
ABSTRACT
Deficits in gastrointestinal (GI) paracellular permeability has been implicated in etiology of Inflammatory Bowel Disease (IBD), and E-cadherin, a key component of the epithelial junctional complex, has been implicated in both barrier function and IBD. We have previously described antibodies against E-cadherin that activate cell adhesion, and in this study, we show that they increase transepithelial electrical resistance in epithelial cell monolayers in vitro. We therefore tested the hypothesis that adhesion activating E-cadherin mAbs will enhance epithelial barrier function in vivo and limit progression of inflammation in IBD. Activating mAbs to mouse E-cadherin were tested in different mouse models of IBD including the IL10-/- and adoptive T cell transfer models of colitis. Previously established histological and biomarker measures of inflammation were evaluated to monitor disease progression. Mouse E-cadherin activating mAb treatment reduced total colitis score, individual histological measures of inflammation, and other hallmarks of inflammation compared to control treatment. Activating mAbs also reduced the fecal accumulation lipocalin2 and albumin content, consistent with enhanced barrier function. Therefore, E-cadherin activation could be a potential strategy for limiting inflammation in UC.
Introduction
Epithelial barrier function plays an important role in gastrointestinal (GI) mucosal immunity and inflammation, and deficits in GI epithelial barrier function is etiologically linked with Inflammatory Bowel Disease (IBD).Citation1–6 An intact GI barrier prevents leakage of antigens derived from intestinal flora and foodstuffs from the intestinal lumen into the interstitial space and the lamina propria where immune cells reside. It also regulates the migration of polymorphonuclear leukocytes (PMN) across the epithelium of the crypts into the lumen, a common occurrence in IBD that is associated with disease symptoms.Citation7–9 Moreover, it regulates the functions of mucosal dendritic cells (DCs) and T-cells through their interactions with epithelial cells.Citation2,Citation10–15 Increased intestinal permeability is even observed in asymptomatic relatives of patients with IBD, suggesting that there may be a genetic component to the barrier defects.Citation16–18 Increased paracellular permeability has also been strongly implicated in the development of IBD in animal models.,Citation1,Citation2,Citation19,Citation20 including SAMP1/YitFc (SAMP) mice, which develop ileitis spontaneously,Citation1 and mice lacking the Interleukin-10 (IL-10) geneCitation19 that develops colitis. The genetic basis for ileitis in the SAMP1 model is not fully known.Citation21–23 Several inflammatory mediators, including TNFα and INFγ are known to increase intestinal epithelial permeability, and both early and late effects on permeability could also be important for promoting disease.Citation24–27 Inflammatory mediators are induced by naïve T cells transferred from healthy donor into lymphophenic mice and adoptive T cell transfer is used as a model of colitis to study the onset and progression of chronic inflammation that mimics human IBD.Citation28–30 Hence, IL10-/- and adoptive T cell transfer are commonly studied mouse models for barrier dysfunction investigation in IBD research.Citation10,Citation11,Citation14,Citation28,Citation30–42
The E-cadherin cell-cell adhesion protein in complex with cytoplasmic catenins is a particularly important component of the epithelial barrier that regulates paracellular permeability. It is the principle adhesive component of the adherens junction and early studies demonstrated its role in facilitating the formation of tight junctionsCitation43 and organizing the entire epithelial junction complex.Citation44 The junctional complex, in association with the contractile actomyosin cytoskeleton has been shown to regulate epithelial paracellular permeability in response to inflammatory mediators.Citation24,Citation45 MLCK1 has been reported to down regulate claudin in response to TNFα in IBD and the therapeutic potential of selective MLCK1 inhibitor Divertin was investigated recently in the IL10-/- and adoptive T cell transfer in RagN12 mice models of IBD.Citation31
Evidence for a role for E-cadherin in the development of IBD includes the GWA studies implicating the E-cadherin gene in UC,Citation3,Citation17,Citation46,Citation47 as well as the finding that the expression of a dominant interfering cadherin mutant in the intestinal epithelium caused IBD in mice.Citation48 Moreover, the cytoplasmic E-cadherin-associated protein p120-catenin has also been functionally implicated in inflammatory disease and IBD. Mice with a tissue specific knockout of p120-catenin in the intestine develop an inflammatory response and IBD-like pathology.Citation49,Citation50 P120-catenin controls both E-cadherin levelsCitation49 as well as the allosteric regulation of E-cadherin adhesive function at the cell surface.Citation51,Citation52
We have developed new insights about the regulation of E-cadherin adhesive function with significant implications for understanding and altering its role in GI barrier function and IBD. Regulation of its adhesive activity occurs at the cell surface in response to various stimuli independent of any changes in its surface levels and may involve changes in its conformation. We have previously identified monoclonal antibodies (mAbs) that bind to E-cadherin and activate adhesion in a variety of epithelial cells.Citation51 We also reported that adhesion activation inhibited tumor metastasis in various in vivo models of metastatic breast cancer.Citation53 In the current study, E-Cadherin activating Fabs rescued barrier function challenge with respiratory syncytial virus in vitro.
In IBD E-cadherin levels have sometimes been reported to decrease near regions of neutrophil migration, but often remains expressed in most of the intestinal tissue.Citation54–56 We hypothesize that the E-cadherin still present may be subject to cell surface regulation in pathophysiological conditions to allow immune cell transmigration or other changes that increase permeability. Therefore E-cadherin activating mAbs may work in these conditions to enhance adhesion and cell junctions. Therefore, we conducted experiments to investigate whether activating E-cadherin mAbs strengthen epithelial barrier function and limit progression of inflammation in mouse models of IBD.
Materials and methods
Cell culture
16HBE14o- cells (Human broncho epithelial cell line) was a generous gift from Dr. Dieter Gruenert lab (California Pacific Medical Center Research Institute, SF, CA) to Dr. Teal Hallstrand lab which were then received by us. T-25 cm2 flasks were coated with fibronectin-collagen solution. 16HBE14o- cells were cultured and differentiated on FNC coated transwells filter. FNC solution was prepared by adding 1 ml Rat tail Type I Collagen (2.9 mg/ml), 250 μl of Fibronectin (1 mg/ml), 2.5 ml BSA (1 mg/ml) in 21.25 ml of D-PBS. 25 ml of FNC solution was then filter sterilized using 0.2 μm filter. 16HBE14o- cells (between passage 12 and 16) were grown up to 80% confluency in MEM (minimum essential medium with Earl’s salt) supplemented with 10% heat inactivated FBS, 2 mM L-glutamine, 1% wt/vol Penicillin/Streptomycin, 1 mM sodium pyruvate, and 0.015 mol/L HEPES. Cells were sub-cultured at 80% confluency. 16HBE14o- cells were grown on 0.4 μm pore size filters (Transwell; Costar;) for transepithelial electrical resistance (TEER) studies and immunofluorescence staining, respectively.
C2BBe1 (clone of Caco2) cells were purchased from ATCC and cultured in T-75 cm2 flasks in DMEM medium supplemented with 0.01 mg/ml human transferrin (sigma-aldrich T5391), 0.015 mol/L HEPES, 10% heat inactivated FBS, and 1% wt/vol Penicillin/Streptomycin.
Trans epithelial electrical resistance (TEER) assay
16HBE14o- cells were plated at a sub confluent density of 1.5 × 105 cells/cmCitation2 on FNC coated inserts (PET membrane, 0.4 μm pore size, 6.5 mm insert, costar 3470) and allowed to reach confluency at liquid-liquid interface culture for 7 days. The medium was changed the next day and followed by every other day up to 7 days. At day 7, the basal TEER across confluent monolayers on insert was measured using EVOM 2 epithelial volt-ohmmeter (World Precision Instruments, Sarasota, Fla) and STX2 chopstick electrodes according to device manufacturer’s protocol. The confluent 16HBE14o- monolayer baseline unit area resistance (TEER) on indicated membrane inserts at 7 day that ranged between 550–1000 Ω x cm2 were used for challenge experiments. Cells grown on inserts were then either pre-treated with control or with activating human E-Cad Fabs (3 ug/ml) on both apical and basal sides for 4 hours and subsequently challenged either with virus or with chemical agents for testing barrier function at 6, 24, and 48 hours. E-Cad Fabs were added both in apical and basal chamber at every 24 hours. The resistance of cell free FNC coated insert was subtracted from each experimental values and data represented either as absolute values (Ω x cm2) or as changes relative to the corresponding control groups. Cell viability was tested during sub-culture by trypan blue exclusion test.
Caco2 cells (1 x 105 cells/cmCitation2) were grown as monolayers on rat tail collagen coated inserts (PET membrane, 0.4 μm pore size, 6.5 mm insert, costar 3470) and allowed to reach confluency at liquid-liquid interface culture for up to 15–20 days to be fully differentiated according to previously established protocol.Citation31 The medium was changed the next day and followed by in every two days for up to 15–20 days. Baseline TEER was measured in fully differentiated monolayers on inserts which were then either treated with control or with activating human E-Cad Fabs (3 ug/ml) on both apical and basal sides for 4, 12, and 16 hours, and TEER was measured with STX2 chopstick electrodes according to device manufacturer’s protocol.
Immunofluorescence staining and cytotoxicity assay:
After the indicated treatment cells were fixed with ice cold methanol for 10 minutes followed by PBS wash, 5% BSA blocking, and overnight staining with primary and corresponding secondary antibody staining for 1 hr at room temperature for junctional proteins. The experimental cell supernatant from TEER assay was used for testing cytotoxicity at indicated time points by measuring release of cytosolic enzyme lactate dehydrogenase (LDH) according to the manufacturer’s protocol.
Respiratory syncytial virus
Human respiratory syncytial virus (RSV strain L19) virus stock (4.7 x 109 pfu/ml) that was propagated in Hep2, PEG concentrated, and quantified by plaque assay in Vero cells was a generous gift from Jason Scott Debley lab, Seattle Children’s Research Institute, WA. RSVL19 stock was used to infect apically on polarized monolayer of 16HBE14o- cells.Citation57
Recombinant E-cadherin antibodies and Fabs
E-cadherin activating and control monoclonal antibodies against mouse and human E-cadherin were established previously.Citation58,Citation51 Briefly, variable regions of heavy and light chain sequence of rabbit activating antibodies to mouse E-cadherin were sequenced by GenScript and cloned into the mouse IgG1 constant region backbone. Recombinant mAbs were expressed in our laboratory in Expi-CHO cells, affinity purified using protein G column and mAb r56-4 was tested for functional activity using an activation assay with E-cadherin expressing Colo205 cellsCitation53 . E-cadherin control mAb r19.1–10 binds to E-cadherin but does not activate. Recombinant Fab fragments for human E-cadherin activating mAb (19A11) or control mAb (46H7) were produced, purified, and screened using similar protocol as described previously.Citation51
Animals
All animal experiments were conducted in accordance with SCRI’s (Seattle Children’s Research Institute) IACUC (Institutional animal care and use committee) compliance. Mice experimental procedures and protocols were reviewed and approved by SCRI IACUC (protocol #0413) and performed in compliance with SCRI’s ethical regulation for animal care and use. Mice were housed and bred under specific pathogen free conditions at SCRI. Mice were obtained commercially either from Jackson laboratory or Taconic biosciences and mated (as required) according to the vendor’s specification.
B6.129P2-IL10tm1Cgn/J (The Jackson laboratory strain #002251) male and female mice were used as IL10KO homozygous breeders in house to generate experimental cohort. Mice were weighed at weaning. Mice were treated twice weekly either with E-Cad activating (r56-4) or control (r19.1–10) mAb (5 mg/kg of body weight) in saline via intraperitoneal route between 8.5 and13.5 weeks of age. Mice were euthanized and colonic tissues were harvested for histology assessment at treatment endpoint.
B6.Cg-Prkdcscid/SzJ (The Jackson laboratory strain #001913) male and female mice were used as immunocompromised homozygous breeders in house to generate experimental cohort. B6.Cg-Prkscscid/SzJ males were used for this experiment. Mice were treated twice weekly either with E-Cad activating (r56-4) or control (r19.1–10) mAb (5 mg/kg of body weight) in saline via intraperitoneal route between 5 and 7 weeks of age prior receiving T cell transfer. Adoptive transfer model of colitis was then established in 7 weeks old B6.Cg-Prkscscid/SzJ male mice by tail vain injection of 400,000 CD4+CD45RBhi T cells isolated from age matched male donor C57BL6/J (The Jackson laboratory strain #000664). CD4+CD45RBhi T cells were prepared for injection by a two-step procedure i.e., CD4+ T cells were purified first from immune sufficient age matched healthy donor male splenocytes via negative isolation protocol using EasysepTM mouse CD4+ T cell isolation kit (STEMCELL technologies) according to manufacturer’s protocol and subsequently purified cells were labeled with surface marker specific antibodies namely, APC anti-mouse CD45RB and PE-Cy7 anti-rat CD4 in FACS buffer for sorting labeled populations into CD4+CD45RBhi and CD4+CD45RBlo sub-populations (supplement 1a). CD4+CD45RBhi T cell transfer recipient mice were continued to receive either E-Cad activating (r56-4) or control (r19.1–10) mAbs at indicated dose and frequency and euthanized after 6 weeks of post T cell transfer. Colonic tissues were harvested for histology assessment at treatment endpoint.
B6.129S6-Rag2tmCitation1Fwa N12 (Taconic Biosciences strain #RAGN12-M) 4 weeks old male mice and corresponding control C57BL6/NTac (Taconic Biosciences strain #B6-M) were commercially obtained as experimental cohort. Mice were allowed to adjust in house SPF facility for 2 weeks before starting any experimental procedure. Mice were treated twice weekly either with E-Cad activating (r56-4) or control (r19.1–10) mAb at 5 mg/kg of body weight and a single dose 50 mg/kg of body weight in saline via intraperitoneal route between 6 and 8 weeks of age prior receiving T cell transfer. Another experimental mouse cohort were treated with anti-rat-IL12 antibody once weekly at 12.5 mg/kg body weight injected via i.p. route as positive treatment control of colitis prior receiving T cell transfer. Adoptive transfer model of colitis was then established in 8 weeks old B6.129S6-Rag2tmCitation1Fwa N12 male mice by intraperitoneal injection of 400,000 CD4+CD45RBhi T cells isolated from age matched male donor C57BL6/NTac (Taconic Biosciences strain #B6-M). CD4+CD45RBhi T cells were prepared for injection by a two-step procedure same as mentioned above for SCID model (Supplemental figure 2a). CD4+CD45RBhi T cell transfer recipient mice were continued to receive either E-Cad activating (r56-4) or control (r19.1–10) mAbs or anti-rat-IL12 antibody at indicated dose and frequency and euthanized after 7 weeks of post T cell transfer. Colonic tissues were harvested for histology assessment at treatment endpoint.
Histology
At experimental endpoint, colonic swiss rolls were prepared according to established protocolsCitation59 and cassettes containing tissue rolls were fixed overnight either in 10% formalin for colons. Samples were processed at CDBRM (center for developmental biology and regenerative medicine) tissue processing core and paraffin embedded. Tissue embedded paraffin blocks were then submitted to HistoTox Labs on a fee-for-service basis for experimental histopathology analysis of IBD progression in experimental models of colitis. HistoTox Labs conducted tissue sectioning, Hematoxylin and Eosin (H&E) staining, imaging, and scoring of inflammatory bowel disease progression according to acceptance criteria established a-priori (as described in the supplemental table). Evaluation of staining was conducted by a board-certified veterinary pathologist at HistoTox Labs.
Lipocalin2 ELISA
Stools and tissue extracts from experimental animals were evaluated for LCN2/NGAL levels using R & D duo set ELISA kit (DY1857) according to manufacturer’s protocol. Briefly, stool samples were prepared by diluting equal number of pellets or wt/vol liquid feces with 500 ul of 0.1% Tween20 in PBS followed by homogenization using lysing matrix E tubes for 40 secs. Stool extracts were then centrifuged for 10 minutes at 12,000 rpm at 4° C. Supernatants were used for ELISA at 1:200 minimum required dilution. Tissue extracts were prepared using Invitrogen Novex tissue extraction reagent according to the kit protocol. Briefly, homogenization buffer with freshly added protease inhibitor cocktail was used proportionately to the tissue weight and homogenized using tissue homogenizer followed by centrifugation at 10,000 rpm for 5 minutes to remove the debris. The supernatant was used for ELISA.
Western blotting (WB)
Stool extract prepared as indicated above and diluted in 2X laemmli sample buffer and heat-denatured at 95°C for 5 minutes for loading onto 4–20% gradient SDS-PAGE. Equal volume of samples was loaded and transferred on to methanol activated PVDF membrane to transfer the protein using Bio-Rad’s semi-dry transfer unit. Western blot running and transfer buffers were commercially purchased, and WB performed according to manufacturer’s protocol. Membrane was then blocked with 5% NFDM in TBST for 1 hour and incubated overnight with anti-albumin antibody (ab207327, Abcam) at 1:1000 concentration in 5% NFDM in TBST followed by three times (5 minutes each) washing with 1X TBST next morning and subsequent 1-hour incubation in 1:5000 goat anti-rabbit HRP conjugated secondary antibody. Membrane was then washed again three times (5 minutes each) washing with 1X TBST followed by substrate incubation and capture of bands using Bio-Rad’s ChemiDoc imaging unit.
Statistics
Statistical analyses were performed using GraphPad Prism9 software (Graphpad Software, Inc., CA, USA). Statistically significant differences between two groups were determined using the nonparametric Mann-Whitney U test or ANOVA as described in the result section. P < .05 was considered significantly different as described in the figure legends.
RESULTS
Human E-cadherin activating antibody Fab increases barrier function in vitro in epithelial cells
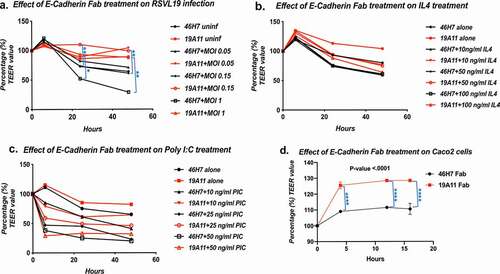
Our previous findings demonstrated that human E-cadherin activating antibody increased cell:cell adhesion in several different epithelial cell lines in vitro.Citation51,Citation53,Citation58,Citation60 Therefore, to determine whether E-cadherin activation at epithelial cell-cell junctions can counteract a loss in barrier function induced by various agents, 16HBE14o- cells (Human broncho epithelial cell line) were cultured, differentiated, and epithelial permeability was assessed using a TEER assay. 16HBE14o- cells were pre-treated either with human E-cadherin activating (19A11) or neutral E-cadherin (46H7) Fabs and subsequently challenged with three different agents which have been previously identified to induce loss in trans epithelial electrical resistance (TEER) in this cell line.Citation57,Citation61,Citation62 Infection with RSV-L19 reduced TEER in 16HBE14o- cells at multiplicity of infection (MOI) of 0.15 and 1 in a time dependent manner. 19A11 activating Fab treatment reduced the loss of TEER in 16HBE14o- cells compared to control 46H7 Fab treatment after 24 hours (p-values .0092 and .0411 respectively) and 48 hours (p-values .0078 and .0021 respectively) post infection, which is statistically significant when analyzed by 2-way ANOVA followed by Sidak’s multiple comparison test (. values indicated in the figure legend). Poly I:C (viral RNA mimic) and IL-4 have also been reported to reduce TEER in 16HBE14o- cells.Citation62 Although we observed that Poly I:C and IL-4 treatment reduced TEER in a dose and time dependent manner, in no condition did E-cadherin activating Fab pre-treatment slow the reduction in TEER ( and ). The unit area TEER values in Ω x cm2 from these experiments are provided in the Supplemental Figure 7a – 7 c. Because E-cadherin activating antibody treatment did not increase TEER compared to untreated 16HBE14o- cells (data not shown), these finding indicate that E-cadherin activation selectively enhanced barrier function against some inflammatory stimuli.
Figure 1. Effect of human E-cadherin activation in epithelial barrier function in vitro.(a-c) Measurement of transepithelial electrical resistance (TEER) in 16HBE14O- cells pre-treated with either with human E-cadherin activating Fab 19A11 or with control neutral Fab treatment followed by (a) RSV challenge at different MOI under indicated time points of post infection, (b) IL4 or (c) poly I:C challenge. Statistical significance was analyzed by ANOVA followed by Sidak’s multiple comparisons test of group mean SD ** = p-value <.005 and * = p-value <.05 (d) Differentiated Caco2 cells were treated either with E-cadherin activating (19A11) or neutral (46H7) Fabs and basal TEER was monitored for indicated time points and TEER values over time was analyzed. ANOVA with Sidak’s multiple comparisons test performed. (group mean SD, *** = p values <.0001 between two groups reported)
We also examined whether the loss in TEER in response to treatments was due to gross junctional disruption or even cell death. In RSVL-19 infected 16HBE14o- cells we did not observe changes in junctional staining of E-cadherin or the tight junction proteins ZO1 as examined by immunostaining (Supplemental Figure 1.a and b). This suggests a more subtle molecular change in cell junctions, both in response to infection and to activating Fab treatment. An LDH release assay revealed no statistically significant cell cytotoxicity due to any of the challenges (Supplemental figure 1. c, d, and e), indicating that the reduction in TEER was not due to gross loss of cells due to cell death.
We also found that E-cadherin activation increase TEER in a cultured intestinal epithelial cell line. A subclone of the Caco2 cell line (human colorectal adenocarcinoma) C2BBe1 was cultured, differentiated and examine by TEER measurements as described in materials and methods. In contrast to the 16HBE14o- cells, E-cadherin activating (19A11) Fabs increased TEER over time in otherwise unchallenged Caco2 compared to 46H7 Fab treated cells. The data were statistically analyzed by 2-way ANOVA followed by Sidak’s multiple comparison test (. values indicated in the figure legend). The unit area TEER values in Ω x cm2 from these experiments are provided in the Supplemental Figure 7d. This suggests that the resting C2BBe1 Caco2 line already has some downregulation or deficit in cell junctions that can be countered by E-cadherin activation under the culture conditions of this experiment.
Mouse E-cadherin activating antibody treatment reduced inflammation progression in spontaneous IL10-/- model of ulcerative colitis
To test the physiological significance of epithelial E-cadherin activation in barrier function in vivo, we studied their effects in mouse models of inflammatory bowel disease (IBD) that implicate intestinal barrier dysfunction. We chose the IL10-/- model because barrier dysfunction has been implicated in the diseaseCitation19,Citation63–65 and because of the spontaneous nature of disease progression may permit examination of whether activating mAbs prevent disease onset. We used the IL10-/- B6.129P2 strain which is the least aggressive model of chronic ulcerative colitis compared to C3H/HeJBir or 129/Sv background.Citation40 The IL10-/- model is also well known to be dependent on intestinal microbiota for disease progressionCitation65 and therefore to minimize any microbial bias due to change in environment during transportation, we generated the IL10-/- experimental cohort in house at SCRI specific pathogen free vivarium facility. B6.129P2-IL10tm1Cgn/J mice cohorts (we will refer the strain as IL10-/- from here on) were bred, weighed and treated by IP injection biweekly with either with mouse E-cadherin activating (r56-4) or neutral (r19.1–10) antibodies. Both sexes were included in the study and male cohorts had statistically significant higher body weight regardless of antibody treatment compared to female cohorts (). Over time, E-cadherin activating antibody (r56-4) treated male and female cohorts had statistically significant higher body weight compared to neutral antibody (r19.1–10) treated male and female cohorts respectively (analyzed by nonparametric Mann-Whitney U test, p-value <0.0001), which indicates that overall health status of r56-4 treated mice cohorts were better than r19.1–10 treated cohort.
Figure 2. Effect of mouse E-cadherin activating antibody in IL10KO BL6 model of ulcerative colitis.(a) Measurement of body weight in IL10-/- mice cohorts (n = 8–10 mice per antibody treatment group) receiving either E-cadherin activating r56- 4 or control neutral r19.1–10 antibody (group mean SEM, Mann-Whitney test p-values <0.0001); (b) Representative microscopic images of H&E stained colonic swiss roll section (scale bar 200 μm). Quantification of inflammation parameters presented for (c) inflammation, (d) sum of histopathology, and (e) sum of colitis (sum of all parameters, group mean SEM); Mann-Whitney test for significance performed; (f) Comparative analysis of colonic length measurement between treatment cohorts at treatment endpoint and (g) Lcn2 content in stool analysis by ELISA. (group mean SEM, Mann-Whitney test p-values reported in the figure)
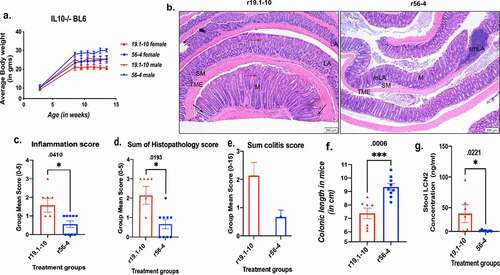
Experimental histopathology evaluation of disease progression was independently conducted by the commercial service HistoTox at the treatment end point of age 13.5 weeks. Examples of histological sections are shown in and histology raw data analysis graphs from HistoTox are reported in supplemental Figure 2. shows that IL10-/- mice had expected histologic lesions of the intestinal tract, including inflammation, hyperplasia, epithelial damage/gland loss, and edema (Supplemental Table 1). Mid and distal colons were evaluated, and inflammation was characterized by infiltration of the mucosa and submucosa by lymphocytes and macrophages with fewer numbers of eosinophils and neutrophils. Epithelial hyperplasia was characterized by elongation of colonic glands, epithelial cell basophilia, and increased numbers of epithelial mitotic figures. Regions of hyperplasia occasionally formed polypoid-like projections of the mucosa with branching and fusion of crypts/glands. Epithelial damage included epithelial infiltration of inflammatory cells, crypt loss, and complete loss of colonic glands; erosions were not observed in this study. Edema was characterized by expansion of the lamina propria and/or submucosa by clear space or pale eosinophilic fluid, variably accompanied by dilation of lymphatic vessels.
Lesion severity was generally minimal to mild in this model, as observed in both the control neutral r19.1–10 treatment group (Group 1) and the r56-4 activating mAb treatment group (Group 2). Nonetheless, average lesion severity scores were lower in the r56-4 activating mAb treatment group (Group 2) in comparison to the r19.1–10 treatment group (Group 1) for sum colitis scores (), mucosal hyperplasia, inflammation, and gland loss scores (Supplemental Figure 2a), edema extent (Supplemental Figure 2b), and neutrophilic infiltration (Supplemental Figure 2c). Significance tested on histologic parameters such as inflammation and sum histopathology score ( and ) were substantially lower in r56-4 activating mAb treated IL10-/- mice compared to r19.1–10 treated cohort. Fecal lipocalin2 (LCN2) was used as a noninvasive biomarker associated with barrier dysfunction, neutrophil migration, and inflammation in IBD.Citation66,Citation67 The level of fecal LCN2 was also significantly reduced in activating mAb r56-4 treated IL10-/- mice when compared to its corresponding control group (). In addition, activating mAb r56-4 treated IL10-/- mice cohort had significantly higher colonic length compared to r19.1–10 treated cohort () as evaluated at treatment end point (analyzed by nonparametric Mann-Whitney U test, P values indicated in the figure legend). These results show that treatment with E-cadherin activating antibody significantly reduced IBD progression in IL10-/- spontaneous model of ulcerative colitis.
In addition to the appearance of LCN2 in the stool, we also measured albumin protein content in mice stool extract (by western blotting) as a hallmark of barrier dysfunction (supplemental figure 5).Citation68 Activating mAb r56-4 treatment (lane 7) in IL10-/- mice reduced stool albumin in protein level compared to r19.1–10 treatment (lane 6) potentially by enhancing barrier function. This suggests that E-cadherin activating antibody may have reduced IBD progression by enhancing barrier function in IL10-/- mice.
Mouse E-cadherin activating antibody treatment reduces progression of experimentally induced inflammatory bowel disease, adoptive T cell transfer model of colitis
Adoptive T cell transfer induces a progressive and chronic experimental model of colitis by direct transfer of isolated naive T-cells from syngeneic WT donor into the age and sex matched recipient lymphophenic animals. This allows for better control of disease initiation and progression and timing of antibody treatment before and during onset of inflammation. Two different immunocompromised strains, SCID and RAG were used as T cell recipients from corresponding age and sex matched healthy donor mice strains.
B6.Cg-Prkscscid/SzJ mice (referred to as B6.SCID from here on) were bred, weighed and treated with either E-cadherin activating (r56-4) or neutral (r19.1–10) antibodies, and then followed by adoptive transfer of CD45RBhi T-cells with continuing antibody treatment. The control for disease induction was CD45RBlow T-cells, which do not cause inflammationCitation28 (, b)). Sorting of these cell populations by FACS is shown in supplemental Figure 3a. The CD45RBhi T-cells caused significant weight loss in control neutral r19.1–10 mAb treated mice compared to those receiving the CD45RBlow T-cells, and treatment with activating mAb r56-4 had significantly higher body weight compared to r19.1–10 treated cohort, almost the same as the CD45RBlow group (Supplemental Figure 3b).
Figure 3. Effect of mouse E-cadherin activating antibody in adoptive T cell transfer SCID model of ulcerative colitis.(a) Representative microscopic images of H&E stained colonic swiss roll section (scale bar 100 μm); (b) Measurement of colonic length in SCID mice cohorts (n = 3– 5 mice per treatment group, group mean SEM, ANOVA with Tukey’s multiple comparisons test, p- values <0.05); Quantification of inflammation parameters presented for (c) hyperplasia, (d) inflammation, (e) gland damage, and (f) sum of histopathology scores (group mean SEM, Mann-Whitney p-values for significance); (g and h)) Lcn2 content analyses in serum and stool by ELISA. (group mean SEM, Mann-Whitney test p-values reported)
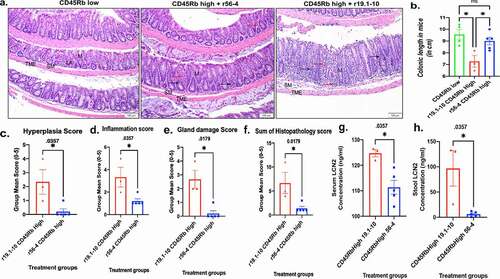
At treatment end point, experimental histo-pathology evaluation of disease progression was independently conducted by HistoTox. Examples of histological sections are shown in and histology raw data analysis graphs from HistoTox is reported in supplementary Figure 4. Mid and distal colons were evaluated and demonstrates that transfer of CD45RBhi T-cells from donor mice to immunocompromised B6.SCID mice produced typical histologic lesions for the adoptive transfer model including inflammation of the mucosa/submucosa, crypt damage, erosions, edema, and epithelial hyperplasia (Supplemental Table 1). These lesions were not observed in the mice receiving the CD45RBlow T-cells. Mid and distal colons were evaluated, and inflammation was characterized by infiltration of neutrophils, lymphocytes, macrophages, and occasional multinucleated giant cells into the mucosa and submucosa. Crypt damage ranged from infiltration of lymphocytes into crypts with distortion of crypt profiles, crypt necrosis or abscessation, or complete loss of colonic glands. Erosions were characterized by necrosis or loss of surface epithelium superficial to the muscularis mucosae. Edema was characterized by expansion of the lamina propria and/or submucosa by clear space or pale eosinophilic fluid, variably accompanied by dilation of lymphatic vessels. Epithelial hyperplasia was characterized by elongation of colonic glands, epithelial cell basophilia, and increased numbers of epithelial mitotic figures. Sum colitis scores (Supplemental Figure 4c), mucosal hyperplasia, inflammation, and gland loss scores (Supplemental Figure 4a), were higher in mice treated with r19.1–10 and CD45RBhi transfer (Group 3) when compared to mice treated with activating mAb r56-4 (Group 2) or CD45Rblow transfer (Group 1); similar scores were observed in the low transfer mice (Group 1) and high transfer r56-4-treated mice (Group 2). Erosion (Supplemental Figure 4a) and edema (Supplemental Figure 4b) were only observed in the mice treated with r19.1–10 and CD45RBhi transfer (Group 3). Mean lymphoid aggregate counts (4.25, 4.20, and 1 for groups 1,2, and 3 respectively) and diameter (225, 160, and 137 μM for groups 1,2, and 3 respective) further supported the observation that activating mAb r56-4 reduced inflammation in CD45RBhi T- cell recipient mice. Statistical significance tested on hallmarks of inflammation parameters such as hyperplasia, inflammation, gland damage, sum of histopathology scores, were significantly higher in r19.1–10 treated B6.SCID mice cohort compared to activating mAb r56-4 treated cohort after 6 weeks of CD45RBhi T cell transfer (, , , ). Moreover, the noninvasive biomarker lipocalin2 (LCN2) levels in serum and stool were significantly higher in r19.1–10 treated B6.SCID mice cohort compared to activating mAb r56-4 treated cohort (, and ) and activating mAb r56-4 treated B6.SCID mice had significantly higher colonic length compared to r19.1–10 treated cohort () after 6 weeks of CD45RBhi T cell transfer. Statistical significance was analyzed by nonparametric Mann-Whitney U test (P values indicated in the figure). These results show that treatment with E-cadherin activating antibody significantly reduced inflammatory bowel disease progression in the adoptive T-cell transfer model using the B6.SCID strain.
We also measured albumin protein content in mice stool extract in T cell transfer model as a hallmark of barrier dysfunction (Supplemental figure 5). Activating mAb r56-4 treatment (lane 4) in CD45RBhi T cell recipient B6.SCID mice reduced stool albumin in protein level compared to r19.1–10 treatment (lane 3). The r56-4 treatment group had similar albumin level as the control or CD45RBlow T cell recipient B6.SCID mice (lane 2) . These results suggest that the reduction of inflammation by the E-cadherin activating antibody may have been due to rescuing barrier dysfunction.
Another lymphophenic model B6.129S6-Rag2tmCitation1Fwa N12 mice (we will refer the strain as RagN12 from here on) were treated with either mouse E-cadherin activating (r56-4), neutral (r19.1–10), or with anti-rat-IL12 antibodies followed by adoptive transfer of CD45RBhi T cells and continuing antibody treatment during the rest of the experiment. Anti-IL12 antibody was used as positive treatment control as published literature demonstrated role for the IL-12 in the initiation of intestinal inflammation caused by epithelial barrier disruption.Citation69,Citation70 By the end of the experiment the RagN12 cohort treated with neutral antibody had lower survival (57%) compared to the cohorts treated with either anti-IL12 antibody treatment (100% survival) or r56-4 activating mAb (85% survival) or animals that received no T-cell transfer (100% survival). For this reason, it was not possible to compare all the experimental groups between each other at same time point of induction in our facility. It’s possibly that this low survival compared to the other facilities was due to a change in microbiome during transfer of RAG1 mice from Taconic Biosciences and their adaptation to the SCRI vivarium affected the baseline disease severity, which is known to occur in a microbiome dependent disease like IBD.Citation71
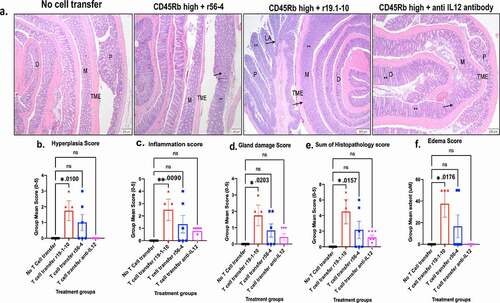
Nonetheless at the treatment end point for the surviving animals (7 weeks post T cell transfer) experimental histo-pathology evaluation of disease progression was conducted using a-priori acceptance criteria as described under the histology section in the supplemental table 1. Mid and distal colons were evaluated, and the findings presented in demonstrate that adoptive T-cell transfer in RagN12 mice produced typical histologic lesions similar to the adoptive transfer model in SCID mice as described in . However, in our experience T cell transfer in RagN12 mice strain in general developed colitis starting at 5 weeks of post CD45RBhi T cell transfer and this model also had aggressive body weight loss in mice starting at 5 weeks in our facility (Supplemental figure 6b). Colitis lesions were generally moderate overall. No lesions of colitis were observed in naïve mice (no T cell transfer, Group 1). No T cell transfer groups were the WT RagN12 mice that did not receive any antibody treatment. The experimental disease groups were compared to this control group to monitor disease progression compared to healthy colon because the survival differences between experimental disease groups made direct comparisons between them inappropriate. Hyperplasia, inflammation, glad damage score, sum of histopathology, and edema scores were increased significantly in mice that received T cell transfer and neutral antibody (r19.1–10, Group 2) as treatment. However, groups that received either anti-IL12 antibody (Group 4) or activating E-cadherin mAb (r56-4, Group 3) did not develop any significant changes with respect to the indicated pathological hallmarks of inflammation (, , , , and ). These findings demonstrate that E-cadherin activating antibody (r56-4) was partially affective in reducing inflammation in the adoptive T cell transfer model of colitis in RagN12 mice, although not as much as the anti-IL12 antibody, which is an established treatment for ulcerative colitis.
Figure 4. Effect of mouse E-cadherin activating antibody in adoptiveT cell transfer Rag-/- model of ulcerative colitis.(a) Representative microscopic images of H&E stained colonic swiss roll section (scale bar 100 μm); Quantification of inflammation parameters presented for (b) hyperplasia, (c) inflammation, (d) gland damage, (e) sum of histopathology, and (f) edema scores (n = 6–8 mice per treatment group, group mean SEM, ANOVA with Dunn’s multiple comparisons test, p-values reported in the figure)
We also measured albumin protein content in stool from these experimental mice (Supplemental figure 5). The CD45RBhi T cell recipient cohort that also received anti-IL12 treatment (lane 12) had low level of stool albumin, comparable to the control no T cell transfer group (lane 9). Although the cohort that received r56-4 (E-cadherin activating antibody) treatment had elevated levels of in the stool (lane 11), the cohort treated with r19.1–10 (neutral antibody) treatment had even higher albumin levels (lane 10). These data suggest that activating mAb was only partially effective in enhancing barrier function in this model.
Discussion
A loss of epithelial barrier function has often been proposed to contribute to the development of mucosal inflammation, especially in the lung and digestive tract. E-cadherin, as the central adhesive component of the epithelial adherens junction specifically has been implicated. However, it has been difficult to elucidate the roles of barrier function and E-cadherin specifically because of the difficulty of assessing and manipulating their functions in the context of disease states. Alterations in E-cadherin function may not be revealed by gross changes in its expression since it can be allosterically regulated at the cell surface in response to various factors. We have developed and characterized novel monoclonal antibodies that bind to E-cadherin and allosterically activate its adhesive function at the cell surface and have used them as tools to manipulate E-cadherin function and examine its role in epithelial barrier function and in the development and progression of IBD.
Previous work with our E-cadherin activating antibodies showed that they enhance E-cadherin mediated adhesion in several cell lines, stimulate epithelialization of colorectal tumor cells, and inhibit metastasis of mammary tumor cells to the lung. Citation51,Citation53,Citation58 In this paper we directly tested their ability to enhance barrier function in differentiated epithelial cell lines. We examined the human airway epithelial line 16HBE14o- because previous studies showed that its permeability, as measured by TEER, could be downregulated by inflammatory stimuli, including infection with respiratory syncytial virus. Indeed, activating Fabs to human E-cadherin prevented the loss of TEER caused by RSV infection. The Fabs did not prevent TEER loss induced by viral RNA mimic polyI:C or inflammatory cytokine IL4. It’s not clear why but these agents could either affect barrier function by another mechanism or have a stronger more persistent effect on E-cadherin function. These activating Fabs also enhanced the basal TEER of the Caco2 intestinal cell line not challenged with other agents. Together these results demonstrate that the activating mAbs not only stimulate cell adhesion but also can enhance epithelial barrier function under some conditions. Therefore, E-cadherin activation have the potential to enhance barrier function in vivo and possibly reduce mucosal inflammation.
We chose to study the role of E-cadherin cell surface regulation in IBD because of the importance of epithelial barrier function in the disease and because several lines of evidence have implicated E-cadherin in IBD.Citation6,Citation17,Citation43,Citation47,Citation72–77 We avoided common chemically induced models of IBD because they are likely to cause serious cellular damage or death, disrupting the epithelium in a way that is unlikely to be directly affected by E-cadherin regulation. Rather we used genetic and/or immune driven models that probably regulate barrier function physiologically, the IL10 gene deletion and adoptive T-cell transfer models of ulcerative colitis. For mouse models we used a different activating mAb to mouse E-cadherin which we previously showed to trigger strong adhesion and epithelialization in cell culture and to be effective in vivo in a mouse model of tumor metastasis.Citation53
IBD developed gradually and asynchronously in the IL10-/- mice in our facility. The rate and extent of IBD in the IL10-/- mice is known to be dependent on the microbiome, and therefore the facility in which they are raised, as well as the specific mouse strain. These mice developed less aggressive inflammation than the adoptive T-cell transfer model (below), but we did observe a significant reduction in the readouts of inflammation in the mice treated with E-cadherin activating mAb. The effect was not due simply to mAb binding to E-cadherin since it was compared to mice treated with control neutral E-cadherin mAb which does not activate adhesion. Thus, the reduction of inflammation is likely due to mAb increased state of adhesion at the cell surface. This suggests therefore, that changes in E-cadherin function due to environmental or inflammatory factors is responsible for the loss of barrier function. The cell culture assays using airway 16HBE14o- and intestinal Caco2 epithelial cells indicate that the activating antibodies work directly to decrease epithelial permeability, although we can’t completely rule out other effects in vivo due to allosteric activation of E-cadherin-associated signaling. Nonetheless, the decreases in serum resident albumin and granulocyte associated marker LCN2 in stool in activating mAb treated animals provide evidence that the mAbs act to decrease intestinal permeability. Thus, loss of barrier function due, at least in part, to E-cadherin misfunction contributes to the development of IBD in IL10-/- mice.
The adoptive T-cell transfer models have the advantage of inducing an acute and rapid IBD, which makes it more advantageous model to synchronize mAb treatment with onset of disease. We observed that E-cadherin activating mAbs significantly reduced inflammation in two immunodeficient mice models using either SCID mice or Rag mice as T-cell recipients (by criteria shown in the supplemental table 1). The activating mAb seems to have reduced intestinal permeability because it decreased the amount of serum resident albumin in the stool compared to control mAb treated mice. The reduction of fecal biomarker LCN2 with E-cadherin activation was significant in the SCID model. This is consistent with the effects of the activating mAb in the IL10-/- model as well as the effect of activating Fabs to human E-cadherin on epithelial permeability in cell culture. Thus, activating mAbs likely reduced IBD severity in adoptive T-cell transfer models by enhancing barrier function. In the adoptive T-cell transfer models it is unlikely that the mAbs act to reverse a barrier defect that precedes the onset of inflammation. Rather, inflammatory factors themselves are likely to cause a loss in barrier function that can be reversed temporarily by activating mAb treatment.
Decreased barrier function has been long suspected as an important factor in the development of IBD but has been difficult to test experimentally. A recent study by Jerrold Turner’s group provided some evidence that a specific barrier enhancing drug acting on the tight junction by diversion of intracellular MLCK1 could reduce inflammatory symptoms in IL10-/- and Rag-/- models of IBD.Citation31 Our findings demonstrate that stimulating E-cadherin adhesive function can also enhance barrier function and reduce inflammation in mouse models of IBD. Although these findings do not demonstrate that these E-cadherin activating antibodies can be used as induction therapy for treating disease, these proof of concept experiments suggests a potential for modulating E-cadherin adhesive function as a means to developing barrier enhancing therapeutic approaches for treating IBD.
Author Contributions
Conceived the idea: CB and BMG. Experimental method design: CB and BMG. Performed the experiments: CB. Antibody generation: LSC. Analyzed the data: CB and BMG. Manuscript figure preparation, writing, editing, and manuscript revision: CB and BMG. Supervision of antibody generation: BMG
Competing Interests:
The authors have declared that no competing interests exist.
Supplemental Material
Download Zip (3.6 MB)Acknowledgments
Animal care and procedural support: SCRI core facility. Crohn’s and Colitis Foundation for visiting research fellowship award to CB. This study was supported by grant R35GM122467 from the National Institutes of Health to BMG.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis. 2011;17:1–16. doi:https://doi.org/10.1002/ibd.21638.
- Silva MA. Intestinal dendritic cells and epithelial barrier dysfunction in crohnʼs disease. Inflamm Bowel Dis. 2009;15(3):436–453. doi:https://doi.org/10.1002/ibd.20660.
- Thompson AI, Lees CW. Genetics of ulcerative colitis. Inflamm Bowel Dis. 2011;17(3):831–848. doi:https://doi.org/10.1002/ibd.21375.
- Strober W, Fuss IJ, Blumberg RS. The Immunology of mucosal m odels of Inflammation. Annu Rev Immunol. 2002;20(1):495–549. doi:https://doi.org/10.1146/annurev.immunol.20.100301.064816.
- Meddings J. Barrier dysfunction and Crohn’s Disease. Ann N Y Acad Sci. 2006;915:333–338.
- Consortium UIG, Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334.
- Lee WY, Chin AC, Voss S, Parkos CA. In vitro neutrophil transepithelial migration. Methods in Molecular Biology (Clifton, N.J.). 2006;341:205–215. doi:https://doi.org/10.1385/1-59745-113-4:205.
- Edens HA, Levi BP, Jaye DL, Walsh S, Reaves TA, Turner JR, Nusrat A, Parkos CA. Neutrophil transepithelial migration: evidence for sequential, contact-dependent signaling events and enhanced paracellular permeability independent of transjunctional migration. J Immunol. 2002;169(1):476–486. doi:https://doi.org/10.4049/jimmunol.169.1.476.
- Nusrat A, Parkos CA, Liang TW, Carnes DK, Madara JL. Neutrophil migration across model intestinal epithelia: monolayer disruption and subsequent events in epithelial repair. Gastroenterology. 1997;113(5):1489–1500. doi:https://doi.org/10.1053/gast.1997.v113.pm9352851.
- Chieppa M, Rescigno M, Huang AY, Germain RN. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203(13):2841–2852. doi:https://doi.org/10.1084/jem.20061884.
- Del Rio M-L, Bernhardt G, Rodriguez-Barbosa J-I, Förster R. Development and functional specialization of CD103+ dendritic cells. Immunol Rev. 2010;234(1):268–281. doi:https://doi.org/10.1111/j.0105-2896.2009.00874.x.
- Rescigno M. Intestinal dendritic cells. Adv Immunol. 2010;107:109–138.
- Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi:https://doi.org/10.1038/86373.
- Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. 1994;372:190–193. doi:https://doi.org/10.1038/372190a0.
- Lamb CA, O’Byrne S, Keir ME, Butcher EC. Gut-selective integrin-targeted therapies for inflammatory bowel disease. J Crohns Colitis. 2018;12(suppl_2):S653–S68. doi:https://doi.org/10.1093/ecco-jcc/jjy060.
- Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. Increased intestinal permeability in patients with Crohn’s disease and their relatives A possible etiologic factor. Ann Intern Med. 1986;105:883–885.
- Fries W, Renda MC, Lo Presti MA, Raso A, Orlando A, Oliva L, Giofre MR, Maggio A, Mattaliano A, Macaluso A, et al. Intestinal permeability and genetic determinants in patients, first-degree relatives, and controls in a high-incidence area of Crohn’s disease in Southern Italy. Am J Gastroenterol. 2005;100(12):2730–2736.
- Hilsden RJ, Meddings JB, Sutherland LR. Intestinal permeability changes in response to acetylsalicylic acid in relatives of patients with Crohn’s disease. Gastroenterology. 1996;110(5):1395–1403. doi:https://doi.org/10.1053/gast.1996.v110.pm8613043.
- Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5:262–270.
- Olson TS, Reuter BK, Scott KG, Morris MA, Wang X-M, Hancock LN, Burcin TL, Cohn SM, Ernst PB, Cominelli F, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203(3):541–552. doi:https://doi.org/10.1084/jem.20050407.
- Komatsu Y, Shimizu Y, Yamano M, Kikuchi M, Nakamura K, Ayabe T, Aizawa T. Disease progression-associated alterations in fecal metabolites in SAMP1/YitFc mice, a Crohn’s disease model. Metabolomics. 2020;16(4):48. doi:https://doi.org/10.1007/s11306-020-01671-5.
- Kozaiwa K, Sugawara K, Smith MF Jr., Carl V, Yamschikov V, Belyea B, Mcewen SB, Moskaluk CA, Pizarro TT, Cominelli F, et al. Identification of a quantitative trait locus for ileitis in a spontaneous mouse model of Crohn’s disease: SAMP1/YitFc. Gastroenterology. 2003;125(2):477–490. doi:https://doi.org/10.1016/S0016-5085(03)00876-X.
- Matsumoto S, Okabe Y, Setoyama H, Takayama K, Ohtsuka J, Funahashi H, Imaoka A, Okada Y, Umesaki Y. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut. 1998;43(1):71–78. doi:https://doi.org/10.1136/gut.43.1.71.
- Utech M, Bruwer M, Nusrat A. Tight junctions and cell-cell interactions. Methods Mol Biol. 2006;341:185–195.
- Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–76.
- Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171(11):6164–6172. doi:https://doi.org/10.4049/jimmunol.171.11.6164.
- Nusrat A, Turner JR, Madara JL. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol. 2000;279(5):G851–7. doi:https://doi.org/10.1152/ajpgi.2000.279.5.G851.
- Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M, Price VH, Grisham MB. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol. 2009;296(2):G135–46. doi:https://doi.org/10.1152/ajpgi.90462.2008.
- Steinbach EC, Gipson GR, Sheikh SZ. Induction of murine intestinal inflammation by adoptive transfer of effector CD4+CD45RBhigh T cells into immunodeficient Mice. J Vis Exp. 2015;98. doi:https://doi.org/10.3791/52533.
- Kanai T, Kawamura T, Dohi T, Makita S, Nemoto Y, Totsuka T, Watanabe M. TH1/TH2-mediated colitis induced by adoptive transfer of CD4+CD45RBhigh T lymphocytes into nude mice. Inflamm Bowel Dis. 2006;12(2):89–99. doi:https://doi.org/10.1097/01.MIB.0000197237.21387.mL.
- Graham WV, He W, Marchiando AM, Zha J, Singh G, Li H-S, Biswas A, Ong MLDM, Jiang Z-H, Choi W, et al. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat Med. 2019;25(4):690–700. doi:https://doi.org/10.1038/s41591-019-0393-7.
- Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58(1):41–48. doi:https://doi.org/10.1136/gut.2008.150888.
- Khare V, Krnjic A, Frick A, Gmainer C, Asboth M, Jimenez K, Lang M, Baumgartner M, Evstatiev R, Gasche C. Mesalamine and azathioprine modulate junctional complexes and restore epithelial barrier function in intestinal inflammation. Sci Rep. 2019;9(1):2842. doi:https://doi.org/10.1038/s41598-019-39401-0.
- Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel J-F. Ulcerative colitis. Lancet. 2017;389(10080):1756–1770. doi:https://doi.org/10.1016/S0140-6736(16)32126-2.
- Sydora BC, Macfarlane SM, Walker JW, Dmytrash AL, Churchill TA, Doyle J, Fedorak RN. Epithelial barrier disruption allows nondisease-causing bacteria to initiate and sustain IBD in the IL-10 gene-deficient mouse. Inflamm Bowel Dis. 2007;13(8):947–954. doi:https://doi.org/10.1002/ibd.20155.
- Andrews C, McLean MH, Durum SK. Cytokine Tuning of Intestinal Epithelial Function. Front Immunol. 2018;9:1270. doi:https://doi.org/10.3389/fimmu.2018.01270.
- Chen P, Bakke D, Kolodziej L, Lodolce J, Weber CR, Boone DL, Toback FG. Antrum mucosal protein-18 peptide targets tight junctions to protect and heal barrier structure and function in models of inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(10):2393–2402. doi:https://doi.org/10.1097/MIB.0000000000000499.
- Clough JN, Omer OS, Tasker S, Lord GM, Irving PM. Regulatory T-cell therapy in Crohn’s disease: challenges and advances. Gut. 2020;69(5):942–952. doi:https://doi.org/10.1136/gutjnl-2019-319850.
- Yamada A, Arakaki R, Saito M, Tsunematsu T, Kudo Y, Ishimaru N. Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2016;22(7):2195–2205. doi:https://doi.org/10.3748/wjg.v22.i7.2195.
- Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98(4):1010–1020. doi:https://doi.org/10.1172/JCI118861.
- Suenaert P, Maerten P, Van Assche G, Van Driessche W, Geboes K, Bulteel V, Simaels J, Augustijns P, Ceuppens JL, Rutgeerts P, et al. Effects of T cell-induced colonic inflammation on epithelial barrier function†. Inflamm Bowel Dis. 2010;16(8):1322–1331. doi:https://doi.org/10.1002/ibd.21211.
- Jayawardena D, Tyagi S, Nazmi A, Olivares-Villagomez D, Dudeja PK. ion transport basis of diarrhea in a mouse model of adoptive T cell transfer colitis. Dig Dis Sci. 2020;65(6):1700–1709. doi:https://doi.org/10.1007/s10620-019-05945-4.
- Gumbiner B, Simons K. A functional assay for proteins involved in establishing an epithelial occluding barrier: identification of a uvomorulin-like polypeptide. J Cell Biol. 1986;102(2):457–468. doi:https://doi.org/10.1083/jcb.102.2.457.
- Gumbiner B, Stevenson B, Grimaldi A. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J Cell Biol. 1988;107(4):1575–1587. doi:https://doi.org/10.1083/jcb.107.4.1575.
- Nusrat A, Turner JR, Madara JL. Molecular physiology and pathophysiology of tight junctions. IV Regulation of Tight Junctions by Extracellular Stimuli: Nutrients, Cytokines, and Immune Cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G851–7.
- Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334.
- Muise AM, Walters TD, Glowacka WK, Griffiths AM, Ngan B-Y, Lan H, Xu W, Silverberg MS, Rotin D. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut. 2009;58(8):1121–1127. doi:https://doi.org/10.1136/gut.2008.175117.
- Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270(5239):1203–1207. doi:https://doi.org/10.1126/science.270.5239.1203.
- Smalley-Freed WG, Efimov A, Burnett PE, Short SP, Davis MA, Gumucio DL, Washington MK, Coffey RJ, Reynolds AB. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J Clin Invest. 2010;120(6):1824–1835. doi:https://doi.org/10.1172/JCI41414.
- Smalley-Freed WG, Efimov A, Short SP, Jia P, Zhao Z, Washington MK, Robine S, Coffey RJ, Reynolds AB, et al. Adenoma formation following limited ablation of p120-catenin in the mouse intestine. PLoS One. 2011;6(5):e19880. doi:https://doi.org/10.1371/journal.pone.0019880.
- Petrova YI, Spano MM, Gumbiner BM. Conformational epitopes at cadherin calcium-binding sites and p120-catenin phosphorylation regulate cell adhesion. Mol Biol Cell. 2012;23:2092–2108.
- Shashikanth N, Petrova YI, Park S, Chekan J, Maiden S, Spano M, Ha T, Gumbiner BM, Leckband DE. Allosteric Regulation of E-Cadherin Adhesion. J Biol Chem. 2015;290(35):21749–21761. doi:https://doi.org/10.1074/jbc.M115.657098.
- Na T-Y, Schecterson L, Mendonsa AM, Gumbiner BM. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc Natl Acad Sci U S A. 2020;117(11):5931–5937. doi:https://doi.org/10.1073/pnas.1918167117.
- Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159(6):2001–2009. doi:https://doi.org/10.1016/S0002-9440(10)63051-9.
- Demetter P, Baeten D, De Keyser F, De Vos M, Van Damme N, Verbruggen G, et al. Subclinical gut inflammation in spondyloarthropathy patients is associated with upregulation of the E-cadherin/catenin complex. Ann Rheum Dis. 2000;59(3):211–216. doi:https://doi.org/10.1136/ard.59.3.211.
- Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermuller N, Otto HF, Autschbach F. Inflammatory bowel disease is associated with changes of enterocytic junctions. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2001;281(1):G216–28. doi:https://doi.org/10.1152/ajpgi.2001.281.1.G216.
- Rezaee F, DeSando SA, Ivanov AI, Chapman TJ, Knowlden SA, Beck LA, Georas SN. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J Virol. 2013;87(20):11088–11095. doi:https://doi.org/10.1128/JVI.01573-13.
- Petrova YI, Schecterson L, Gumbiner BM. Roles for E-cadherin cell surface regulation in cancer. Mol Biol Cell. 2016;27(21):3233–3244. doi:https://doi.org/10.1091/mbc.E16-01-0058.
- Bialkowska AB, Ghaleb AM, Nandan MO, Yang VW. Improved swiss-rolling technique for intestinal tissue preparation for immunohistochemical and immunofluorescent analyses. J Vis Exp. 2016;113. doi:https://doi.org/10.3791/54161.
- Mendonsa AM, Bandyopadhyay C, Gumbiner BM. p120-catenin phosphorylation status alters E-cadherin mediated cell adhesion and ability of tumor cells to metastasize. PLoS One. 2020;15(6):e0235337. doi:https://doi.org/10.1371/journal.pone.0235337.
- Rezaee F, Meednu N, Emo JA, Saatian B, Chapman TJ, Naydenov NG, De Benedetto A, Beck LA, Ivanov AI, Georas SN, et al. Polyinosinic:polycytidylic acid induces protein kinase D–dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J Allergy Clin Immunol. 2011;128(6):1216–24 e11. doi:https://doi.org/10.1016/j.jaci.2011.08.035.
- Saatian B, Rezaee F, Desando S, Emo J, Chapman T, Knowlden S, Georas SN. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. 2013;1(2):e24333. doi:https://doi.org/10.4161/tisb.24333.
- Scheinin T, Butler DM, Salway F, Scallon B, Feldmann M. Validation of the interleukin-10 knockout mouse model of colitis: antitumour necrosis factor-antibodies suppress the progression of colitis. Clin Exp Immunol. 2003;133(1):38–43. doi:https://doi.org/10.1046/j.1365-2249.2003.02193.x.
- Kennedy RJ, Hoper M, Deodhar K, Erwin PJ, Kirk SJ, Gardiner KR. Interleukin 10-deficient colitis: new similarities to human inflammatory bowel disease. Br J Surg. 2002;87(10):1346–1351. doi:https://doi.org/10.1046/j.1365-2168.2000.01615.x.
- Keubler LM, Buettner M, Hager C, Bleich A. A multihit model: colitis lessons from the interleukin-10-deficient mouse. Inflamm Bowel Dis. 2015;21(8):1967–1975. doi:https://doi.org/10.1097/MIB.0000000000000468.
- Thorsvik S, Damas JK, Granlund AV, Flo TH, Bergh K, Ostvik AE, Sandvik AK. Fecal neutrophil gelatinase-associated lipocalin as a biomarker for inflammatory bowel disease. J Gastroenterol Hepatol. 2017;32(1):128–135. doi:https://doi.org/10.1111/jgh.13598.
- Wells JM, Brummer RJ, Derrien M, MacDonald TT, Troost F, Cani PD, Theodorou V, Dekker J, Méheust A, de Vos WM, et al. Homeostasis of the gut barrier and potential biomarkers. Am J Physiol Gastrointest Liver Physiol. 2017;312(3):G171–G93. doi:https://doi.org/10.1152/ajpgi.00048.2015.
- Helke K, Angel P, Lu P, Garrett-Mayer E, Ogretmen B, Drake R, Voelkel-Johnson C. Ceramide synthase 6 deficiency enhances inflammation in the DSS model of Colitis. Sci Rep. 2018;8(1):1627. doi:https://doi.org/10.1038/s41598-018-20102-z.
- Castro-Mejia J, Jakesevic M, Krych Ł, Nielsen DS, Hansen LH, Sondergaard BC, Kvist PH, Hansen AK, Holm TL. Treatment with a monoclonal anti-IL-12p40 antibody induces substantial gut microbiota changes in an experimental colitis model. Gastroenterol Res Pract. 2016;2016:4953120. doi:https://doi.org/10.1155/2016/4953120.
- Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25(2):309–318. doi:https://doi.org/10.1016/j.immuni.2006.05.017.
- Roy U, Galvez EJC, Iljazovic A, Lesker TR, Blazejewski AJ, Pils MC, Heise U, Huber S, Flavell RA, Strowig T, et al. Distinct microbial communities trigger colitis development upon intestinal barrier damage via innate or adaptive immune cells. Cell Rep. 2017;21(4):994–1008. doi:https://doi.org/10.1016/j.celrep.2017.09.097.
- Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134(4):1031–1049. doi:https://doi.org/10.1083/jcb.134.4.1031.
- Bruewer M, Samarin S, Nusrat A. Inflammatory bowel disease and the apical junctional complex. Ann N Y Acad Sci. 2006;1072(1):242–252. doi:https://doi.org/10.1196/annals.1326.017.
- Ivanov AI, Nusrat A, Parkos CA. The epithelium in inflammatory bowel disease: potential role of endocytosis of junctional proteins in barrier disruption. Novartis Found Symp. 2004;263:115–118. discussion 24-32, 211-8.
- Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177(2):512–524. doi:https://doi.org/10.2353/ajpath.2010.100168.
- Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–76.
- Meddings J. Barrier dysfunction and Crohn’s disease. Ann N Y Acad Sci. 2006;915:333.