
ABSTRACT
Sphingosine 1-phosphate (S1P) is a multifaceted lipid signaling molecule that activates five specific G protein-coupled S1P receptors. Despite the fact that S1P is known as one of the strongest barrier-enhancing molecules for two decades, no medical application is available yet. The reason for this lack of translation into clinical practice may be the complex regulatory network of S1P signaling, metabolism and transportation.
In this review, we will provide an overview about the physiology and the network of S1P signaling with the focus on endothelial barrier maintenance in inflammation. We briefly describe the physiological role of S1P and the underlying S1P signaling in barrier maintenance, outline differences of S1P signaling and metabolism in inflammatory diseases, discuss potential targets and compounds for medical intervention, and summarize our current knowledge regarding the role of S1P in the maintenance of specialized barriers like the blood-brain barrier and the placenta.
S1P in embryonic development and physiology
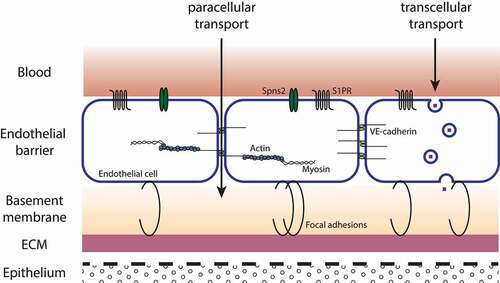
Blood vessels are lined by a layer of endothelial cells (ECs), which are responsible for the movement of fluid, proteins or circulating cells between the blood and the interstitium. The EC barrier is attached to the basement membrane, and via focal adhesions (FAs), ECs are tethered to the underlying extracellular matrix (ECM).Citation1, Citation2 Adjacent ECs are joined by interendothelial junctions, namely adherens junctions (AJs), tight junctions (TJs) and gap junctions (GJs). Next to the regulation of the paracellular passage of fluid and solutes into the interstitium, cell-cell junctions are also involved in cell-growth or cell-cell interactions. Alterations in expression of cell-cell junctions lead to changes in permeability of the EC barrier.Citation1,Citation2 Macromolecules greater than 3 nm in size are transported via the transcellular pathway. This includes mainly the transport by vesicles ().Citation1–3 Inflammation and inflammation associated diseases like sepsis are often marked by an increase of vascular permeability. Due to changes in interendothelial junctions, the EC barrier is disrupted and water and proteins infiltrate the extravascular compartment, causing damage to tissue and organs, and breakdown of the mean arterial pressure due to fluid leakage. Despite resuscitation and vasopression, one additional strategy for treatment includes the enhancement of the EC barrier to restrict the movement of fluid or proteins and maintain tissue homeostasis. The sphingolipid sphingosine 1-phosphate (S1P) has the ability to regulate the integrity of the vascular endotheliumCitation4 and is an interesting candidate to protect the function of the EC barrier.
Figure 1. Schematic picture of the vascular EC barrier. ECs line blood vessels and restrict the movement of water and solutes between the blood and interstitium. Attached to the basement membrane, ECs are tethered to the ECM by FAs. Interendothelial junctions connect neighboring ECs and regulate the paracellular passage of water and solutes. Macromolecules are transported via the transcellular pathway by the formation of vesicles.Citation1–3 ECM, extracellular matrix; FA, focal adhesion; S1PR, sphingosine 1-phosphate receptor; Spns2, spinster homolog 2
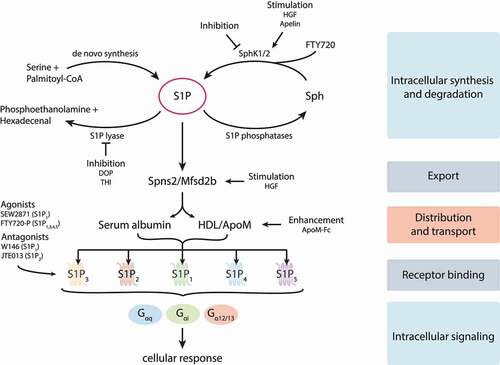
In its function as extra- and intracellular messenger S1P regulates processes relevant for the progression of inflammatory diseases like sepsis. S1P for instance is involved in the maintenance of the vasculature, the inflammatory response and the circulation of lymphocytes.Citation5–8 Produced intracellularly by sphingosine kinases 1 or 2 (SphK1/2),Citation9–12 S1P is either exported by spinster homolog 2 (Spns2)Citation13,Citation14 or major facilitator superfamily transporter 2b (Mfsd2b),Citation15 cleaved by the S1P degrading enzyme S1P lyase (SGPL1)Citation16 or dephosphorylated to sphingosine (Sph) by S1P phosphatases.Citation17 Outside the cells high concentrations of S1P are found in plasma, where it is bound to high-density lipoprotein (HDL) or albumin for transport to its specific G-protein coupled receptors S1P1-5Citation18 ().
Figure 2. Network of S1P metabolism, distribution and signaling. S1P is produced by sphingosine kinases 1 and 2 (SphK1/2) and irreversibly degraded by the S1P-lyase. The specific S1P transporters spinster homolog 2 (Spns2) and major facilitator superfamily transporter 2b (Mfsd2b) release S1P out of cells, where it is associated with its transporters serum albumin and apolipoprotein M (apoM) containing high-density lipoprotein (HDL). Five specific G protein-coupled S1P receptors (S1P1-5) can be activated by S1P and transduce signals inside cells via coupling to different trimeric G proteins. Synthesis, degradation, release, transportation and receptor signaling of S1P can be pharmacologically modified. Further details are provided in the text. HGF, hepatocyte growth factor; DOP, 4-deoxypyridoxine; THI, 2-acetyl-4-tetrahydroxybutyl imidazole; Sph, sphingosine; FTY720-P, FTY720-phosphate
The importance of S1P signaling for the integrity of the vascular system was first demonstrated in S1P1−/- mice, which showed a major defect in the formation of blood vessels during embryogenesis.Citation19 Vasculogenesis and angiogenesis are essential for blood vessel formation and maintenance. In vasculogenesis, new blood vessels are formed out of differentiated ECs. In angiogenesis, the new vascular system is stabilized by the differentiation of vascular smooth muscle cells (VSMCs) to cover new vessels.Citation20–23 S1P1−/- mouse embryos died between week 12.5 and 14.5 because of severe bleeding,Citation19 which occurred due to the incomplete coverage of the developed blood vessels by smooth muscle cells, leading to their instability and death of the embryos.Citation20 Besides S1P1, additional deletion of S1P2 and S1P3 in double and triple knock-out mice increased the severity of the vascular phenotype, indicating cooperative functions of S1P receptors in vascular development.Citation24
Mice lacking S1P in plasma (pS1Pless) displayed vascular leakages and a higher sensitivity to leak-inducing agents compared to wild-type mice. These effects were counteracted either by transfusion of erythrocytes to replenish missing S1P, or by the application of S1P1 agonists, which indicates the important role of S1P and its receptor S1P1 to maintain the vascular integrity.Citation2,Citation6 Important for the generation of S1P are the sphingosine kinases SphK1 and SphK2. Genetic deletion of one of the enzyme isoforms showed no obvious phenotype in mice, whereas the knock-out of both enzymes simultaneously led to embryonic lethality.Citation25 This suggests that both isoforms compensate for each other, and that S1P produced by them is needed for embryonic development. Deletion of both sphingosine kinases in hematopoietic cells and ECs led to increased vascular leakage and impaired survival after anaphylaxis.Citation6
2. Barrier maintenance via the S1P-S1PR signaling pathway
Of great interest are the mechanisms behind S1P promoted barrier protection. Under physiological conditions the barrier function is maintained by the balance of S1P receptors on ECs. ECs express predominantly S1P1 and S1P3,Citation26,Citation27 expression of S1P2 was also reported, and the expression of S1P5 is restricted to ECs of the blood-brain barrier.Citation28–30 All receptors are accessible for S1P and enable different signal transduction pathways.Citation31
Measurements of transendothelial electrical resistance (TEER) in human and bovine ECs showed the barrier enhancing properties of S1P. Treatment of ECs with up to 1 µM S1P led to increased EC barrier stability and even after barrier disruption by edemagenic agents, S1P was able to stabilize the endothelial monolayer.Citation4 Early studies identified S1P signaling via S1P1 and Gαi-protein as primary source of the regulation of EC barrier function, and an involvement of the family of Rho-GTPases in the S1P-S1PR mediated cytoskeletal rearrangement and barrier enhancement. The maintenance of barrier integrity via the S1P/S1P1 axis was conducted by protein kinase B (Akt) signaling. Due to the activation of Akt, activity of Rac and Cdc42 was forwarded, leading to the stimulation and formation of TJs in ECs including the redistribution of zona occludens-1 (ZO-1) and claudin-5 as well as the enrichment of platelet-endothelial cell adhesion molecule-1 (PECAM) and junctional adhesion molecule (JAM) at cell contact sides to promote barrier stability.Citation32,Citation33
Ras-related C3 botulinum toxin substrate 1 guanosine 5ʹ-triphosphatase (Rac-GTPase) was activated and in turn activated p21-associated Ser/Thr kinase (PAK-1) and the LIM kinase. After the phosphorylation of cofilin, actin severing was reduced, leading to lamellipodia formation and thickening of cortical actin.Citation4,Citation34 Furthermore, Singleton et al. identified the recruitment of S1P1 to caveolin-enriched microdomains (CEMs) as an essential step to maintain the EC barrier.Citation35 Activation of S1P1 by S1P in CEMs induced the formation of phosphatidylinositol-3,4,5-triphosphate (PIP3) by the PI3 Kinase, leading to the recruitment of T-lymphoma invasion and metastasis gene 1 (Tiam1) to the CEM. Tiam1 on the one hand modulated the translocation of α-actinin 1 and 4 and, on the other hand, catalyzed the formation of Rac1-GTP, which was an important factor in S1P-S1P1 mediated barrier enhancement and reduced the barrier permeability by cortical actin cytoskeletal rearrangement.Citation35 The enhancement of EC barriers by oxidized phospholipids like oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (OxPAPC) in CEMs was mediated via Src/Akt/S1P1 signaling. OxPAPC facilitated the activation of Src/Fyn, which in turn partially phosphorylated Akt, leading to the phosphorylation and activation of S1P1. S1P1 set of signaling cascades via the mammalian target of rapamycin (mTOR) and the phosphoinositide 3-kinase (PI3K) and promoted further phosphorylation of Akt and the cytoskeletal rearrangement and barrier enhancement by Rac1 activation.Citation36
An interesting feature of S1P in the maintenance of EC barrier was the remodeling of FAs. In comparison to thrombin, which caused intercellular gaps and promoted stress fiber formation, S1P showed barrier enhancing properties. Acting via the receptor S1P1 and the activation of the Rac-GTPase, S1P expedited the rearrangement of FAs by FA Kinase (FAK), paxillin and G protein-coupled receptor kinase-interacting proteins (GITs). Existing FAs were disassembled by S1P, and FAKs and paxillin translocated to the cell periphery, mediated by GITs. Finally, new FAs were formed and linked to the cortical actin ring, stabilizing and protecting the EC barrier.Citation4,Citation37,Citation38 Additionally, Rac was also able to promote the activation of Src, which resulted in the activation of FAK in the cytosol. On the one hand, this increased the association of FAK with GIT1 during FA assembly and led to the formation of new focal contacts by FAK, paxillin and GIT2. On the other hand, activated FAK, either by Rac or by Src, promoted the release of Arp-3 and the formation of lamellipodia.Citation37–39 Furthermore, Rac activated c-Abl, a cytoskeletal effector that forwarded the recruitment of the myosin light chain kinase (MLCK) and cortactin to form lamellipodia.Citation39,Citation40 Interestingly, Akt and Src were associated and maintained a mutual connection to regulate barrier integrity. Gao et al. showed that due to the long-term activation of Akt by the vascular endothelial growth factor (VEGF) or angiopoietin-1 (Ang-1), the activity of Src was modulated by Akt to stabilize TJs and the EC barrier. In opposition to the activation of Akt, its long-term inhibition led to uncontrolled activation of Src and the breakdown of EC barriers.Citation41 This was further confirmed by a recent study showing that the short-time activation of Src led to reorganization of AJs and transient accumulation of VE-cadherin and, thus, promoted barrier integrity.Citation42
The S1P-S1P1 axis was not only responsible for the rearrangement of actin in ECs, but also for the expression of surface molecules like PECAM-1 and vascular endothelial (VE)-cadherin, which were important for cell adhesion and cell-cell contacts. Long-term down-regulation of S1P1 led to diminished expression of PECAM-1 and VE-cadherin on ECs, influencing the cell-cell-interactions to strengthen EC barriers. Additionally, the expression of the adhesion molecule E-selectin, which was part of the immune response after the exposure to cytokines like tumor necrosis factor α (TNFα), was reduced in S1P1 knock-down cells.Citation43
S1P1 is the dominant receptor on ECs, and recent studies indicate that under pathophysiological conditions vascular permeability and inflammation were promoted by a shift of receptor expression toward S1P2. S1P2 and its counteractive relation to S1P1 are currently in focus to explain the mechanisms behind disruption of the EC barrier.Citation44
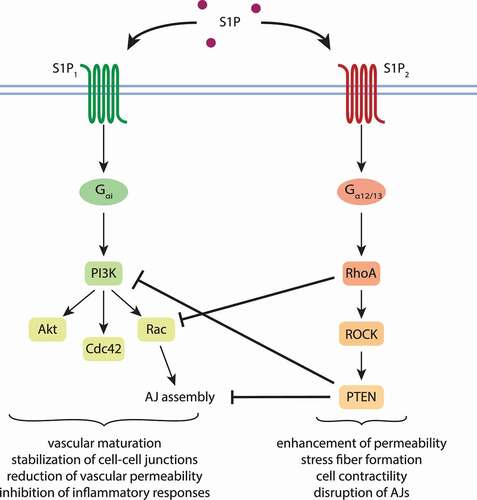
The barrier stabilizing and anti-inflammatory effects of S1P1 are mediated via the Gαi protein, whereas the antagonistic effects of S1P2 are regulated by Gα12/13. Key players in the S1P2 mediated signaling cascade interfere at several points with S1P1 regulated signaling. Sanchez et al. investigated the S1P2 downstream effectors Ras homolog gene family, member A (RhoA), Rho-associated protein kinase (ROCK) and phosphatase and tensin homolog (PTEN).Citation44 Rho and PTEN were able to disrupt AJs, promoted stress fiber formation and endothelial permeability. By inhibiting Rac1 signaling, RhoA prevented AJ assembly, which was further detained by the phosphorylation of VE-cadherin by PTEN ().Citation44 Under inflammatory conditions, inflammation was also promoted by S1P2 signaling. Due to the activation of ROCK/nuclear factor kappa B (NFкB) and stress-activated protein kinase (SAPK), pro-inflammatory responses were evoked.Citation45 The inhibition of S1P2 by specific antagonists was able to rescind the adverse effects. Additionally, the simultaneous stimulation of S1P1 by S1P acted like a feedback mechanism to attenuate the effects of S1P2 and promoted the maintenance of the EC barrier.Citation44,Citation45 This hypothesis was further confirmed by Reinhard et al.Citation46 Under healthy conditions, S1P bound to its receptors S1P1 and S1P2 and activated the distinct G proteins. Furthermore, S1P activated Rac1, followed by S1P2 mediated activation of RhoA. The latter induced local contractions in cells and suppressed Rac1. But in addition to Rac, S1P1- Gαi signaling also led to activation of cell division control protein 42 (Cdc42), which abrogated the effects of RhoA and stabilized cell junctions ().Citation46
Figure 3. Influence of S1P1 and S1P2 on endothelial barrier integrity. Signaling via S1P1 promotes the stability of the endothelial barrier, whereas signaling transduced by S1P2 enhances vascular permeability. Under physiological conditions, S1P binds equally well both receptors, of which S1P1 is broader expressed. Subsequently, distinct sets of G proteins are activated. Firstly, Rac is activated, secondly, RhoA, which suppresses Rac. Downstream effectors of RhoA further inhibit S1P1 signaling, leading to cell contraction and permeability. S1P1-Gαi signaling also leads to the activation of Cdc42, which reverses the effects of RhoA and promotes barrier stability.Citation44–46 Akt, protein kinase B; Cdc42, cell division control protein 42; Rac, Ras-related C3 botulinum toxin substrate 1; RhoA, Ras homolog gene family, member A; ROCK, Rho-associated protein kinase; PTEN, phosphatase and tensin homolog; S1P, sphingosine 1-phosphate
3. S1P signaling and metabolism in inflammatory diseases
Triggered by infection or tissue injury, inflammation is an adaptive response of the body to restore homeostasis.Citation47 A dysregulated host response, however, may cause inflammatory tissue damage, sepsis, organ dysfunction, and acute respiratory distress syndrome (ARDS). Even in the novel coronavirus disease 2019 (COVID-19), which is caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), 29% of infected patients with a severe course of the disease develop ARDS.Citation48 COVID-19 patients had decreased serum and plasma levels of S1P compared to healthy controls with no significant changes of S1P levels in red blood cells.Citation49–51 The SphK2 inhibitor Opaganib is currently tested in a clinical trial for the treatment of COVID-19 patients (NCT04467840, ), and the sphingosine analog and immune modulator FTY720 was planned for a clinical study in COVID-19 patients as well, but the trial was withdrawn (NCT04280588, ). A main cause of mortality is a hyperinflammatory response of the body leading to septic shock, which is additionally facilitated by the viral infection of ECs and the following dysfunction of the EC barrier.Citation52–55 Breakdown of the EC barrier is a common trait in inflammatory diseases. S1P signaling and metabolism could be a valuable target to treat inflammation and to promote barrier stability.
Table 1. Clinical status of current S1P-related drug candidates
3.1 Sepsis
Sepsis is characterized by systemic inflammation, which affects the whole organ system, often ultimately leading to organ failure and death. In prospective studies, septic patients or patients suffering from ARDS showed decreased concentrations of S1P in serum. The loss of S1P in serum correlated with the severity of the disease.Citation56,Citation57 Since disruption of the EC barrier and an uncontrollable inflammatory response are hallmarks of sepsis, the involvement of S1P metabolism and signaling in the progression of sepsis and other inflammatory diseases is likely. The Albumin Italian Outcome Sepsis Trial (ALBIOS) investigated the concentrations of S1P in plasma in correlation with the proteoglycan syndecan-1 (SYN-1), which covers ECs and stabilizes the EC barrier. The authors demonstrated an inverse relation between plasma S1P and SYN-1 and observed decreased levels of S1P in plasma of patients with septic shock with concomitantly elevated levels of SYN-1 and VE-cadherin. This correlation supported the hypothesis of an involvement of S1P in EC barrier stabilization since S1P inhibited the degradation of SYN-1 by metalloproteases.Citation58
Hemdan et al. examined the effects of pharmacologically modulated S1P signaling in a mouse model of sepsis.Citation59 First, administration of 4-deoxypyridoxine (DOP), an inhibitor of the S1P-degrading enzyme S1P-lyase, resulted in elevated levels of S1P. The second approach was the treatment with the sphingosine analog FTY720, which is phosphorylated by cells to the S1P bioequivalent FTY720-phosphate and able to activate all S1P receptors except S1P2. Mice were pre-treated with either DOP or FTY720, and after sepsis induction both compounds were able to reduce the disruption of the vascular barrier in lung and liver.Citation59 Furthermore, those mice showed declined levels of the barrier disruptive molecule VEGF-A in plasma, and the infiltration of immune cells in the liver was significantly reduced, proposing a beneficial effect of S1P modulating agents on the EC barrier by improving its stability, which ultimately led to improved survival.Citation59
Next to indirect modulation of S1P signaling via the inhibition of S1P-degradation, the application of agonists and/or antagonists provides the opportunity to target specific signaling pathways. The activation of S1P1 by its specific agonist SEW2871 and the simultaneous antagonization of S1P2 by JTE013 showed beneficial effects in a mouse model of sepsis-induced acute kidney injury (AKI).Citation60 Pre-treatment of mice with both compounds led to a dose-dependent reduction of microvascular permeability. To depict close-to-clinic conditions, the compounds were administered 6 h after sepsis induction. At that time of sepsis and AKI, vascular permeability was already increased. Interestingly, SEW2871 was able to diminish vascular permeability and to improve kidney function, showing preventive as well as curative effects, whereas JTE013 showed only preventive effects.Citation60 But the administration of SEW2871 to amplify signaling via S1P1 and therefore to promote barrier stability in sepsis is controversial. In a clinically relevant sepsis model, Flemming et al. treated mice 12 h after sepsis induction with SEW2871.Citation61 Septic mice, but not the healthy control group, suffered from severe cardiac side effects, leading to a higher lethality due to prolonged administration of SEW2871. Additionally, no beneficial effects on barrier stability were detected. Since SEW2871 showed barrier promoting effects at lower dosages and when applied 1 h after sepsis induction, the authors suspected that the effects of SEW2871 depended on the dosage and time point of application. Another possibility could also be functional antagonism of SEW2871 toward S1P1 after repeated administration, leading to the inhibition of S1P1 signaling.Citation61
3.2 ARDS/ALI
ARDS or acute lung injury (ALI) is often experienced by patients suffering from sepsis. ALI develops upon failure of the respiratory system, which is marked by a breakdown of vascular barriers and accumulation of fluid in the lungs.Citation62 As a consequence, patients with ALI are mechanically ventilated, causing in turn ventilator-induced lung injury (VILI) due to mechanical stress.Citation63–65 Mechanical ventilation (MV) stretches endothelial and epithelial cells, induces vascular leakage and triggers an inflammatory response.Citation64,Citation65
Based on the beneficial effects of S1P on EC barrier integrity in in vitro studies, Peng et al. conducted experiments in an in vivo model of ALI.Citation62 Mice suffering from lipopolysaccharide (LPS)-induced sub-lethal lung inflammation were treated intravenously with S1P one hour after LPS challenge. S1P’s barrier modulating properties became apparent in the reduction of vascular leakages, in the prevention of phagocyte infiltration in the lung and in the increase of neutrophils in bronchoalveolar lavage fluid (BALF). Furthermore, intraperitoneal administration of the sphingosine analogue FTY720 reduced systemic inflammation and displayed the same beneficial effects as S1P.Citation62 Besides rodents, the positive effects of S1P in inflammatory lung injuries were shown in a canine model of ALI. Here, intravenous S1P was able to maintain oxygenation of the lung and reduced lung edema formation. Additionally, it prevented an increase in lung tissue volume following accumulation of fluid.Citation63 Those studies showed the therapeutic potential of S1P and the sphingosine analogue FTY720 in in vivo models of ALI. But the way of administration also turned out to be important. Intratracheal administration was tested next to intravenous injection in a rodent model of ALI. S1P was either beneficial or harmful in ALI dependent on the dosage. Physiological concentrations of S1P and also of the S1P1 agonist SEW2871 (< 0.3 mg/kg), applied intravenously or intratracheally, were able to reduce the permeability of lung tissue and inflammation. Higher dosages (0.5 mg/kg) of S1P and SEW2871 turned out to be barrier disruptive when given intratracheally, and S1P induced pulmonary edema and led to increased protein levels. Two mg/kg S1P given intratracheally turned out to be lethal due to pulmonary edema.Citation66 Next to the administration of S1P to treat ALI and barrier disruption, Zhao et al. suggested the inhibition of SGPL1 as a therapeutic option. In LPS induced murine ALI, genetic deletion of SGPL1 (S1PL± mice) and the inhibition of SGPL1 by 2-acetyl-4-tetrahydroxybutyl imidazole (THI) led to increased intracellular S1P levels and attenuated inflammation in the lung. The elevated intracellular S1P levels protected the EC barrier from LPS-induced disruption, which was regulated by signaling via S1P1 and the activation of Rac1.Citation67
3.3. Diabetes and Obesity
Around 463 million people worldwide suffer from diabetes. 90% of those cases are afflicted with type 2 diabetes mellitus (T2D), which is caused mainly by obesity.Citation68,Citation69 Insulin, a hormone responsible for the maintenance of blood glucose levels, regulation of lipogenesis and protein synthesis, is secreted by β-cells of Langerhans islets in the pancreas. In T2D, the resistance of cells against insulin disturbed normal metabolism and led to endothelial dysfunction, amongst others.Citation69,Citation70 Even before the onset of diabetes, patients suffering from metabolic syndrome (MetS) showed signs of endothelial dysfunction, heightened activity of RhoA/ROCK and elevated levels of markers of oxidative stress and inflammation.Citation71,Citation72 In T2D, oxidative stress was responsible for the breakdown of vascular barriers, facilitated by mitochondrial collapse and an increased production of reactive oxygen species (ROS) as well as enhanced inflammatory stress response.Citation73,Citation74 The subsequent change in glucose metabolism was responsible for altered expression of TJ proteins like occludin, ZO-1 or VE-cadherin. Furthermore, it caused the expression of inflammatory markers like vascular cell adhesion molecule-1 (VCAM-1) and led to the breakdown of EC barriers in brain or retina.Citation74–76 The current research provides a mixed picture about the role of S1P in obesity and diabetes. The generation of extracellular S1P by SphK1 provided a protective role and promoted glucose tolerance, insulin secretion and the survival of pancreatic β-cells.Citation69,Citation77–80 SphK2 and SphK2 generated intracellular S1P possess a more complex role. On the one hand, SphK2 promoted insulin resistance, lipotoxicity and β-cell dysfunction and death via the activation of the mitochondrial apoptotic pathway.Citation69,Citation81,Citation82 On the other hand, SphK2 preserved insulin signaling in the liver via phosphorylation of Sph to S1P and consequently prevented the inhibition of the PI3K pathway by Sph.Citation83 Extracellular S1P occupies a dual role. Depending on its transport in plasma and its signaling pathway, S1P either promoted or prevented insulin resistance. A recent study showed that signaling via S1P1/3 diminished insulin resistance by the activation of the Akt pathway and the maintenance of mitochondrial functions. Furthermore, this effect was sustained by ApoM, which facilitated the transport of S1P to its receptors, and by FTY720, which promoted the Akt pathway by activating S1P1/3 signaling cascades.Citation84–87 However, S1P signaling via S1P2 impaired insulin signaling by inhibition of Akt phosphorylation, which was abrogated by the S1P2 antagonist JTE-013.Citation87 Given the prevalence of MetS and T2D, strategies to prevent and/or treat those diseases are needed. Despite the controversial role of S1P metabolism and signaling in obesity and diabetes, it may nonetheless be an interesting and important target to affect insulin signaling and to attenuate the adverse effects of obesity on EC barriers.
4. Potential targets and treatment strategies
4.1 Activation of S1P1 with FTY720 analogs
Knowing the influence of S1P1 on important functions of ECs, the S1P receptor agonist FTY720-phosphate might show unwanted side effects regarding the stability of cell barriers. FTY720-phosphate was beneficial under VEGF-induced conditions with enhanced vascular permeability,Citation88 and several studies evaluated the immunosuppressive effects of FTY720 due to the down-regulation of S1P1 expression and signaling in lymphocytes, which proved beneficial for the treatment of autoimmune diseases like multiple sclerosis.Citation89,Citation90
In the central nervous system, FTY720 and its active form FTY720-phosphate displayed beneficial effects. Pre-treatment of brain microvascular ECs (BMECs) with FTY720-phosphate increased the expression of claudin-5 and led to enhanced TEER values. Furthermore, FTY720-phosphate was able to diminish the VCAM-1 expression and resultant barrier disruption caused by sera from MS patients, which pointed to a positive modification of the BBB by FTY720-phosphate to prevent the passage of lymphocytes.Citation91 Also, the blood-nerve barrier (BNB) was positively affected by FTY720, which was shown after the pre-treatment of human peripheral nerve microvascular ECs (HPnMECs) with FTY720-phosphate. FTY720-phosphate decreased the barrier permeability in a dose-dependent manner. Barrier disruption was prevented by up-regulating claudin-5 expression, which also led to increased TEER values.Citation92 In vivo, the effects of FTY720 on the BBB were investigated in 20 patients with MS. It reduced the passage of immunocompetent and autoreactive cells across the BBB by enhancing the functional activity of the BBB, which was demonstrated by decreased levels of circulating TJ proteins and reduced expression of S1P1.Citation93 A recent study by Wang et al. indicated that the barrier enhancing effects of FTY720 were not caused by an alteration in the expression of AJ or TJ proteins, but rather by the activation of the nonreceptor tyrosine kinase c-Abl and the subsequent change in cytoskeletal structure.Citation94
In ECs, therapeutic concentrations of phosphorylated FTY720 compete with S1P for the binding to S1P1. FTY720 is phosphorylated to FTY720-phosphate by SphK2. Short-term exposure to FTY720 disrupted the assembly of cortical actin and the migration of ECs. Long time exposure to FTY720 led to a reduced expression of S1P1 and inhibited angiogenesis and wound healing.Citation95 Those detrimental effects of long-term exposure to FTY720 were also observed in a mouse model of bleomycin induced lung injury. The sustained activation of S1P1 not only by FTY720-phosphate, but also by other agonists like AUY954, SEW2871 and even exogenously added S1P inhibited signaling via S1P1 and led to vascular leakage and exacerbated lung injury.Citation96,Citation97 Depending on the dosage and time of exposure, S1P1 agonists applied to support the maintenance of EC barrier might develop long-term opposing effects and act as functional antagonists.Citation96 The natural feedback regulatory mechanism of receptor activation, internalization and proteasomal degradation was intensified by functional antagonists, resulting in reduced S1P1 expression and signaling, which resulted in EC barrier destabilization.Citation97,Citation98 To activate S1P1 without mounting the devastating proteolytic cascade, which ultimately leads to enhanced EC barrier permeability, several analogs of FTY720 were screened and tested in vitro and in vivo for their effects on S1P1 mediated cellular response: The analog (S)-FTY720-phosphonate displayed promising results and showed EC barrier-promoting properties.Citation99 Furthermore (S)-FTY720-phosphonate was able to maintain the S1P1 expression, to reduce vascular leakage and to blunt the inflammatory reaction in bleomycin induced ALI. Activation of S1P1 without induction of the β-arrestin pathway exerted barrier promoting effects and prevented degradation of S1P1.Citation97 Additional studies revealed barrier enhancing properties of the FTY720 analogs (R)-methoxy-FTY720 ((R)-OMe-FTY), (R)/(S)-fluoro-FTY720 (FTY-F) and beta-glucuronide-FTY720 (FTY-G) in vitro.Citation100 According to TEER measurements after stimulation with S1P1 agonists and inhibitors, FTY720-phosphate and its analogs induce S1P1- Gαi signaling and barrier protection.Citation100 FTY720 analogs are therefore a promising approach for the treatment of inflammatory diseases like sepsis or ARDS/ALI and are able to prevent unwanted side effects like inhibition of S1P1 signaling due to prolonged treatment.
4.2 Maintenance of plasma S1P concentrations by SphKs
S1P metabolism involves not only its degrading enzyme S1P-lyase, but also the SphK1/2, which help to maintain the concentration of S1P in blood and tissues by generating S1P. The S1P-lyase reinforces inflammation and tissue damage, whereas the SphK1 acts protective. In mechanical stress-induced VILI, SphK1−/- mice had elevated levels of cytokines, proteins and cells in BALF and were more susceptible to lung injuries compared to WT mice. In Sgpl± mice those effects were attenuated.Citation64 The importance of the SphK1 in maintaining EC barriers was demonstrated in Sphk1−/- mice, which showed increased barrier permeability.Citation101,Citation102 SphK1 was activated by several different stimuli. On the one hand VEGF and TNFα activated SphK1 and induced pro-inflammatory responses, whereas on the other hand Ang-1 stimulated anti-inflammatory pathways.Citation102 The activation of SphK1 by Ang-1 proved to be important for the maintenance of the EC barrier. It resulted in increased levels of S1P, but inhibition of the S1P receptors 1 to 3 did not diminish the positive effects of Ang-1 on cell permeability, suggesting that Ang-1 mediated its effects via intracellular S1P.Citation102 Disruption of EC barriers can either be achieved via thrombin induced protease-activated receptor 1 (PAR-1) activation or by LPS. In both cases wild-type mice were able to recover from the impact after two hours, whereas the damage to the endothelium in SphK1−/- mice was persistent.Citation102 Furthermore, in vitro studies with small interfering RNA (siRNA) to inhibit the expression of SphK1 demonstrated the importance of SphK1-derived S1P to recover from EC barrier disruption and the involvement of S1P1 signaling to maintain EC barrier function.Citation101 Normal levels of SphK2 in those mice had no effect, suggesting that the EC barrier was mainly maintained by S1P that was generated by SphK1, which may be due to its location at the plasma membrane.Citation9,Citation101 Both SphK1 and SphK2 proved to be essential for the inflammatory response. The down-regulation as well as the inhibition of both enzymes caused elevated levels of the pro-inflammatory cytokine IL-6 in the cells.Citation67 Studies regarding the knock-out or inhibition of SphK2 are often contradictory since the loss or inhibition of SphK2 may be compensated by increased expression and activity of SphK1.Citation103,Citation104 A discrepancy between in vivo and in vitro studies regarding the knock-out of SphK2 was also observed by Dimasi et al.Citation104 In vitro, the inhibition of SphK2 caused a disruption of the EC barrier. On the other hand, SphK2−/- mice showed elevated levels of S1P in plasma and were able to maintain the EC barrier integrity in vivo. This was in contrast to the SphK1−/- mice that showed significantly reduced plasma S1P levels and suffered from vascular leakage.Citation104 Application of SphK2 inhibitors like SLR080811 demonstrated increased levels of S1P in blood, serum and lymphatic fluids. The authors proposed an increased activity of SphK1 to maintain S1P levels.Citation105
4.3 The role of S1P transporters for barrier protection
Zhao et al. showed that intracellular S1P is involved in the protection of the EC barrier under inflammatory conditions.Citation67 Their findings support the hypothesis that ECs produce and transport S1P autonomously via the Spns2 transporter to maintain vascular barrier integrity.Citation67 The transport of intracellular S1P out of cells is conducted by two transporters: Spns2 and Mfsd2b. Located on ECs, Spns2 is involved in the establishment of the S1P gradient between tissue and circulation. Spns2-deficient mice showed decreased levels of S1P in plasma and impaired egress of T and B cells from lymphoid organs,Citation14,Citation106 suggesting an involvement of Spns2 in inflammatory and autoimmune diseases. Furthermore, Spns2 was not only necessary for the transport of S1P, but also for the export of its analog FTY720-phosphate, and thus, it may play an important role in the treatment of diseases with S1P analogues.Citation13 Since many inflammatory diseases are associated with the disruption of the EC barrier, the release of S1P and therefore the preservation of the S1P gradient between plasma and tissues by Spns2 may be involved in the regulation of the vascular barrier integrity. The down-regulation of Spns2 resulted in defective blood vessel formation and disrupted EC barriers, confirming the importance of Spns2 in the maintenance of vascular integrity.Citation26,Citation107–109
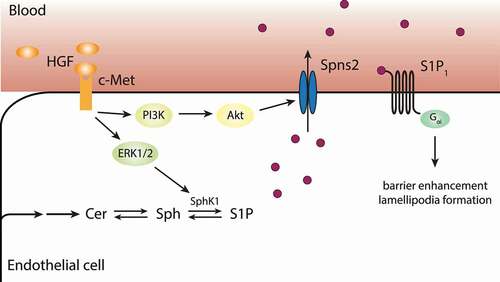
The hepatocyte growth factor (HGF) promoted EC barrier function.Citation110 Ephstein et al. demonstrated that the HGF/tyrosine-protein kinase Met (c-Met) and S1P1 formed a signaling complex, which was involved in the maintenance of vascular barriers.Citation111 Due to the activation of c-Met by HGF, the S1P1 receptor was trans-activated and led to the transduction of important signals for barrier protection.Citation111 Subsequent studies were able to connect the maintenance of vascular barrier by HGF with S1P metabolism and signaling. HGF/c-Met stimulated extracellular signal-regulated protein kinase 1/2 (ERK1/2), which promoted the activity of SphK1 and led to elevated levels of S1P.Citation107,Citation112 Furthermore, the PI3K/Akt pathway was also activated by HGF/c-Met, which stimulated the transporter Spns2 and promoted the secretion of intracellular S1P to stimulate S1P1 and enhanced barrier stability via the formation of lamellipodia ().Citation107,Citation113 Interestingly, not only HGF, but also apelin, a peptide involved in angiogenesis and the maintenance of cardiac lymphatic vasculature, interfered with S1P metabolism and transport. It stimulated the expression of SphK2 and Spns2 on lymphatic ECs (LECs) and caused elevated levels and secretion of S1P to maintain EC barrier integrity of the lymphatic endothelium.Citation108
Figure 4. Transactivation of S1P1 and autonomous production of S1P. Expression of Spns2 on ECs enables the cells to transport S1P outside the cells, where it can act in an autocrine or paracrine manner to activate signaling via S1P1. Furthermore, HGF binding to c-Met facilitates the production of S1P by stimulating SphK1 and promotes the autonomous sustenance of ECs with S1P by stimulating its transport via Spns2. This leads to the activation of S1P1 by S1P and the enhancement of barrier stability.Citation26,Citation107,Citation111 Akt, protein kinase B; c-Met, tyrosine-protein kinase Met; Cer, ceramide; ERK, extracellular signal-regulated protein kinase; HGF, hepatocyte growth factor; PI3K, phosphoinositide 3-kinase; S1P, sphingosine 1-phosphate; Sph, sphingosine; Spns2, spinster homolog 2
Spns2 was particularly expressed in vascular ECs, which led to the hypothesis that ECs might be able to produce barrier stabilizing S1P autonomously. This hypothesis was supported by the findings of Jeya Paul et al.Citation26 Cell culture experiments with primary human umbilical vein ECs (HUVECs) showed that HUVECs depend on S1P1 and its continuous stimulation by S1P for the integrity of the EC barrier. Moreover, HUVECs expressed the transporter Spns2 to ensure constant export of S1P for the maintenance of extracellular S1P levels and EC barrier integrity. The expression of Spns2 was attenuated under inflammatory conditions, which were mimicked by cytokines and LPS. Inflammatory conditions led to barrier disruption due to declined concentrations of extracellular S1P and resulted in EC barrier disruption. Interestingly the expression of the S1P1 receptor was not affected by cytokine and LPS treatment, leaving the option to increase barrier stability under inflammatory conditions via the stimulation of S1P1.Citation26
Mfsd2b is the second S1P transporter helping to maintain S1P concentration in plasma. Mfsd2b is expressed on erythrocytes and platelets.Citation15,Citation114 Similar to the knock-out of Spns2, the knock-out of Mfsd2b led to significantly reduced levels of S1P in plasma. But unlike the deletion of Spns2, deletion of Mfsd2b did not affect egress and trafficking of lymphocytes. Furthermore, the integrity of blood vessels was not affected by the loss of Mfsd2b, yet the transporter seemed to be involved in protection against anaphylactic shock.Citation15 This led to the suggestion that not the overall amount of S1P in plasma might be important for the protective effect of S1P in inflammatory diseases, but the environment in which S1P is released by Spns2 or Mfsd2b.Citation15
4.4 The role of apoM associated S1P for barrier protection
As a hydrophobic molecule, S1P depends on carrier proteins for efficient transport. More than 60% of S1P in plasma was bound to HDL and the remaining S1P was transported by albumin.Citation18 A recent study investigated the connection between the severity of sepsis and S1P in plasma either bound to albumin or to HDL.Citation115 Interestingly, a shift of S1P from albumin to HDL was observed in patients with systemic inflammation, which was presumably an adaptive response of the body to maintain S1P levels in plasma. Further loss of S1P in septic shock correlated with the loss of HDL and vice versa. Elevated levels of HDL led to increased levels of HDL-S1P, supporting the therapeutic strategy of HDL administration to treat patients with septic shock.Citation115 Compared to albumin-S1P, HDL-S1P was able to preserve the stability of the EC barrier longer, which was facilitated by S1P1 and the activation of the PI3K-Akt-endothelial nitric oxide synthase (eNOS) pathway.Citation116,Citation117 Additionally the S1P1 receptor was recycled after the binding of HDL-S1P and expressed at higher numbers on the cell surface, promoting the barrier enhancing effects of HDL-S1P.Citation116,Citation117 Further studies revealed the necessity of apolipoprotein M (apoM) as a chaperone for S1P for its binding to HDL.Citation118,Citation119
The importance of apoM for S1P transport and its function as effector molecule became apparent in experiments with apoM−/- mice. Those mice had approximately 60% less S1P in plasma, namely S1P bound to HDL, and only albumin-bound S1P was circulating.Citation118 Moreover, apoM−/- mice displayed decreased EC barrier function in the lung. This result suggested that apoM was necessary for the EC barrier stabilizing function of S1P, and that the vasoprotective effects of apoM-bound S1P were mediated via the activation and internalization of S1P1.Citation118,Citation119 Christoffersen et al. even hypothesized that the vascular EC barrier could be protected from injury via an increase of apoM+ HDL.Citation118 Also, the anti-inflammatory properties of S1P were reinforced when it was bound to HDL with apoM. This apoM+-HDL suppressed inflammation through the activation of S1P1 via the inhibition of TNFα induced NFкB activation and via the reduction of VCAM-1 to promote barrier stability.Citation120,Citation121 These results were confirmed in an animal model of LPS induced ALI. Mice lacking apoM had elevated concentrations of pro-inflammatory cytokines in serum as well as more severe lung injury compared to wild-type mice. Treatment with the S1P1 antagonist W146 under inflammatory conditions interfered with the apoM-S1P mediated improvement of lung injury and pointed to a beneficial role of S1P-S1P1 signaling in ALI.Citation122 Septic rats showed decreased levels of HDL-S1P and apoM in plasma according to the severity of sepsis. The treatment of those animals with HDL-S1P was able to prevent LPS induced ALI via reduction of pulmonary edema, vascular leakage and the expression of inflammatory factors.Citation123 Furthermore, the administration of HDL-S1P and the S1P1 agonist SEW2871 enhanced the activation of eNOS and protected the EC barrier by supporting the proliferation and migration of ECs. Those beneficial effects were counteracted by the administration of the S1P1 inhibitor W146, again confirming the important role of the transport of S1P by HDL and the signaling via S1P1 under inflammatory conditions.Citation123
Treatment strategies to enhance the barrier stability in inflammatory diseases like sepsis are needed. Swendeman et al. engineered a soluble carrier for S1P, apoM-Fc, to transport S1P, activate S1P1 signaling and to promote EC barrier stability.Citation119,Citation124 In vitro experiments confirmed the impact of apoM-Fc on barrier stability due to the activation of S1P1 and S1P3. In vivo experiments were able to prove that administration of apoM-Fc could improve S1P levels in plasma and activate S1P1 without causing lymphopenia, which led to the conclusion that S1P1 in lymphatic tissues is not accessible for apoM-Fc. apoM-Fc-S1P revealed anti-hypotensive effects via S1P1. After ischemia or reperfusion injuries, neutrophil accumulation at the infarcted site was attenuated, and endothelial homeostasis was maintained. Activation of S1P1 by apoM-Fc also promoted the integrity of the blood-brain-barrier and suppressed neuronal damage after stroke in mice.Citation124
Inflammatory diseases like systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) are also characterized by EC barrier dysfunction due to an abnormal immune reaction. The stimulation of S1P1 with apoM-Fc and agonists showed beneficial effects under inflammatory conditions due to the enhancement of the EC barrier. Stimulation of S1P1 prevented the degradation of VE-cadherin as well as the phosphorylation of myosin light chain 2 (MLC-2), leading to less cell contraction and the stabilization of adherent junctions.Citation125 In comparison to receptor modulators like FTY720-phosphate, the engineered carrier apoM-Fc proved to be beneficial. Depending on the dosage and frequency of administration, FTY720-phosphate could enhance leakage of the EC barrier, probably due to binding to S1P1 and subsequent internalization and proteasomal degradation of the receptor.Citation96,Citation98 The carrier apoM-Fc on the other hand transported S1P to its receptor and prevented excessive internalization of S1P1. Furthermore, the half-life of apoM-Fc was about four days, providing another advantage compared to short-lived agonists like SEW2871. In conclusion, apoM-Fc proved to be a useful tool for enhancing S1P in plasma and to enforce its transport and binding to its receptors, mainly S1P1, to stabilize vascular EC barrier integrity without causing negative side-effects like lymphopenia.Citation124,Citation125
5. S1P in specialized endothelial barriers
5.1 Blood-brain barrier
The blood-brain barrier (BBB) is a specialized vascular structure, which seals the central nervous system (CNS) and maintains homeostasis in the brain by controlling the exchange of substances between the blood and CNS. This physiological barrier consists of specialized ECs, which form a monolayer connected via tight junctions to restrict the transport of substances to transcellular pathwaysCitation126,Citation127 (). Several diseases like amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS) or epilepsy are marked by a breakdown of the BBB. Increased permeability of the EC barrier and abnormal flux of molecules and cells of the immune system arise from its degradation. As a consequence, neuroinflammation, dysfunction and degeneration occur.Citation127
Van Doorn et al. identified S1P5 as a key player to maintain the stability of the EC barrier and immunoquiescence.Citation128 ECs of the BBB expressed S1P1-3,5 at different levels, while S1P1 and S1P5 dominated. Moreover, the expression of S1P5 was restricted to the brain.Citation28–30 As a consequence of a knock-down of S1P5 in brain ECs (hCMEC/D3), the expression of S1P1-3 was significantly reduced and the integrity of the EC barrier was compromised. This led to a pro-inflammatory condition of the brain, which was characterized by increased migration of monocytes, adhesion of leukocytes via VCAM-1 and intercellular adhesion molecule-1 (ICAM-1), and production of pro-inflammatory cytokines and chemokines. This state could be attenuated by the activation of S1P5 via a specific agonist or FTY720-phosphate, emphasizing the important role of S1P5 to maintain the BBB and modulate inflammatory processes.Citation128 S1P1 was prominently expressed in brain endothelial cells,Citation128 and targeting S1P1 in acute ischemic stroke (AIS) prevented neuroinflammation and breakdown of the BBB.Citation129 S1pr1iECKO mice, which lack S1P1 in ECs of the brain, suffered from BBB leakiness for small molecules. The permeability of the BBB was regulated by S1P1, and leakage of the barrier was probably due to a changed localization of tight junction proteins as well as a change in the structure and function of AJ proteins.
The modulation of EC barrier integrity by S1P1 is a new approach to target diseases of the CNS. By pharmacological modulation of S1P1, a reversible opening of the BBB could be achieved and provided the possibility to deliver drugs to the CNS.Citation130 Inflammatory conditions reduced the integrity of EC barriers, which could be measured by a reduction of TEER. In an in vitro BBB model, Siponimod (BAF-312) was able to activate S1P1 and S1P5, and the maintenance of EC barrier integrity by both receptors was enhanced under inflammatory conditions. Depending on the duration of Siponimod treatment, the interaction of both receptors enhanced and promoted barrier stabilization by maintaining ZO-1 and claudin-5 expression.Citation131
Next to the modulation of S1P signaling, the modulation of S1P metabolism is an interesting approach to strengthen and modify the BBB. The investigation of the role of intracellular S1P (iS1P) in hCMEC/D3 cells revealed opposing effects of the knock-down of SGPL1 (SPL-kd). SPL-kd enhanced the levels of iS1P in hCMEC/D3, and a barrier stabilizing or disruptive effect was observed depending on the conditions. Under inflammatory conditions, enhanced levels of iS1P were beneficial and led to the stabilization of the EC barrier, whereas, under normal conditions, a knock-down of the SGPL1 promoted the disruption of the barrier.Citation132 Especially the beneficial effects of iS1P on the expression of AJ proteins like VE-cadherin, PECAM1 or β-catenin promoted the stabilization of EC barriers under inflammatory conditions. Furthermore, upregulation of signaling molecules like protein kinase C (PKC), adenosine monophosphate-activated protein kinase (AMPK) and p38-mitogen-activated protein kinase (MAPK) were observed together with downregulation of pro-inflammatory cytokines and VCAM1.Citation132
5.2 Placenta
The placenta is a complex and specialized organ and composed of fetal and maternal tissue. The placenta plays a crucial role in the development of the fetus and is responsible for the sustenance of the fetus with nutrients, oxygen, vitamins and other important metabolites, and for the removal of fetal waste products.Citation133 The maternal circulation is separated from the fetal by the placental membrane, which initially consists of four layers. One layer of the placental membrane is the feto-placental endothelium, and disruption and dysfunction of the vasculature peaks in diseases like preeclampsia (PE), intrauterine growth restriction (IUGR) and other inflammatory diseases, posing a risk to mother and fetus.Citation133,Citation134
So far, the influence of S1P on placental development and EC barrier maintenance was not thoroughly investigated. Many pregnancy-related and inflammatory diseases are marked by a reduction of barrier function. S1P is involved in maintaining EC function and presents a new approach to target placental dysfunction. Placental arteries expressed the receptors S1P1-3,5. S1P was involved in regulating the vascular tone of the placenta by Rho-associated kinases and Ca2+ sensitization. Additionally, S1P activated NOS3, which was responsible for vasoconstriction.Citation135 Recently, Del Gaudio et al. examined the sphingolipid metabolism in the feto-placental vasculature in PE. Alterations in the sphingolipid metabolism occurred on several sites. On the one hand, serine palmitoyltransferase (SPT), S1P phosphatase (SPP) and SGPL1 expression and activity were increased and caused decreased levels of S1P and enhanced levels of dihydrosphingosine (dhSph). Furthermore, expression of S1P1 was significantly reduced, whereas expression of S1P2 was upregulated, which resulted in endothelial dysfunction.Citation136,Citation137 Decreased levels of endothelial S1P together with decreased expression of S1P1 were one possibility for placental endothelial dysfunction. Therefore, optimization and support of signaling via the S1P-S1P1 axis might be an option to maintain stability and integrity of the placental EC barrier. As mentioned previously, S1P-apoM bound to HDL displayed barrier enhancing effects. Neonatal HDL (nHDL) was also able to carry the S1P-apoM complex. In vitro, nHDL delivered S1P was able to induce signaling via phospholipase C (PLC) and extracellular signal-regulated protein kinases (ERK) and strengthened the EC barrier function. Furthermore, upregulation of eNOS was observed and pointed to the involvement of S1P in placental vasorelaxation and – dilation.Citation134 In an in vitro model of PE, apoM-HDL was reduced and led to degradation of the EC barrier. Vascular inflammation was diminished following administration of nHDL-S1P, depicted by reduced expression of inflammatory markers and surface molecules like ICAM and VCAM. Additionally, nHDL-S1P provided protection from oxidative stress by negatively regulating angiotensin II induced reactive oxygen species (ROS) production and decreasing the expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1.Citation138 Although our knowledge about the maintenance of the placental EC barrier is still scarce, the contribution of S1P to EC barrier maintenance in the placenta may offer the opportunity to treat placental dysfunction and other inflammatory diseases.
6. Conclusion
While barrier maintenance is a complex process that requires organ-specific approaches for fine-tuning and stabilization, data from the literature clearly indicate the high potential of S1P for medical intervention. Some compounds like FTY720 or apoM-Fc are already available and tested in different disease entities, additional applications such as inhibition of the S1P lyase or modulation of SphKs and S1P transporters may lead to even more options in the future. Although targeting signaling and metabolic pathways of S1P is a challenging task, the herein presented data from the literature demonstrate that continuous research in this field could result in highly valuable treatment options in the future. S1P is probably the molecule with the highest potential for EC barrier stabilizing applications in the future.
Acknowledgments
We thank Ralf A. Claus from the Department of Anesthesiology and Intensive Care Medicine of the Jena University Hospital for critical reading.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
- Rodrigues SF, Granger DN. Blood cells and endothelial barrier function. Tissue Barriers. 2015;3(1–2):1. doi:https://doi.org/10.4161/21688370.2014.978720.
- Jernigan PL, Makley AT, Hoehn RS, Edwards MJ, Pritts TA. The role of sphingolipids in endothelial barrier function. Biol Chem. 2015;396(6–7):681–21. doi:https://doi.org/10.1515/hsz-2014-0305.
- Xiong Y, Hla T. S1P control of endothelial integrity. Curr Top Microbiol Immunol. 2014;378:85–105. doi:https://doi.org/10.1007/978-3-319-05879-5_4.
- Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108(5):689–701. doi:https://doi.org/10.1172/JCI12450.
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4(5):397–407. doi:https://doi.org/10.1038/nrm1103.
- Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119(7):1871–1879. doi:https://doi.org/10.1172/jci38575.
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi:https://doi.org/10.1038/nature02284.
- Walsh KB, Teijaro JR, Wilker PR, Jatzek A, Fremgen DM, Das SC, Watanabe T, Hatta M, Shinya K, Suresh M, et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci U S A. 2011;108(29):12018–12023. doi:https://doi.org/10.1073/pnas.1107024108.
- Pitson SM. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003;22(20):5491–5500. doi:https://doi.org/10.1093/emboj/cdg540.
- Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S. Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J Biol Chem. 2007;282:12058–12065. doi:https://doi.org/10.1074/jbc.M609559200.
- Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280(44):37118–37129. doi:https://doi.org/10.1074/jbc.M502207200.
- Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat Rev Immunol. 2011;11(6):403–415. doi:https://doi.org/10.1038/nri2974.
- Hisano Y, Kobayashi N, Kawahara A, Yamaguchi A, Nishi T. The sphingosine 1-phosphate transporter, SPNS2, functions as a transporter of the phosphorylated form of the immunomodulating agent FTY720. J Biol Chem. 2011;286(3):1758–1766. doi:https://doi.org/10.1074/jbc.M110.171116.
- Hisano Y, Kobayashi N, Yamaguchi A, Nishi T, Holowka D. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS One. 2012;7(6):e38941. doi:https://doi.org/10.1371/journal.pone.0038941.
- Vu TM, Ishizu A-N, Foo JC, Toh XR, Zhang F, Whee DM, Torta F, Cazenave-Gassiot A, Matsumura T, Kim S, et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature. 2017;550(7677):524–528. doi:https://doi.org/10.1038/nature24053.
- Van Veldhoven PP. Sphingosine-1-phosphate lyase. Methods Enzymol. 2000;311:244–254.
- Mandala SM. Sphingosine-1-phosphate phosphatases. Prostaglandins Other Lipid Mediat. 2001;64(1–4):143–156. doi:https://doi.org/10.1016/S0090-6980(01)00111-3.
- Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, UI M, Okajima F. Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J. 2000;352(3):809–815. doi:https://doi.org/10.1042/bj3520809.
- Liu Y, Wada R, Yamashita T, Mi Y, Deng C-X, Hobson JP, Rosenfeldt HM, Nava VE, Chae S-S, Lee M-J, et al. Edg-1, the G protein–coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106(8):951–961. doi:https://doi.org/10.1172/JCI10905.
- Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102(10):3665–3667. doi:https://doi.org/10.1182/blood-2003-02-0460.
- Allende ML. Sphingosine-1-phosphate receptors and the development of the vascular system. Biochim Biophys Acta. 2002;1582(1–3):222–227. doi:https://doi.org/10.1016/S1388-1981(02)00175-0.
- Darland DC, D’Amore PA. Blood vessel maturation: vascular development comes of age. J Clin Invest. 1999;103(2):157–158. doi:https://doi.org/10.1172/JCI6127.
- Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507–521. doi:https://doi.org/10.1016/S0008-6363(00)00281-9.
- Kono M, Mi Y, Liu Y, Sasaki T, Allende ML, Wu YP, Yamashita T, Proia RL. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279(28):29367–29373. doi:https://doi.org/10.1074/jbc.M403937200.
- Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25(24):11113–11121. doi:https://doi.org/10.1128/MCB.25.24.11113-11121.2005.
- Jeya Paul J, Weigel C, Muller T, Heller R, Spiegel S, Graler MH. Inflammatory Conditions Disrupt Constitutive Endothelial Cell Barrier Stabilization by Alleviating Autonomous Secretion of Sphingosine 1-Phosphate. Cells. 2020;9(4):928. doi:https://doi.org/10.3390/cells9040928.
- Sensken SC, Staubert C, Keul P, Levkau B, Schoneberg T, Graler MH. Selective activation of G alpha i mediated signalling of S1P3 by FTY720-phosphate. Cell Signal. 2008;20(6):1125–1133. doi:https://doi.org/10.1016/j.cellsig.2008.01.019.
- Allard J, Barron S, Diaz J, Lubetzki C, Zalc B, Schwartz JC, Sokoloff P. A rat G protein-coupled receptor selectively expressed in myelin-forming cells. Eur J Neurosci. 1998;10(3):1045–1053. doi:https://doi.org/10.1046/j.1460-9568.1998.00117.x.
- Im D-S, Heise CE, Ancellin N, O’Dowd BF, Shei G-J, Heavens RP, Rigby MR, Hla T, Mandala S, McAllister G, et al. Characterization of a novel sphingosine 1-phosphate receptor, Edg-8. J Biol Chem. 2000;275(19):14281–14286. doi:https://doi.org/10.1074/jbc.275.19.14281.
- Im DS, Clemens J, Macdonald TL, Lynch KR. Characterization of the human and mouse sphingosine 1-phosphate receptor, S1P5 (Edg-8): structure-activity relationship of sphingosine1-phosphate receptors. Biochemistry. 2001;40:14053–14060. doi:https://doi.org/10.1021/bi011606i.
- Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55(8):1596–1608. doi:https://doi.org/10.1194/jlr.R046300.
- Lee J-F, Zeng Q, Ozaki H, Wang L, Hand AR, Hla T, Wang E, Lee M-J. Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. J Biol Chem. 2006;281(39):29190–29200. doi:https://doi.org/10.1074/jbc.M604310200.
- Argraves KM, Gazzolo PJ, Groh EM, Wilkerson BA, Matsuura BS, Twal WO, Hammad SM, Argraves WS. High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function. J Biol Chem. 2008;283(36):25074–25081. doi:https://doi.org/10.1074/jbc.M801214200.
- McVerry BJ, Garcia JG. In vitro and in vivo modulation of vascular barrier integrity by sphingosine 1-phosphate: mechanistic insights. Cell Signal. 2005;17(2):131–139. doi:https://doi.org/10.1016/j.cellsig.2004.08.006.
- Singleton PA, Dudek SM, Chiang ET, Garcia JG. Regulation of sphingosine 1-phosphate-induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005;19:1646–1656. doi:https://doi.org/10.1096/fj.05-3928com.
- Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JGN, Birukov KG. Akt-Mediated Transactivation of the S1P1 Receptor in Caveolin-Enriched Microdomains Regulates Endothelial Barrier Enhancement by Oxidized Phospholipids. Circ Res. 2009;104(8):978–986. doi:https://doi.org/10.1161/CIRCRESAHA.108.193367.
- Shikata Y, Birukov KG, Birukova AA, Verin A, Garcia JG. Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J. 2003;17(15):2240–2249. doi:https://doi.org/10.1096/fj.03-0198com.
- Shikata Y, Birukov KG, Garcia JG. S1P induces FA remodeling in human pulmonary endothelial cells: role of Rac, GIT1, FAK, and paxillin. J Appl Physiol. 1985;2003:1193–1203.
- Belvitch P, Dudek SM. Role of FAK in S1P-regulated endothelial permeability. Microvasc Res. 2012;83(1):22–30. doi:https://doi.org/10.1016/j.mvr.2011.08.012.
- Dudek SM, Chiang ET, Camp SM, Guo Y, Zhao J, Brown ME, Singleton PA, Wang L, Desai A, Arce FT, et al. Abl tyrosine kinase phosphorylates nonmuscle Myosin light chain kinase to regulate endothelial barrier function. Mol Biol Cell. 2010;21(22):4042–4056. doi:https://doi.org/10.1091/mbc.e09-10-0876.
- Gao F, Sabbineni H, Artham S, Somanath PR. Modulation of long-term endothelial-barrier integrity is conditional to the cross-talk between Akt and Src signaling. J Cell Physiol. 2017;232(10):2599–2609. doi:https://doi.org/10.1002/jcp.25791.
- Klomp JE, Shaaya M, Matsche J, Rebiai R, Aaron JS, Collins KB, Huyot V, Gonzalez AM, Muller WA, Chew T-L, et al. Time-Variant SRC Kinase Activation Determines Endothelial Permeability Response. Cell Chem Biol. 2019;26(8):1081–94 e6. doi:https://doi.org/10.1016/j.chembiol.2019.04.007.
- Krump-Konvalinkova V, Yasuda S, Rubic T, Makarova N, Mages J, Erl W, Vosseler C, Kirkpatrick CJ, Tigyi G, Siess W, et al. Stable Knock-Down of the Sphingosine 1-Phosphate Receptor S1P 1 Influences Multiple Functions of Human Endothelial Cells. Arterioscler Thromb Vasc Biol. 2005;25(3):546–552. doi:https://doi.org/10.1161/01.ATV.0000154360.36106.d9.
- Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of Vascular Permeability by the Sphingosine-1-Phosphate Receptor–2 (S1P2R) and its Downstream Effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27(6):1312–1318. doi:https://doi.org/10.1161/ATVBAHA.107.143735.
- Zhang G, Yang L, Kim GS, Ryan K, Lu S, O’Donnell RK, Spokes K, Shapiro N, Aird WC, Kluk MJ, et al. Critical role of sphingosine-1-phosphate receptor 2 (S1PR2) in acute vascular inflammation. Blood. 2013;122(3):443–455. doi:https://doi.org/10.1182/blood-2012-11-467191.
- Reinhard NR, Mastop M, Yin T, Wu Y, Bosma EK, Gadella TWJ Jr., Goedhart J, Hordijk PL. The balance between Gαi-Cdc42/Rac and Gα12/13-RhoA pathways determines endothelial barrier regulation by sphingosine-1-phosphate. Mol Biol Cell. 2017;28(23):3371–3382. doi:https://doi.org/10.1091/mbc.e17-03-0136.
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi:https://doi.org/10.1038/nature07201.
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:https://doi.org/10.1016/S0140-6736(20)30183-5.
- Marfia G, Navone S, Guarnaccia L, Campanella R, Mondoni M, Locatelli M, Barassi A, Fontana L, Palumbo F, Garzia E, et al. Decreased serum level of sphingosine-1-phosphate: a novel predictor of clinical severity in COVID-19. EMBO Mol Med. 2021;13(1):e13424. doi:https://doi.org/10.15252/emmm.202013424.
- Song J-W, Lam SM, Fan X, Cao W-J, Wang S-Y, Tian H, Chua GH, Zhang C, Meng F-P, Xu Z, et al. Omics-Driven Systems Interrogation of Metabolic Dysregulation in COVID-19 Pathogenesis. Cell Metab. 2020;32(2):188–202 e5. doi:https://doi.org/10.1016/j.cmet.2020.06.016.
- Thomas T, Stefanoni D, Dzieciatkowska M, Issaian A, Nemkov T, Hill RC, Francis RO, Hudson KE, Buehler PW, Zimring JC, et al. Evidence of Structural Protein Damage and Membrane Lipid Remodeling in Red Blood Cells from COVID-19 Patients. J Proteome Res. 2020;19(11):4455–4469. doi:https://doi.org/10.1021/acs.jproteome.0c00606.
- Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun. 2020;109:102433. doi:https://doi.org/10.1016/j.jaut.2020.102433.
- Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239–1242. doi:https://doi.org/10.1001/jama.2020.2648.
- Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418. doi:https://doi.org/10.1016/S0140-6736(20)30937-5.
- McGowan EM, Haddadi N, Nassif NT, Lin Y. Targeting the SphK-S1P-SIPR Pathway as a Potential Therapeutic Approach for COVID-19. Int J Mol Sci. 2020;21(19):21. doi:https://doi.org/10.3390/ijms21197189.
- Winkler MS, Nierhaus A, Holzmann M, Mudersbach E, Bauer A, Robbe L, Zahrte C, Geffken M, Peine S, Schwedhelm E, et al. Decreased serum concentrations of sphingosine-1-phosphate in sepsis. Critical Care. 2015;19(1):372. doi:https://doi.org/10.1186/s13054-015-1089-0.
- Zhao J, Tan Y, Wang L, Su X, Shi Y. Serum sphingosine-1-phosphate levels and Sphingosine-1-Phosphate gene polymorphisms in acute respiratory distress syndrome: a multicenter prospective study. J Transl Med. 2020;18:156. doi:https://doi.org/10.1186/s12967-020-02322-y.
- Piotti A, Novelli D, Meessen J, Ferlicca D, Coppolecchia S, Marino A, Salati G, Savioli M, Grasselli G, Bellani G, et al. Endothelial damage in septic shock patients as evidenced by circulating syndecan-1, sphingosine-1-phosphate and soluble VE-cadherin: a substudy of ALBIOS. Critical Care. 2021;25(1):113. doi:https://doi.org/10.1186/s13054-021-03545-1.
- Hemdan NY, Weigel C, Reimann C-M, Graler MH. Modulating sphingosine 1-phosphate signaling with DOP or FTY720 alleviates vascular and immune defects in mouse sepsis. Eur J Immunol. 2016;46(12):2767–2777. doi:https://doi.org/10.1002/eji.201646417.
- Wang Z, Sims CR, Patil NK, Gokden N, Mayeux PR. Pharmacologic targeting of sphingosine-1-phosphate receptor 1 improves the renal microcirculation during sepsis in the mouse. J Pharmacol Exp Ther. 2015;352(1):61–66. doi:https://doi.org/10.1124/jpet.114.219394.
- Flemming S, Burkard N, Meir M, Schick MA, Germer C-T, Schlegel N. Sphingosine-1-Phosphate Receptor-1 Agonist Sew2871 Causes Severe Cardiac Side Effects and Does Not Improve Microvascular Barrier Breakdown in Sepsis. Shock. 2018;49(1):71–81. doi:https://doi.org/10.1097/SHK.0000000000000908.
- Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JGN. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169(11):1245–1251. doi:https://doi.org/10.1164/rccm.200309-1258OC.
- McVerry BJ, Peng X, Hassoun PM, Sammani S, Simon BA, Garcia JG. Sphingosine 1-phosphate reduces vascular leak in murine and canine models of acute lung injury. Am J Respir Crit Care Med. 2004;170(9):987–993. doi:https://doi.org/10.1164/rccm.200405-684OC.
- Suryadevara V, Fu P, Ebenezer DL, Berdyshev E, Bronova IA, Huang LS, Harijith A, Natarajan V. Sphingolipids in Ventilator Induced Lung Injury: role of Sphingosine-1-Phosphate Lyase. Int J Mol Sci. 2018;19(1):114. doi:https://doi.org/10.3390/ijms19010114.
- Spieth PM, Bluth T, Gama De Abreu M, Bacelis A, Goetz AE, Kiefmann R. Mechanotransduction in the lungs. Minerva anestesiologica. 2014;80:933–941.
- Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain A, Moitra J, et al. Differential Effects of Sphingosine 1–Phosphate Receptors on Airway and Vascular Barrier Function in the Murine Lung. Am J Respir Cell Mol Biol. 2010;43(4):394–402. doi:https://doi.org/10.1165/rcmb.2009-0223OC.
- Zhao Y, Gorshkova IA, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B, et al. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol Biol. 2011;45(2):426–435. doi:https://doi.org/10.1165/rcmb.2010-0422OC.
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. 2019;157:107843. doi:https://doi.org/10.1016/j.diabres.2019.107843.
- Wigger D, Schumacher F, Schneider-Schaulies S, Kleuser B. Sphingosine 1-phosphate metabolism and insulin signaling. Cell Signal. 2021;82:109959. doi:https://doi.org/10.1016/j.cellsig.2021.109959.
- Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98(4):2133–2223. doi:https://doi.org/10.1152/physrev.00063.2017.
- Leguina-Ruzzi A, Pereira J, Pereira-Flores K, Valderas JP, Mezzano D, Velarde V, Sáez CG. Increased RhoA/Rho-Kinase Activity and Markers of Endothelial Dysfunction in Young Adult Subjects with Metabolic Syndrome. Metab Syndr Relat Disord. 2015;13(9):373–380. doi:https://doi.org/10.1089/met.2015.0061.
- Alberti KG, Zimmet P, Shaw J. Metabolic syndrome-a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23(5):469–480. doi:https://doi.org/10.1111/j.1464-5491.2006.01858.x.
- Singh A, Ramnath RD, Foster RR, Wylie EC, Friden V, Dasgupta I, Haraldsson B, Welsh GI, Mathieson PW, Satchell SC, et al. Reactive oxygen species modulate the barrier function of the human glomerular endothelial glycocalyx. PLoS One. 2013;8(2):e55852. doi:https://doi.org/10.1371/journal.pone.0055852.
- Shi Y, Vanhoutte PM. Macro- and microvascular endothelial dysfunction in diabetes. J Diabetes. 2017;9:434–449.
- Sajja RK, Prasad S, Cucullo L. Impact of altered glycaemia on blood-brain barrier endothelium: an in vitro study using the hCMEC/D3 cell line. Fluids and Barriers of the CNS. 2014;11(1):8. doi:https://doi.org/10.1186/2045-8118-11-8.
- Tien T, Barrette KF, Chronopoulos A, Roy S. Effects of high glucose-induced Cx43 downregulation on occludin and ZO-1 expression and tight junction barrier function in retinal endothelial cells. Invest Ophthalmol Vis Sci. 2013;54(10):6518–6525. doi:https://doi.org/10.1167/iovs.13-11763.
- Laychock SG, Sessanna SM, Lin M-H, Mastrandrea LD. Sphingosine 1-Phosphate Affects Cytokine-Induced Apoptosis in Rat Pancreatic Islet β-Cells. Endocrinology. 2006;147(10):4705–4712. doi:https://doi.org/10.1210/en.2006-0456.
- Hasan NM, Longacre MJ, Stoker SW, Kendrick MA, Druckenbrod NR, Laychock SG, Mastrandrea LD, MacDonald MJ. Sphingosine kinase 1 knockdown reduces insulin synthesis and secretion in a rat insulinoma cell line. Arch Biochem Biophys. 2012;518(1):23–30. doi:https://doi.org/10.1016/j.abb.2011.11.016.
- Qi Y, Chen J, Lay A, Don A, Vadas M, Xia P. Loss of sphingosine kinase 1 predisposes to the onset of diabetes via promoting pancreatic β-cell death in diet-induced obese mice. FASEB J. 2013;27(10):4294–4304. doi:https://doi.org/10.1096/fj.13-230052.
- Liu Y, Harashima S-I, Wang Y, Suzuki K, Tokumoto S, Usui R, Tatsuoka H, Tanaka D, Yabe D, Harada N, et al. Sphingosine kinase 1–interacting protein is a dual regulator of insulin and incretin secretion. FASEB J. 2019;33(5):6239–6253. doi:https://doi.org/10.1096/fj.201801783RR.
- Ravichandran S, Finlin BS, Kern PA, Ozcan S. Sphk2−/− mice are protected from obesity and insulin resistance. Biochim Biophys Acta Mol Basis Dis. 2019;1865(3):570–576. doi:https://doi.org/10.1016/j.bbadis.2018.12.012.
- Song Z, Wang W, Li N, Yan S, Rong K, Lan T, Xia P. Sphingosine kinase 2 promotes lipotoxicity in pancreatic β-cells and the progression of diabetes. FASEB J. 2019;33(3):3636–3646. doi:https://doi.org/10.1096/fj.201801496R.
- Aji G, Huang Y, Ng ML, Wang W, Lan T, Li M, Li Y, Chen Q, Li R, Yan S, et al. Regulation of hepatic insulin signaling and glucose homeostasis by sphingosine kinase 2. Proc Natl Acad Sci U S A. 2020;117(39):24434–24442. doi:https://doi.org/10.1073/pnas.2007856117.
- Fang H, Feng Q, Shi Y, Zhou J, Wang Q, Zhong L. Hepatic insulin resistance induced by mitochondrial oxidative stress can be ameliorated by sphingosine 1-phosphate. Mol Cell Endocrinol. 2020;501:110660. doi:https://doi.org/10.1016/j.mce.2019.110660.
- Kurano M, Hara M, Tsuneyama K, Sakoda H, Shimizu T, Tsukamoto K, Ikeda H, Yatomi Y. Induction of insulin secretion by apolipoprotein M, a carrier for sphingosine 1-phosphate. Biochim Biophys Acta. 2014;1841(9):1217–1226. doi:https://doi.org/10.1016/j.bbalip.2014.05.002.
- Kurano M, Tsukamoto K, Shimizu T, Kassai H, Nakao K, Aiba A, Hara M, Yatomi Y. Protection Against Insulin Resistance by Apolipoprotein M/Sphingosine-1-Phosphate. Diabetes. 2020;69(5):867–881. doi:https://doi.org/10.2337/db19-0811.
- Fayyaz S, Henkel J, Japtok L, Kramer S, Damm G, Seehofer D, Püschel GP, Kleuser B. Involvement of sphingosine 1-phosphate in palmitate-induced insulin resistance of hepatocytes via the S1P2 receptor subtype. Diabetologia. 2014;57(2):373–382. doi:https://doi.org/10.1007/s00125-013-3123-6.
- Sanchez T, Estrada-Hernandez T, Paik J-H, Wu M-T, Venkataraman K, Brinkmann V, Claffey K, Hla T. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278(47):47281–47290. doi:https://doi.org/10.1074/jbc.M306896200.
- Kappos L, Antel J, Comi G, Montalban X, O’Connor P, Polman CH, Haas T, Korn AA, Karlsson G, Radue EW, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–1140. doi:https://doi.org/10.1056/NEJMoa052643.
- Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med. 2012;366(4):339–347. doi:https://doi.org/10.1056/NEJMct1101691.
- Nishihara H, Shimizu F, Sano Y, Takeshita Y, Maeda T, Abe M, Koga M, Kanda T. Fingolimod prevents blood-brain barrier disruption induced by the sera from patients with multiple sclerosis. PLoS One. 2015;10(3):e0121488. doi:https://doi.org/10.1371/journal.pone.0121488.
- Nishihara H, Maeda T, Sano Y, Ueno M, Okamoto N, Takeshita Y, Shimizu F, Koga M, Kanda T. Fingolimod promotes blood-nerve barrier properties in vitro. Brain and Behavior. 2018;8(4):e00924. doi:https://doi.org/10.1002/brb3.924.
- Annunziata P, Cioni C, Masi G, Tassi M, Marotta G, Severi S. Fingolimod reduces circulating tight-junction protein levels and in vitro peripheral blood mononuclear cells migration in multiple sclerosis patients. Sci Rep. 2018;8(1):15371. doi:https://doi.org/10.1038/s41598-018-33672-9.
- Wang L, Chiang ET, Simmons JT, Garcia JG, Dudek SM. FTY720-induced human pulmonary endothelial barrier enhancement is mediated by c-Abl. Eur Respir J. 2011;38(1):78–88. doi:https://doi.org/10.1183/09031936.00047810.
- Krump-Konvalinkova V, Chwalla I, Siess W. FTY720 inhibits S1P-mediated endothelial healing: relationship to S1P1-receptor surface expression. Biochem Biophys Res Commun. 2008;370(4):603–608. doi:https://doi.org/10.1016/j.bbrc.2008.03.144.
- Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged Exposure to Sphingosine 1–Phosphate Receptor-1 Agonists Exacerbates Vascular Leak, Fibrosis, and Mortality after Lung Injury. Am J Respir Cell Mol Biol. 2010;43(6):662–673. doi:https://doi.org/10.1165/rcmb.2009-0345OC.
- Wang L, Sammani S, Moreno-Vinasco L, Letsiou E, Wang T, Camp SM, Bittman R, Garcia JGN, Dudek SM. FTY720 (s)-phosphonate preserves sphingosine 1-phosphate receptor 1 expression and exhibits superior barrier protection to FTY720 in acute lung injury. Crit Care Med. 2014;42(3):e189–99. doi:https://doi.org/10.1097/CCM.0000000000000097.
- Oo ML, Chang S-H, Thangada S, Wu M-T, Rezaul K, Blaho V, Hwang S-I, Han DK, Hla T. Engagement of S1P1-degradative mechanisms leads to vascular leak in mice. J Clin Invest. 2011;121(6):2290–2300. doi:https://doi.org/10.1172/JCI45403.
- Camp SM, Bittman R, Chiang ET, Moreno-Vinasco L, Mirzapoiazova T, Sammani S, Lu X, Sun C, Harbeck M, Roe M, et al. Synthetic analogs of FTY720 [2-amino-2-(2-[4-octylphenyl]ethyl)-1,3-propanediol] differentially regulate pulmonary vascular permeability in vivo and in vitro. J Pharmacol Exp Ther. 2009;331(1):54–64. doi:https://doi.org/10.1124/jpet.109.153544.
- Camp SM, Chiang ET, Sun C, Usatyuk PV, Bittman R, Natarajan V, Garcia JGN, Dudek SM. Pulmonary endothelial cell barrier enhancement by novel FTY720 analogs: methoxy-FTY720, fluoro-FTY720, and beta-glucuronide-FTY720. Chem Phys Lipids. 2015;191:16–24. doi:https://doi.org/10.1016/j.chemphyslip.2015.08.004.
- Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB, Mehta D, et al. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res. 2008;103(10):1164–1172. doi:https://doi.org/10.1161/01.RES.0000338501.84810.51.
- Li X, Stankovic M, Bonder CS, Hahn CN, Parsons M, Pitson SM, Xia P, Proia RL, Vadas MA, Gamble JR, et al. Basal and angiopoietin-1-mediated endothelial permeability is regulated by sphingosine kinase-1. Blood. 2008;111(7):3489–3497. doi:https://doi.org/10.1182/blood-2007-05-092148.
- Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang W-C, Hait N, Allegood J, Price M, Avni D, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23(1):107–120. doi:https://doi.org/10.1016/j.ccr.2012.11.013.
- Kharel Y, Raje M, Gao M, Gellett AM, Tomsig JL, Lynch KR, Santos W. Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem J. 2012;447(1):149–157. doi:https://doi.org/10.1042/BJ20120609.
- Dimasi DP, Pitson SM, Bonder CS. Examining the Role of Sphingosine Kinase-2 in the Regulation of Endothelial Cell Barrier Integrity. Microcirculation. 2016;23(3):248–265. doi:https://doi.org/10.1111/micc.12271.
- Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K, Ishida J, et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest. 2012;122(4):1416–1426. doi:https://doi.org/10.1172/JCI60746.
- Fu P, Ebenezer DL, Berdyshev EV, Bronova IA, Shaaya M, Harijith A, Natarajan V. Role of Sphingosine Kinase 1 and S1P Transporter Spns2 in HGF-mediated Lamellipodia Formation in Lung Endothelium. J Biol Chem. 2016;291(53):27187–27203. doi:https://doi.org/10.1074/jbc.M116.758946.
- Tatin F, Renaud-Gabardos E, Godet AC, Hantelys F, Pujol F, Morfoisse F, Calise D, Viars F, Valet P, Masri B, et al. Apelin modulates pathological remodeling of lymphatic endothelium after myocardial infarction. JCI Insight. 2017;2(12):2. doi:https://doi.org/10.1172/jci.insight.93887.
- Spiegel S, Maczis MA, Maceyka M, Milstien S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J Lipid Res. 2019;60(3):484–489. doi:https://doi.org/10.1194/jlr.S091959.
- Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM, et al. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119(3):629–641. doi:https://doi.org/10.1083/jcb.119.3.629.
- Ephstein Y, Singleton PA, Chen W, Wang L, Salgia R, Kanteti P, Dudek SM, Garcia JGN, Jacobson JR. Critical Role of S1PR1 and Integrin β4 in HGF/c-Met-mediated Increases in Vascular Integrity. J Biol Chem. 2013;288(4):2191–2200. doi:https://doi.org/10.1074/jbc.M112.404780.
- Duan H-F, Wu C-T, Lu Y, Wang H, Liu H-J, Zhang Q-W, Jia -X-X, Lu -Z-Z, Wang L-S. Sphingosine kinase activation regulates hepatocyte growth factor induced migration of endothelial cells. Exp Cell Res. 2004;298(2):593–601. doi:https://doi.org/10.1016/j.yexcr.2004.04.049.
- Fu P, Shaaya M, Harijith A, Jacobson JR, Karginov A, Natarajan V. Sphingolipids Signaling in Lamellipodia Formation and Enhancement of Endothelial Barrier Function. Curr Top Membr. 2018;82:1–31.
- Kobayashi N, Kawasaki-Nishi S, Otsuka M, Hisano Y, Yamaguchi A, Nishi T. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci Rep. 2018;8(1):4969. doi:https://doi.org/10.1038/s41598-018-23300-x.
- Winkler MS, Martz KB, Nierhaus A, Daum G, Schwedhelm E, Kluge S, Gräler MH. Loss of sphingosine 1-phosphate (S1P) in septic shock is predominantly caused by decreased levels of high-density lipoproteins (HDL). J Intensive Care. 2019;7(1):23. doi:https://doi.org/10.1186/s40560-019-0376-2.
- Wilkerson BA, Grass GD, Wing SB, Argraves WS, Argraves KM. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: high density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J Biol Chem. 2012;287(53):44645–44653. doi:https://doi.org/10.1074/jbc.M112.423426.
- Wilkerson BA, Argraves KM. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim Biophys Acta. 2014;1841(10):1403–1412. doi:https://doi.org/10.1016/j.bbalip.2014.06.012.
- Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108(23):9613–9618. doi:https://doi.org/10.1073/pnas.1103187108.
- Bisgaard LS, Christoffersen C. Apolipoprotein M/sphingosine-1-phosphate: novel effects on lipids, inflammation and kidney biology. Curr Opin Lipidol. 2019;30(3):212–217. doi:https://doi.org/10.1097/MOL.0000000000000606.
- Galvani S, Sanson M, Blaho VA, Swendeman SL, Obinata H, Conger H, Dahlbäck B, Kono M, Proia RL, Smith JD, et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal. 2015;8(389):ra79. doi:https://doi.org/10.1126/scisignal.aaa2581.
- Ruiz M, Frej C, Holmer A, Guo LJ, Tran S, Dahlback B. High-Density Lipoprotein–Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler Thromb Vasc Biol. 2017;37(1):118–129. doi:https://doi.org/10.1161/ATVBAHA.116.308435.
- Zhu B, Luo G-H, Feng Y-H, Yu -M-M, Zhang J, Wei J, Yang C, Xu N, Zhang X-Y. Apolipoprotein M protects against lipopolysaccharide-induced acute lung injury via sphingosine-1-phosphate signaling. Inflammation. 2018;41(2):643–653. doi:https://doi.org/10.1007/s10753-017-0719-x.
- Fan Y, Chen J, Liu D, Li W, Wang H, Huang Y, Gao C. HDL-S1P protects endothelial function and reduces lung injury during sepsis in vivo and in vitro. Int J Biochem Cell Biol. 2020;126:105819. doi:https://doi.org/10.1016/j.biocel.2020.105819.
- Swendeman SL, Xiong Y, Cantalupo A, Yuan H, Burg N, Hisano Y, Cartier A, Liu CH, Engelbrecht E, Blaho V, et al. An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci Signal. 2017;10(492):eaal2722. doi:https://doi.org/10.1126/scisignal.aal2722.
- Burg N, Swendeman S, Worgall S, Hla T, Salmon JE. Sphingosine 1-Phosphate Receptor 1 Signaling Maintains Endothelial Cell Barrier Function and Protects Against Immune Complex-Induced Vascular Injury. Arthritis Rheumatol. 2018;70(11):1879–1889. doi:https://doi.org/10.1002/art.40558.
- Abbott NJ, Ronnback L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi:https://doi.org/10.1038/nrn1824.
- Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–1596. doi:https://doi.org/10.1038/nm.3407.
- Van Doorn R, Lopes Pinheiro MA, Kooij G, Lakeman K, van het Hof B, Van Der Pol SM, Geerts D, van Horssen J, van der Valk P, van der Kam E, et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflammation. 2012;9(1):133. doi:https://doi.org/10.1186/1742-2094-9-133.
- Li Y-J, Shi SX, Liu Q, Shi F-D, Gonzales RJ. Targeted role for sphingosine-1-phosphate receptor 1 in cerebrovascular integrity and inflammation during acute ischemic stroke. Neurosci Lett. 2020;735:135160. doi:https://doi.org/10.1016/j.neulet.2020.135160.
- Yanagida K, Liu CH, Faraco G, Galvani S, Smith HK, Burg N, Anrather J, Sanchez T, Iadecola C, Hla T, et al. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc Natl Acad Sci U S A. 2017;114(17):4531–4536. doi:https://doi.org/10.1073/pnas.1618659114.
- Spampinato SF, Merlo S, Sano Y, Kanda T, Sortino MA. Protective effect of the sphingosine-1 phosphate receptor agonist siponimod on disrupted blood brain barrier function. Biochem Pharmacol. 2021;186:114465. doi:https://doi.org/10.1016/j.bcp.2021.114465.
- Stepanovska B, Lange AI, Schwalm S, Pfeilschifter J, Coldewey SM, Huwiler A. Downregulation of S1P Lyase Improves Barrier Function in Human Cerebral Microvascular Endothelial Cells Following an Inflammatory Challenge. Int J Mol Sci. 2020;21(4):1240. doi:https://doi.org/10.3390/ijms21041240.
- Gude NM, Roberts CT, Kalionis B, King RG. Growth and function of the normal human placenta. Thromb Res. 2004;114(5–6):397–407. doi:https://doi.org/10.1016/j.thromres.2004.06.038.
- Del Gaudio I, Sreckovic I, Zardoya-Laguardia P, Bernhart E, Christoffersen C, Frank S, Marsche G, Illanes SE, Wadsack C. Circulating cord blood HDL-S1P complex preserves the integrity of the feto-placental vasculature. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(4):158632. doi:https://doi.org/10.1016/j.bbalip.2020.158632.
- Hemmings DG, Hudson NK, Halliday D, O’Hara M, Baker PN, Davidge ST, Taggart MJ. Sphingosine-1-phosphate acts via rho-associated kinase and nitric oxide to regulate human placental vascular tone. Biol Reprod. 2006;74(1):88–94. doi:https://doi.org/10.1095/biolreprod.105.043034.
- Del Gaudio I, Sasset L, Lorenzo AD, Wadsack C. Sphingolipid Signature of Human Feto-Placental Vasculature in Preeclampsia. Int J Mol Sci. 2020;21(3):1019. doi:https://doi.org/10.3390/ijms21031019.
- Dobierzewska A, Palominos M, Sanchez M, Dyhr M, Helgert K, Venegas-Araneda P, Tong S, Illanes SE. Impairment of Angiogenic Sphingosine Kinase-1/Sphingosine-1-Phosphate Receptors Pathway in Preeclampsia. PLoS One. 2016;11(6):e0157221. doi:https://doi.org/10.1371/journal.pone.0157221.
- Del Gaudio I, Hendrix S, Christoffersen C, Wadsack WC. Neonatal HDL Counteracts Placental Vascular Inflammation via S1P–S1PR1 Axis. Int J Mol Sci. 2020;21(3):789. doi:https://doi.org/10.3390/ijms21030789.