vivo studies were reviewed and approved for compliance with the rules and regulations of the MD BioSciences Committee for Ethical Conduct in the Care and Use of Laboratory Animals. All other inflammation animal model studies were approved by the Animal Care Committee of QLT, Inc., Vancouver, Canada, as follows: LPS systemic inflammation studies, Protocol No. ACC05-013; PMA inflammation studies, Protocol No. ACC05-002; FITC-contact hypersensitivity studies, Protocol No. ACC06-011.
In vitro DRM02 activity profiling
DRM02 (10 μM) biochemical profiling assays were conducted under contract, using The Safety Profile panel (Eurofins Cerep, Poitiers, France) against 76 enzyme and 112 receptor binding targets. Further, the activity of 355 different kinases assessed DRM02 at 10 μM using the KINOMEscan™ (Eurofins DiscoveRx Corporation, San Diego, CA). The compound (1, 10 μM) was also tested against selected 57 active kinase and three phosphatase targets by SignalChem Lifesciences Corporation (Richmond, Canada). Following initial determination, DRM02 was re-assayed against U-937 cell-derived PDE4 (Eurofins Cerep) to obtain definitive IC50 values. DRM02 was subsequently tested against available representative isoforms of different human PDE families, using recombinant human PDE proteins, except for PDE5 and PDE6 which were prepared from human and bovine tissues, respectively (Eurofins Cerep).
ILK biochemical assays and selectivity profiling studies were conducted with recombinant ILK prepared as a glutathione-s-transferase (GST)-fusion protein expressed in Hi-5 insect cells using a baculovirus expression system, Citation61 and are detailed in the Supplemental Information section.
Human cell inflammatory response studies
For all cell culture experiments, DRM02, FK506 and dexamethasone were solubilized in 100% tissue culture-grade dimethyl sulfoxide (DMSO).
To obtain PBMC, blood was drawn from healthy adult human male and female volunteers under informed consent on the day of experiments. PBMC were isolated using Ficoll-Hypaque Plus (Cat. No. 17–1440-02, GE Healthcare, Little Chalfont, UK) density centrifugation and re-suspended in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin/streptomycin, 1% GlutaMAX, 20 mM HEPES, 1 mM sodium pyruvate and 0.05 mM 2-mercaptoethanol (2-ME). PBMC (1.5 x 106 cells/ml) were added to 96-well flat-bottom plates (0.1 ml/well). To stimulate monocyte cytokine production, PBMC were treated with LPS (10 ng/ml) plus IFNγ (33 ng/ml). To activate T lymphocytes, PBMC were incubated with phytohemagglutinin (1 μg/ml). Test compounds were solubilized in DMSO. Culture wells contained 0.15% DMSO, using an assay volume of 0.2 ml per well. Triplicate determinations were conducted for each test concentration. Incubations were carried out at 37°C for 48 h under 5% CO2.
THP-1 monocytes (American Type Culture Collection) were maintained in RPMI medium supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, 1% GlutaMAX, 20 mM HEPES buffer and 0.05 mM 2-ME. For experiments to measure cytokine production, THP-1 cells (1 x 105 cells/ml) were seeded into 96-well flat-bottom plates (0.2 ml per well). To stimulate cytokine production, cells were treated with E. coli 0111:B-4 LPS (10 ng/ml) plus IFNγ (100 ng/ml) for 48 h. To assess effects on ICAM-1 expression, THP-1 cells (1 x 105 cells/ml) were seeded into 12-well plates (1 ml/well) and incubated with DRM02 (10 μM) for 1 h. TNFα (10 ng/ml), IFNγ (10 ng/ml) or Toll-like receptor (TLR) agonists PGN (TLR2, 1 μg/ml), poly I: C (TLR3, 5 μg/ml) or LPS (TLR4, 1 μg/ml) were then added, with a final DMSO concentration of 0.25% in 1 ml. After 48 h of culture, cells were washed twice with PBS and stained with phycoerythrin (PE)-conjugated mouse anti-human ICAM-1 (CD54) monoclonal antibody (HA58 antibody from eBiosciences; ThermoFisher Cat. No. 12–0549-42). A FACScan flow cytometer (BD Biosciences) was used to acquire signals from 10,000–15,000 cells per sample, analyzed using CellQuest software, and mean channel fluorescence intensity (MCFI) values were obtained. To account for inter-experiment variability, THP-1 ICAM-1 levels were expressed as a ratio of the MCFI value obtained for stimulated cells, relative to the MCFI result determined for non-stimulated cells.
Normal human epidermal keratinocytes (NHEK) (Cat. No. 00192907, Lonza, Basel, Switzerland) were cultured in serum-free medium (KGM) prepared using KC basal medium (KBM) supplemented with KGM SingleQuots and 2 µM hydrocortisone. The medium was changed every 2–3 days and cells were subcultured when 80–90% confluence was attained. For experiments, NHEK at passage 2–4 were seeded at 8 × 103 cells/ml into 96-well microtiter plates in 0.2 ml per well. The final culture concentration for DMSO was 0.15%. Cells were maintained without hydrocortisone 24 h prior to and throughout the experimental phase.
HaCaT cells, Citation62 kindly provided by Dr. N.E. Fusenig (German Cancer Research Center), were maintained in DMEM supplemented with 10% FBS, 1 mM sodium pyruvate and 20 mM HEPES buffer. Cells were passaged when 80–95% confluence was reached, and plated into 12-well plates at 5 × 105 cells/well for experiments. For surface ICAM-1 expression studies, test compounds were added 1 h before stimulation with TNFα (10 ng/ml) or TNFα plus IFNγ (2.5 ng/ml) with a final DMSO concentration of 0.5%. The effects of DRM02 on surface expression of ICAM-1 induced by IFNγ (2.5 ng/ml), IFNγ plus IL-1β (100 ng/ml), as well as IL-1β plus TNFα, were also evaluated. After 48 h of treatment, cells were washed twice with PBS, detached using trypsin, stained with HA58 PE-conjugated mouse anti-human ICAM-1 antibody and analyzed by flow cytometry. For each experimental condition, at least 5,000–10,000 cells were analyzed. HaCaT cell surface ICAM-1 levels were expressed as a percentage of the result for drug-treated cytokine-stimulated cells versus the MCFI determination obtained for vehicle-treated cytokine-stimulated cells, evaluated in parallel.
Phorbol-induced dermatitis skin inflammation studies
Methods employed for testing against systemic LPS-induced endotoxemia and skin inflammation triggered by phorbol ester were as described. Citation63 While under isoflurane anesthesia, baseline ear thickness measurements were taken from female BALB/c mice (Charles River Canada, St. Constant, Canada) of approximately 8 weeks of age, using a micrometer (Mitutoyo Corporation, Mississauga, Canada). Ten μL of a 0.02% PMA solution in 100% acetone were applied onto each side of the test ear (4 μg PMA/ear). Three hours later, animals received 10 μL of the test agent diluted in 3% DMSO/97% acetone on each side of the ear. Ear thickness was re-measured 24 h after PMA application. Technical staff blinded to the identity of the test articles applied conducted all treatments and measurements.
Contact hypersensitivity (CHS) skin inflammation studies
A mouse Th2-type FITC-CHS response model was employed, Citation27,Citation64 with minor modifications. Female BALB/c mice of approximately 8 weeks of age were used. The abdominal region of each mouse was shaved and 400 μL of 0.5% fluorescein isothiocyanate (FITC) dissolved in acetone:dibutyl phthalate (1:1 v/v, hereafter termed FITC solution) were applied onto the abdomen on Day 0 and Day 1. FITC sensitization was repeated on Days 14 and 15, using 400 μL of FITC solution. Prior to FITC challenge (Day 20), mice were anesthetized using isoflurane, baseline ear thickness measurements were obtained and 10 μL of FITC solution maintained at 4°C were applied to each side of one ear. Two hours later, each animal received 5 μL of the control vehicle (15% DMSO in acetone) or test agent (dissolved in the same mixture of DMSO and acetone) on each side of the same ear. Ear thickness was re-measured 24 h following FITC challenge. All treatments and measurements were conducted by personnel blinded to the identity of test substances applied.
Imiquimod-induced skin inflammation model
Development of psoriasis-like inflammation driven by the IL-23/IL-17 immune axis is produced in mouse skin with repeat topical application of the TLR 7/8 agonist imiquimod. Citation29,Citation30 For these experiments, conducted by MD Biosciences (Zurich, Switzerland) under contract, the backs of 9–10 weeks old female BALB/c mice were shaved and 5% imiquimod cream (62.5 mg; Aldara Cream, Meda, Dubai, UAE) was applied to an area of approximately 10 cm2, once daily for three days. The anti-inflammatory treatments consisted in ~0.3 ml of placebo gel, 0.25% DRM02 gel, or clobetasol propionate (0.05% Dermovate cream; GlaxoSmithKline, Uxbridge, UK). Clobetasol served as a comparative anti-inflammatory treatment, and was selected on the basis that this potent corticosteroid is an approved and clinically effective topical agent to treat psoriasis. Topical clobetasol effectively suppresses disease activity in the imiquimod psoriasis-like mouse model. Citation65 For biomarker studies, imiquimod cream was applied at t = 2, 26, and 50 h, whereas control gel, 0.25% DRM02 gel or clobetasol cream (0.3 ml per 10 cm2 surface area) were given 2 h prior to each imiquimod application. Animals were euthanized 6 h after the final imiquimod dose. One section from treated skin was placed in RNAlater for mRNA analyses. qPCR was conducted using commercial primer pairs (ThermoFisher Scientific) with skin mRNA levels expressed by normalization of sample values to the qPCR result obtained in each sample for the large sub-unit of ribosomal protein P0 (Rplpo) housekeeping gene. A 2.5-fold or greater increase in the relative level of a given mRNA species in imiquimod-treated versus naïve skin was considered significant. A second skin section was snap-frozen in liquid nitrogen for subsequent protein biomarker analysis using a Multiplex system (Luminex Corporation, Austin, TX, USA) or by ELISA (IL-22, Cat. No. LS-F26459, eBioscience Reagents, ThermoFisher Scientific). Biomarker levels were normalized to skin extract protein concentration as determined with Pierce Micro BCA protein assays (Cat. No. 23235, ThermoFisher Scientific).
To evaluate treatment effects on clinical symptoms, 62.5 mg of imiquimod cream was applied once daily starting on Day 0 onto pre-shaved back skin. To induce ear edema, 10 μL of imiquimod cream was applied on to the right ear daily. Right ear thickness measurements were conducted on Day 0 before the first imiquimod application and on Day 6, using a digital caliper. The control gel was given twice daily, 0.25% DRM02 gel applied either once or twice daily and clobetasol cream once daily at a volume of 300 μL for the dorsal skin site and 10 μL for the ear. For mice receiving test items twice daily, these treatments were approximately 6 h apart with the first application given 2 h prior to and the second 4 h after each imiquimod dose. Body weights, as well as skin clinical plaque (0–7) and erythema (0–4) scores were recorded daily, using an established scale (MD BioSciences, Zurich, Switzerland). On Day 6, animals were euthanized by CO2 overdose 6 h after the final imiquimod application. Spleen and inguinal lymph node weights of individual mice were recorded.
Additional procedures
The Supplemental Information section describes the following: suppliers of growth factors, cell stimulants, other inhibitors and chemicals; methods for isolation and cytokine stimulation of primary mouse epidermal keratinocytes; assays to determine human cell viability and proliferation; cultured human cell cytokine/chemokine quantification; NF-κB reporter assays; systemic LPS endotoxemia mouse model studies. All mandatory laboratory health and safety procedures were followed in the course of the experiments reported herein.
Author contributions
Conceived and designed the experiments: DH, IAI, LD
Conducted some experiments: IAI
Analyzed the data: DH, IAI, LD
Wrote the paper: DH, LD
Disclosure of interest
During the course of some of this work, DH was an employee of Dermira, Inc. IAI and LD report no conflict of interest.
Supplemental Material
Download PDF (1.4 MB)Acknowledgments
The authors thank Dr. Hans Hofland for numerous helpful discussions. We gratefully acknowledge Dr. Jamie Harden for her thorough review and many helpful suggestions on the manuscript. The integral contribution of the QLT preclinical pharmacology group is also recognized. This work was funded by Dermira, Inc. and QLT, Inc.
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Petersen TK. In vivo pharmacological disease models for psoriasis and atopic dermatitis in drug discovery. Basic Clin Pharmacol Toxicol. 2006;99:1–20. doi:10.1111/j.1742-7843.2006.pto_298.x.
- Dainichi T , Hanakawa S , Kabashima K . Classification of inflammatory skin diseases: a proposal based on the disorders of the three-layered defense systems, barrier, innate immunity and acquired immunity. J Dermatol Sci. 2014;76:81–89. doi:10.1016/j.jdermsci.2014.08.010.
- Naik S , Larsen SB , Gomez NC , Alaverdyan K , Sendoel A , Yuan S , Polak L , Kulukian A , Chai S , Fuchs E , et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature. 2017;550(7677):475–480. doi:10.1038/nature24271.
- Eichenfield LF , Tom WL , Berger TG , Krol A , Paller AS , Schwarzenberger K , Bergman JN , Chamlin SL , Cohen DE , Cooper KD , et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116–132. doi:10.1016/j.jaad.2014.03.023.
- Zebda R , Paller AS . Phosphodiesterase 4 inhibitors. J Am Acad Dermatol. 2018;78(3):S43–s52. doi:10.1016/j.jaad.2017.11.056.
- Jin SL , Lan L , Zoudilova M , Conti M . Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J Immunol. 2005;175(3):1523–1531. doi:10.4049/jimmunol.175.3.1523.
- Schafer PH , Parton A , Gandhi AK , Capone L , Adams M , Wu L , Bartlett JB , Loveland MA , Gilhar A , Cheung Y-F , et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159(4):842–855. doi:10.1111/j.1476-5381.2009.00559.x.
- Chujor CS , Hammerschmid F , Lam C . Cyclic nucleotide phosphodiesterase 4 subtypes are differentially expressed by primary keratinocytes and human epidermoid cell lines. J Invest Dermatol. 1998;110(3):287–291. doi:10.1046/j.1523-1747.1998.00114.x.
- Schafer P . Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem Pharmacol. 2012;83(12):1583–1590. doi:10.1016/j.bcp.2012.01.001.
- Nazarian R , Weinberg JM . AN-2728, a PDE4 inhibitor for the potential topical treatment of psoriasis and atopic dermatitis. Curr Opin Invest Drugs. 2009;10:1236–1242.
- Sadrai Z , Stevenson W , Okanobo A , Chen Y , Dohlman TH , Hua J , Amparo F , Chauhan SK , Dana R . PDE4 inhibition suppresses IL-17-associated immunity in dry eye disease. Invest Ophthalmol Vis Sci. 2012;53:3584–3591. doi:10.1167/iovs.11-9110.
- Schafer PH , Parton A , Capone L , Cedzik D , Brady H , Evans JF , Man H-W , Muller GW , Stirling DI , Chopra R , et al. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal. 2014;26:2016–2029. doi:10.1016/j.cellsig.2014.05.014.
- Gooderham M , Papp K . Selective phosphodiesterase inhibitors for psoriasis: focus on apremilast. BioDrugs. 2015;29:327–339. doi:10.1007/s40259-015-0144-3.
- Girolomoni G , Pastore S . The role of keratinocytes in the pathogenesis of atopic dermatitis. J Am Acad Dermatol. 2001;45:S25–8. doi:10.1067/mjd.2001.117021.
- Nakrieko KA , Welch I , Dupuis H , Bryce D , Pajak A , St Arnaud R , Dedhar S , D’Souza SJA , Dagnino L . Impaired hair follicle morphogenesis and polarized keratinocyte movement upon conditional inactivation of integrin-linked kinase in the epidermis. Mol Biol Cell. 2008;19:1462–1473. doi:10.1091/mbc.e07-06-0526.
- Kashem SW , Haniffa M , Kaplan DH . Antigen-Presenting Cells in the Skin. Annu Rev Immunol. 2017;35:469–499. doi:10.1146/annurev-immunol-051116-052215.
- Tedesco S , De Majo F , Kim J , Trenti A , Trevisi L , Fadini GP , Bolego C , Zandstra PW , Cignarella A , Vitiello L , et al. Convenience versus biological significance: are PMA-differentiated THP-1 cells a reliable substitute for blood-derived macrophages when studying in Vitro polarization? Front Pharmacol. 2018;9:71. doi:10.3389/fphar.2018.00071.
- Frank DA , Mahajan S , Ritz J . Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling. Nat Med. 1999;5:444–447. doi:10.1038/7445.
- Lawrence T . The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi:10.1101/cshperspect.a001651.
- Gerlo S , Kooijman R , Beck IM , Kolmus K , Spooren A , Haegeman G . Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol Life Sci. 2011;68:3823–3841. doi:10.1007/s00018-011-0757-8.
- Tobe M , Isobe Y , Tomizawa H , Nagasaki T , Takahashi H , Fukazawa T , Hayashi H . Discovery of quinazolines as a novel structural class of potent inhibitors of NF-kappa B activation. Bioorg Med Chem. 2003;11(3):383–391. doi:10.1016/S0968-0896(02)00440-6.
- Krutmann J , Grewe M . Involvement of cytokines, DNA damage, and reactive oxygen intermediates in ultraviolet radiation-induced modulation of intercellular adhesion molecule-1 expression. J Invest Dermatol. 1995;105(1):67s–70s. doi:10.1038/jid.1995.14.
- Dang LH , Michalek MT , Takei F , Benaceraff B , Rock KL . Role of ICAM-1 in antigen presentation demonstrated by ICAM-1 defective mutants. J Immunol. 1990;144:4082–4091.
- Trotta A , Velasquez LN , Milillo MA , Delpino MV , Rodriguez AM , Landoni VI , Giambartolomei GH , Pozner RG , Barrionuevo P . Platelets promote brucella abortus monocyte invasion by establishing complexes with monocytes. Front Immunol. 2018;9:1000. doi:10.3389/fimmu.2018.01000.
- Itharat A , Hiransai P . Dioscoreanone suppresses LPS-induced nitric oxide production and inflammatory cytokine expression in RAW 264.7 macrophages by NF-kappaB and ERK1/2 signaling transduction. J Cell Biochem. 2012;113(11):3427–3435. doi:10.1002/jcb.24219.
- Sweet MJ , Hume DA . Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60(1):8–26. doi:10.1002/jlb.60.1.8.
- Takeshita K , Yamasaki T , Akira S , Gantner F , Bacon KB . Essential role of MHC II-independent CD4+ T cells, IL-4 and STAT6 in contact hypersensitivity induced by fluorescein isothiocyanate in the mouse. Int Immunol. 2004;16(5):685–695. doi:10.1093/intimm/dxh073.
- Kabashima K , Nomura T . Revisiting murine models for atopic dermatitis and psoriasis with multipolar cytokine axes. Curr Opin Immunol. 2017;48:99–107. doi:10.1016/j.coi.2017.08.010.
- van der Fits L , Mourits S , Voerman JS , Kant M , Boon L , Laman JD , Cornelissen F , Mus A-M , Florencia E , Prens EP , et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182(9):5836–5845. doi:10.4049/jimmunol.0802999.
- Van Belle AB , de Heusch M , Lemaire MM , Hendrickx E , Warnier G , Dunussi-Joannopoulos K , Fouser LA , Renauld J-C , Dumoutier L . IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J Immunol. 2012;188(1):462–469. doi:10.4049/jimmunol.1102224.
- Carlin CS , Feldman SR , Krueger JG , Menter A , Krueger GG . A 50% reduction in the psoriasis area and severity index (PASI 50) is a clinically significant endpoint in the assessment of psoriasis. J Am Acad Dermatol. 2004;50(6):859–866. doi:10.1016/j.jaad.2003.09.014.
- Ahmed AU , Yim HCH , Alorro M , Ernst M , Williams BRG . Integrin-linked kinase expression in myeloid cells promotes inflammatory signaling during experimental colitis. J Immunol. 2017;199(6):2128–2139. doi:10.4049/jimmunol.1700125.
- Liu E , Sinha S , Williams C , Cyrille M , Heller E , Snapper SB , Georgopoulos K , St-Arnaud R , Force T , Dedhar S , et al. Targeted deletion of integrin-linked kinase reveals a role in T-cell chemotaxis and survival. Mol Cell Biol. 2005;25(24):11145–11155. doi:10.1128/MCB.25.24.11145-11155.2005.
- Van Belle K , Herman J , Waer M , Sprangers B , Louat T . OSU-T315 as an interesting lead molecule for novel B cell-specific therapeutics. J Immunol Res. 2018;2018:2505818. doi:10.1155/2018/2505818.
- Baumer W , Hoppmann J , Rundfeldt C , Kietzmann M . Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets. 2007;6(1):17–26. doi:10.2174/187152807780077318.
- Weidinger S , Beck LA , Bieber T , Kabashima K , Irvine AD . Atopic dermatitis. Nat Rev Dis Primers. 2018;4(1):1. doi:10.1038/s41572-018-0001-z.
- Feng X , Wang H , Ye M , Xu XT , Xu Y , Yang W , Zhang H-T , Song G , Ke H . Identification of a PDE4-specific pocket for design of selective inhibitors. Biochemistry. 2018;57(30):4518–4525. doi:10.1021/acs.biochem.8b00336.
- Parry GC , Mackman N . Role of cyclic AMP response element-binding protein in cyclic AMP inhibition of NF-kappaB-mediated transcription. J Immunol. 1997;159:5450–5456.
- Tan Y , Watkins AA , Freeman BB , Meyers JA , Rifkin IR , Lerner A . Inhibition of type 4 cyclic nucleotide phosphodiesterase blocks intracellular TLR signaling in chronic lymphocytic leukemia and normal hematopoietic cells. J Immunol. 2015;194(1):101–112. doi:10.4049/jimmunol.1401854.
- Youn GS , Kwon DJ , Ju SM , Choi SY , Park J . Curcumin ameliorates TNF-alpha-induced ICAM-1 expression and subsequent THP-1 adhesiveness via the induction of heme oxygenase-1 in the HaCaT cells. BMB Rep. 2013;46(8):410–415. doi:10.5483/BMBRep.2013.46.8.014.
- Kawai T , Akira S . TLR signaling. Cell Death Differ. 2006;13(5):816–825. doi:10.1038/sj.cdd.4401850.
- Pastore S , Fanales-Belasio E , Albanesi C , Chinni LM , Giannetti A , Girolomoni G . Granulocyte macrophage colony-stimulating factor is overproduced by keratinocytes in atopic dermatitis. Implications for sustained dendritic cell activation in the skin. J Clin Invest. 1997;99(12):3009–3017. doi:10.1172/JCI119496.
- Mascia F , Cataisson C , Lee TC , Threadgill D , Mariani V , Amerio P , Chandrasekhara C , Souto Adeva G , Girolomoni G , Yuspa SH , et al. EGFR regulates the expression of keratinocyte-derived granulocyte/macrophage colony-stimulating factor in vitro and in vivo. J Invest Dermatol. 2010;130(3):682–693. doi:10.1038/jid.2009.336.
- Werfel T . The role of leukocytes, keratinocytes, and allergen-specific IgE in the development of atopic dermatitis. J Invest Dermatol. 2009;129(8):1878–1891. doi:10.1038/jid.2009.71.
- Nograles KE , Zaba LC , Guttman-Yassky E , Fuentes-Duculan J , Suarez-Farinas M , Cardinale I , Khatcherian A , Gonzalez J , Pierson KC , White TR , et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159(5):1092–1102. doi:10.1111/j.1365-2133.2008.08769.x.
- Eyerich S , Eyerich K , Cavani A , Schmidt-Weber C . IL-17 and IL-22: siblings, not twins. Trends Immunol. 2010;31(9):354–361. doi:10.1016/j.it.2010.06.004.
- Takahashi T , Koga Y , Kainoh M . Anti-IL-12/IL-23p40 antibody ameliorates dermatitis and skin barrier dysfunction in mice with imiquimod-induced psoriasis-like dermatitis. Eur J Pharmacol. 2018;828:26–30. doi:10.1016/j.ejphar.2018.03.018.
- Psomadakis CE , Han G . New and emerging topical therapies for psoriasis and atopic dermatitis. J Clin Aesthet Dermatol. 2019;12:28–34.
- Carr WW . Topical calcineurin inhibitors for atopic dermatitis: review and treatment recommendations. Paediatr Drugs. 2013;15(4):303–310. doi:10.1007/s40272-013-0013-9.
- Hengge UR , Ruzicka T , Schwartz RA , Cork MJ . Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol. 2006;54(1):1–15. doi:10.1016/j.jaad.2005.01.010. quiz 6-8
- Dong C , Virtucio C , Zemska O , Baltazar G , Zhou Y , Baia D , Jones-Iatauro S , Sexton H , Martin S , Dee J , et al. Treatment of skin inflammation with benzoxaborole phosphodiesterase inhibitors: selectivity, cellular activity, and effect on cytokines associated with skin inflammation and skin architecture changes. J Pharmacol Exp Ther. 2016;358(3):413–422. doi:10.1124/jpet.116.232819.
- Pariser DM , Kircik LH , Stein Gold LF . Treating psoriasis: patient assessment, treatment goals, and treatment options. Cutis. 2019;103:S4–s7.
- Woodby B , Sticozzi C , Pambianchi E , Villetti G , Civelli M , Valacchi G , Facchinetti F . The PDE4 inhibitor CHF6001 affects keratinocyte proliferation via cellular redox pathways. Arch Biochem Biophys. 2020;685:108355. doi:10.1016/j.abb.2020.108355.
- Moretto N , Caruso P , Bosco R , Marchini G , Pastore F , Armani E , Amari G , Rizzi A , Ghidini E , De Fanti R , et al. CHF6001 I: a novel highly potent and selective phosphodiesterase 4 inhibitor with robust anti-inflammatory activity and suitable for topical pulmonary administration. J Pharmacol Exp Ther. 2015;352(3):559–567. doi:10.1124/jpet.114.220541.
- Kitahara Y , Hojo S , Nomoto M , Onozuka D , Furue M , Hagihara A . Pharmacokinetic disposition of topical phosphodiesterase-4 inhibitor E6005 in patients with atopic dermatitis. J Dermatolog Treat. 2019;30(5):466–470. doi:10.1080/09546634.2018.1530439.
- Akama T , Baker SJ , Zhang YK , Hernandez V , Zhou H , Sanders V , Freund Y , Kimura R , Maples KR , Plattner JJ , et al. Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg Med Chem Lett. 2009;19(8):2129–2132. doi:10.1016/j.bmcl.2009.03.007.
- Eichenfield LF , Call RS , Forsha DW , Fowler J Jr. , Hebert AA , Spellman M , Stein Gold LF , Van Syoc M , Zane LT , Tschen E , et al. Long-term safety of crisaborole ointment 2% in children and adults with mild to moderate atopic dermatitis. J Am Acad Dermatol. 2017;77(4):641–9.e5. doi:10.1016/j.jaad.2017.06.010.
- Zane LT , Kircik L , Call R , Tschen E , Draelos ZD , Chanda S , Van Syoc M , Hebert AA . Crisaborole topical ointment, 2% in patients ages 2 to 17 years with atopic dermatitis: a phase 1b, open-label, maximal-use systemic exposure study. Pediatr Dermatol. 2016;33(4):380–387. doi:10.1111/pde.12872.
- Schlessinger J , Shepard JS , Gower R , Su JC , Lynde C , Cha A , Ports WC, Purohit V, Takiya L, Werth JL, et al. Safety, effectiveness, and pharmacokinetics of crisaborole in infants aged 3 to < 24 Months with mild-to-moderate atopic dermatitis: a phase IV open-label study (CrisADe CARE 1). Am J Clin Dermatol. 2020;21:275–284. doi:10.1007/s40257-020-00510-6.
- Bissonnette R , Pavel AB , Diaz A , Werth JL , Zang C , Vranic I , Purohit VS , Zielinski MA , Vlahos B , Estrada YD , et al. Crisaborole and atopic dermatitis skin biomarkers: an intrapatient randomized trial. J Allergy Clin Immunol. 2019;144(5):1274–1289. doi:10.1016/j.jaci.2019.06.047.
- Persad S , Attwell S , Gray V , Mawji N , Deng JT , Leung D , Yan J , Sanghera J , Walsh MP , Dedhar S , et al. Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase. Critical roles for kinase activity and amino acids arginine 211 and serine 343. J Biol Chem. 2001;276(29):27462–27469. doi:10.1074/jbc.M102940200.
- Boukamp P , Petrussevska RT , Breitkreutz D , Hornung J , Markham A , Fusenig NE . Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106(3):761–771. doi:10.1083/jcb.106.3.761.
- Alford JG , Stanley PL , Todderud G , Tramposch KM . Temporal infiltration of leukocyte subsets into mouse skin inflamed with phorbol ester. Agents Actions. 1992;37(3–4):260–267. doi:10.1007/BF02028118.
- Tang A , Judge TA , Nickoloff BJ , Turka LA . Suppression of murine allergic contact dermatitis by CTLA4Ig. Tolerance Induction of Th2 Responses Requires Additional Blockade of CD40-ligand. J Immunol. 1996;157:117–125.
- Sun J , Dou W , Zhao Y , Hu J . A comparison of the effects of topical treatment of calcipotriol, camptothecin, clobetasol and tazarotene on an imiquimod-induced psoriasis-like mouse model. Immunopharmacol Immunotoxicol. 2014;36(1):17–24. doi:10.3109/08923973.2013.862542.