
ABSTRACT
The intestinal barrier function relies on the presence of a single layer of epithelial cells. Barrier dysfunction is associated with the inflammatory bowel diseases (IBD). Understanding the mechanisms involved in intestinal wound healing in order to sustain the barrier function has a great therapeutic potential. Activation of protease-activated receptor-2 (PAR2) induces COX-2 expression in intestinal epithelial cells via EGFR transactivation. COX-2 is well known for its protective effects in the gastrointestinal tract. Therefore, we hypothesized that PAR-2 activation induces a wound healing response in intestinal epithelial cells through COX-2-derived lipid mediators and EGFR transactivation. Immunofluorescence and calcium assay were used to characterize CMT-93 mouse colonic epithelial cell line for PAR2 expression and its activity, respectively. Treatment with PAR2 activating peptide 2-furoyl-LIGRLO-NH2 (2fLI), but not by its inactive reverse-sequence peptide (2fO) enhanced wound closure in scratch wounded monolayers. The EGFR tyrosine kinase inhibitor (PD153035), broad-spectrum matrix metalloproteinase inhibitor (GM6001) and Src tyrosine kinase inhibitor (PP2) inhibited PAR2-induced wound healing. However, PAR2 activation did not induce COX-2 expression in CMT-93 cells and inhibition of COX-2 by COX-2 selective inhibitor (NS-398) did not alter PAR2-induced wound healing. In conclusion, PAR2 activation drives wound healing in CMT-93 cells via EGFR transactivation. Matrix metalloproteinases and Src tyrosine kinase activity may involve in EGFR transactivation and PAR2-induced wound healing is independent of COX-2 activity. These findings provide a mechanism whereby PAR2 can participate in the resolution of intestinal wounds in gastrointestinal inflammatory diseases.
Introduction
The intestinal epithelium is comprised of a single layer of columnar epithelial cells which forms a semipermeable barrier allowing selective absorption of nutrients, electrolytes and water while limiting the exposure of the mucosa to commensal bacteria, pathogens and other immunogenic substances.Citation1,Citation2 Compromised epithelial barrier function is characteristic of gastrointestinal diseases, such as the inflammatory bowel diseases (IBD), Crohn’s disease (CD) and ulcerative colitis (UC), and leads to uncontrolled ingress of luminal antigens ultimately resulting in chronic inflammation and mucosal damage.Citation3 Despite the availability of new therapeutics, the prevalence of IBD continues to rise in adults and children,Citation4 causing significant morbidity and having a major impact on patients’ quality of life.Citation5,Citation6 In addition, current therapies are ineffective for some patients or lose their efficacy over time. Many, such as the biologics, are costly, and many are associated with serious side effects. As mucosal healing is now considered the ultimate goal of IBD treatment, as it is better associated with sustained remission than is symptom relief,Citation7–9 new therapies to safely drive mucosal healing are needed. Nevertheless, the mechanisms by which mucosal healing occurs are complex and not completely understood.
During homeostasis, intestinal epithelial cells are continuously being replenished by stem cells, which reside near the base of the crypts. The progeny of these stem cells acquire differentiated phenotypes as they migrate from the crypt to the mucosal surface.Citation10 However, adaptive cellular responses are initiated during wound healing that allow the epithelium to migrate to adjacent damaged areas in order to reestablish the epithelial barrier function. Epithelial cells at the wound edge lose their columnar polarity and take on a flattened morphology, which is associated with cytoskeletal reorganization and formation of lamellipodial extensions.Citation11 These atypical epithelial cells, known as wound-associated epithelial (WAE) cellsCitation12,Citation13 migrate collectively into the wound area within minutes to hours. The initial restitution phase depends on cell migrationCitation14 and the subsequent phase requires enhanced proliferation of stem cells in the crypts adjacent to the wound to increase the availability of cells to migrate over the wound area.Citation15 Ultimately, these cells differentiate into mature columnar epithelial cells restoring the morphology and function of the intestinal epithelium.Citation13
Several factors including cytokines, growth factors, bioactive peptides, metalloproteinases and lipid mediators can influence intestinal epithelial restitution by enhancing cell migration and proliferation.Citation16,Citation17 In addition to these factors, the inflamed intestinal mucosa is exposed to a plethora of proteolytic enzymes.Citation18 How these proteases influence intestinal epithelial restitution is not well understood. A potential route by which serine proteases can alter epithelial function is through the protease-activated receptors (PARs). PARs are a subfamily of G protein-coupled receptors (GPCRs) with four members (PAR1-4). The unique mechanism of receptor activation involves proteolytic cleavage of the extracellular N-terminus revealing a “tethered ligand” domain which subsequently binds and auto-activates the receptor.Citation19 PAR2 is widely expressed in the gastrointestinal tract and has been associated with IBD.Citation20 Elevated levels of mast cell tryptase, an endogenous PAR2 agonist, were reported in both CD and UC patientsCitation21 and the expression of PAR2 was increased in colons of UC patients.Citation22 Interestingly, PAR2 activation promotes cell migration and proliferation in intestinal epithelial cells implicating its involvement in wound healing.Citation23–25 However, the underlying mechanisms that mediate PAR2-induced cell proliferation and migration are not fully elucidated and require further investigation.
PAR2 activation induces cyclooxygenase (COX)-2 expression in intestinal epithelial cell lines, and is accompanied by a COX-2 dependent increase in prostaglandin E2 (PGE2) production in Caco-2 cells.Citation26,Citation27 Epidermal growth factor receptor (EGFR) transactivation is required for PAR2-induced COX-2 expression in the Caco-2 intestinal epithelial cell line.Citation27 COX-2 expression is increased in IBD patientsCitation28 and it has long been known that tissue PGE2 levels are increased in Crohn’s diseaseCitation29 and ulcerative colitis.Citation30 However, despite the role of PGE2 in inflammation, it has also been shown that COX-2 deficient mice had defects in healing of colonic wounds induced endoscopically.Citation31 In addition, PGE2 drives wound closure in intestinal wounds by promoting the formation of WAE cells.Citation12 Inhibition of COX-2 leads to delay in ulcer healing in models of gastric mucosal injury.Citation32
Based on this evidence, the objective of this study was to investigate the mechanisms underlying intestinal epithelial wound healing, focusing on PAR2-induced COX-2 expression and EGFR transactivation, using the mouse intestinal epithelial cell line, CMT-93, as the experimental model. We showed that PAR2 activation induces a wound healing response in colonic epithelial cells independently of COX-2-derived lipid mediators, but which is dependent upon MMP activity, Src signaling, and EGFR transactivation.
Materials and methods
Cell culture
CMT-93 cells (derived from a mouse rectum carcinoma), originally from ATTC (Manassas, VA), were kindly provided by Dr Paul Beck (University of Calgary). All experiments were performed with cells from passages 10 to 25. CMT-93 cell culture media consisted of DMEM High Glucose supplemented with 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY), 1 mM sodium pyruvate, 4 mM L-glutamine, 10,000 U/ml Penicillin, 100 µg/ml Streptomycin, and 5 μg/ml Plasmocin (InvivoGen, San Diego, CA). Cell culture reagents were purchased from Hyclone (Logan, UT) unless otherwise stated. Cells were incubated at 37°C in 5% CO2, and media were replaced every other day and subcultured using Trypsin-EDTA (T4174, Sigma-Aldrich, Oakville, ON, Canada) when cells reached 90% confluency. Cells were periodically checked for mycoplasma contamination using the PCR-based Venor GeM Mycoplasma detection kit (Sigma).
RNA isolation and reverse transcriptase PCR
CMT-93 cells were grown to confluence in 6-well plates, and PAR2 mRNA expression was assessed by RT-PCR. RNA isolation was performed using the Qiagen RNeasy Mini Kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. RNA concentration was measured at 260 nm using the NanoDrop 2000c spectrophotometer (ThermoFisher Scientific, Wilmington, DE) and genomic DNA was digested using gDNA Wipeout Buffer (Qiagen, Germantown, MD). First strand cDNA was synthesized from 700 ng of total RNA using Invitrogen Superscript™ III Reverse Transcriptase (Carlsbad, CA) according to manufacturer’s protocols. Primers for mouse PAR2 (NM_007974.4: Mus musculus coagulation factor II (thrombin) receptor-like 1 (F2rl1)) were designed using Primer-BLAST data base and synthesized commercially (Integrated DNA Technologies, Toronto, ON). PAR2 primers consisted of 5´-GAGTAGGGCTCCGAGTTTCG-3´ (forward) and 5´-TACTGTTGTTGCGTCCCGGT-3´ (reverse). RT-PCR was carried out using Qiagen HotstarTaq™ Master Mix, according to the manufacturer’s instructions and performed using the Bio-Rad T100™ Thermal Cycler (Bio-Rad Laboratories, Mississauga, ON, Canada). PCR reaction included an initial 95°C denaturation step for 15 min, followed by 35 cycles of 95°C for 35 s, 60°C for 30 s, and 72°C for 30 s. A final elongation step was performed at 72°C for 5 min. Samples were mixed with loading dye (0.25% bromophenol blue, 30% glycerol) and were run on a 1.5% agarose gel containing 0.1% ethidium bromide. Gels were imaged on a ChemiDoc XRS system (Bio-Rad) using QuantityOne software. Sizes of bands were determined by comparing to the 1 kb Plus DNA Ladder (Invitrogen). PCR products were purified using the Qiagen QIAquick PCR Purification Kit (Qiagen, Germantown, MD) according to manufacturer’s instructions. Purified DNA samples were sent for sequencing at the University of Calgary Core DNA services lab.
Immunofluorescence staining
To determine the cellular localization of PAR2, sequential immunofluorescence confocal microscopy was conducted as previously described.Citation26 CMT-93 cells were grown to post-confluence on 0.4 µm pore diameter semipermeable transparent membrane supports (Greiner Bio-One International, Kremsmünster, Austria) and fixed with 100% ice-cold methanol for 20 min. Cells were blocked and permeabilized in 5% w/v donkey serum in phosphate-buffered saline (PBS) containing 0.1% Triton X-100 for 2 h at room temperature on a rocking platform. Tight junction protein Zonula Occludens-1 (ZO-1) was used as an apical cell marker in order to determine the cellular localization of PAR2. Cells were incubated with anti-ZO-1 goat IgG antibodies (1:200 dilution, Ab190085 Abcam, Cambridge, United Kingdom) overnight at 4°C and on the following day cells were incubated with donkey anti-goat IgG (1:250 dilution, Alexa Flour 647; A21447, Invitrogen) secondary antibodies for 2 h at room temperature. For PAR2 staining anti-PAR2 antibody (1:200 dilution, A5, provided by Dr M. Hollenberg, University of Calgary) was added to cells and incubated at 4°C overnight with subsequent exposure to goat anti-rabbit IgG secondary antibodies (1:250 dilution, Alexa Flour 488; A11034, Invitrogen) for 2 h at room temperature. Finally, cells were incubated with DAPI (1;50,000 dilution, D1306: Invitrogen) for 30 min to visualize nuclei and membranes were mounted with FluorSave (34,789 Calbiochem, Darmstadt, Germany) and imaged with an Olympus IX81 FV1000 Laser Scanning Confocal Microscope. Isotype antibody controls were incubated with goat polyclonal IgG (1:200 dilution, Sc2028: Santa Cruz Biotechnology, Dallas, TX) and rabbit polyclonal IgG (1:200 dilution, 015–000-003: Jackson ImmunoResearch Laboratories, West Grove, PA) isotype antibodies followed by the respective secondary antibody incubations. Secondary antibody controls were incubated with donkey anti-goat IgG (1:250 dilution, Alexa Flour 647; A21447, Invitrogen) and goat anti-rabbit IgG (1:250 dilution, Alexa Flour 488; A11034, Invitrogen) secondary antibodies without any primary antibodies (Supplementary Figure S1).
PAR2 activation
PAR2 was activated using 2-furoyl-LIGRLO-NH2 (2fLI), a small potent and selective PAR2 activating peptide with sequence similarity to the tryptic proteolysis site of the receptor N-terminal domain.Citation33 The inactive reverse-sequence peptide 2-furoyl-OLRGIL-NH2 (2fO) was used as a control.Citation34 Peptides were solubilized in HEPES buffer and subsequent dilutions were made in PBS lacking magnesium or calcium (HyClone). Initially, 2fLI and 2fO peptides were synthesized by solid-phase methods at the Peptide Synthesis Facility, University of Calgary, Faculty of Medicine, Calgary and were kindly provided by Dr M. Hollenberg. Subsequently, the peptides were purchased from EZBiolab Inc (EZBiolab Inc., Carmel, IN). CMT-93 cells responded similarly to 2fLI from both sources (data not shown).
Calcium assay
Intracellular Ca2+ levels are elevated by PAR2 activation.Citation19,Citation35 To assess PAR2 activation in CMT-93 cells, a fluorescence-based Ca2+ assay was conducted as previously described.Citation34,Citation36 Briefly, cells were grown in T75 flasks to reach 70% confluency and cells were harvested using enzyme free cell dissociation buffer (HyClone HyQTase cell detachment reagent, GE Healthcare, Chicago, IL). Cells were incubated with Fluo-4 acetoxymethyl ester (Molecular Probes, Eugene, OR) for 30 min at room temperature and were then resuspended in calcium buffer (150 mM NaCl, 3 mM KCl, 1.5 mM CaCl2, 20 mM HEPES, 10 mM glucose). Fluorescence measurements, reflecting elevation of intracellular Ca2+, were measured using a Perkin-Elmer fluorescence spectrometer (Perkin-Elmer, Waltham, MA; excitation wavelength of 480 nm and emission wavelength 530 nm). The responses that resulted from addition of different concentrations of the PAR2 activating peptide, 2fLI, and the inactive reverse-sequence peptide, 2fO, were standardized relative to the fluorescence peak elicited by the addition of 7.5 µM of the calcium ionophore, A23187 (Sigma-Aldrich).
Protein assay and western blotting
Cells were washed twice with ice-cold PBS (without magnesium or calcium, Hyclone, Logan, UT) and ice-cold SDS lysis buffer containing NaCl (100 mM), Tris-HCl (20 mM pH 8.0), SDS (0.1%), EDTA (0.9 mM), protease inhibitor cocktail (2%, Sigma-Aldrich, p2714), Triton-X (0.5%), activated Na3VO4 (2 mM), and NaF (50 mM). Cells were then scraped and transferred to microcentrifuge tubes and frozen at −80°C for further lysis. On the following day, whole-cell lysates were thawed on ice and disrupted by probe sonication (Fisher Scientific F60 Sonic Dismembrator, Pittsburgh, PA), then centrifuged at 20,817 × g for 10 min at 4°C. The supernatants were transferred to clean microcentrifuge tubes and protein concentrations were determined using the DC Protein Assay (Bio-Rad). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using 20 μg of protein per lane in 26-well CriterionTM XT precast 4–12% Bis-Tris gels, and proteins were transferred onto nitrocellulose membranes. The membranes were then washed with 0.1% Tween-20-Tris-buffered saline (TBST- 0.1% Tween-20, 50 mM Tris-HCl, 137 mM NaCl, 2.7 mM KCl) and blocked with either 5% milk or 5% BSA in TBST for 1 h at room temperature and incubated in primary antibody at 4°C overnight. The primary antibody for COX-2 (160,126, Cayman Chemical) was diluted 1:2,000 in 5% BSA. The primary antibody for β-actin (AC5441, Sigma-Aldrich) was diluted 1:5,000 in 5% skim milk. Blots were then washed and incubated in appropriate horseradish peroxidase-conjugated secondary antibody (either anti-rabbit or anti-mouse HRP-linked; Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature, followed by three additional washes in TBST. Bands were visualized using Clarity Western Enhanced Chemiluminescent Substrate ECL (Bio-Rad) and a Bio-Rad Chemidoc. Band density was analyzed using Image Lab software (Bio-Rad).
Scratch wound healing assay
CMT-93 cells were seeded in 96-well plates at a density of 5 × 103 cells/well and grown under standard culture conditions for 3-5-days post-confluence. A scratch wound was made in each well using the WoundMakerTM tool (Essen Biosciences, Ann Arbor, MI). After wounding, cells were washed twice in serum-free DMEM High Glucose supplemented media to remove any cell debris remaining inside the wound area. Cells were treated (0 h) and then imaged using the IncuCyte Zoom live-cell imaging system maintained in 37°C humidified incubator with 5% CO2. Whole-well phase contrast images were taken at every 2 h for 24 h. Following image acquisition, the IncuCyte ZOOM software was used to create confluency masks to highlight the wound area and the wound area was calculated using ImageJ software (National Institutes of Health, Bethesda, MD). Results were expressed as percentage wound area remaining.
Concentrations of 1.0 µM to 10.0 µM 2fLI were used in the concentration-response curve in 10% serum and serum-free media conditions. Dimethyl sulfoxide (DMSO, Sigma-Aldrich; 0.1%) was used as the solvent for pharmacological inhibitors. The effect of COX-1 and COX-2 enzymes on wound healing were investigated using a COX-2 selective inhibitor NS-398 (Cayman Chemical, Ann Arbor, MI; 10 µM) and a nonselective COX inhibitor, indomethacin (Sigma-Aldrich; 10 µM). The involvement of EGFR activation in PAR2-induced wound healing was assessed using the EGFR tyrosine kinase inhibitor, PD153035 (4-[(3-bromophenyl)amino]-6,7-dimethoxyquinazoline hydrochloride, Tocris Bioscience, Oakville, ON, Canada) in wound healing assays. Human epidermal growth factor (EGF, 5 and 10 nM; GF-010-8 Cedarlane, Burlington, ON) was used as the positive control for EGFR activation. Cells were pre-treated with 10 nM PD153035 or the vehicle (0.02% DMSO) for 30 min before treatments and 10 nM of PD153035 was used in combination with 10 µM 2fLI or 5 ng/ml EGF. PP2 (Biomol, Hamburg, Germany: AG-CR1-3563-M005) was used as a Src tyrosine kinase inhibitor and GM6001 (Cayman Chemical: 14,533) was used as a broad-spectrum MMP inhibitor. Cells were pre-treated with 10 µM GM6001, 100 nM PP2 or the vehicle (0.02% DMSO) for 30 min. Inhibitors were used in combination with 10 µM 2fLI.
Statistical analysis
Data are expressed as mean standard error and were graphed and analyzed using GraphPad Prism version 9.2.0 (GraphPad Software, La Jolla, CA). All N values represent experimental replicates, with each individual experimental replicate including at least three technical replicates.
Non-parametric data were analyzed using Kruskal-Wallis test followed by Dunn’s multiple comparison test. Comparisons of more than two groups were made using two-way ANOVA followed by Tukey multiple comparison tests. A probability value of p < .05 was considered significant.
Results
PAR2 expression and activation in CMT-93 cells
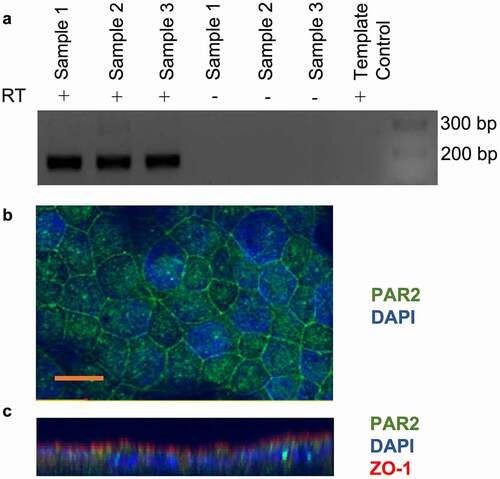
The expression of PAR2 by the CMT-93 cell line has not been previously demonstrated. We showed that CMT-93 cells constitutively expressed PAR2 mRNA and protein as shown by RT-PCR () and immunofluorescence microscopy (), respectively. RT-PCR products were sequenced using the Sanger sequencing method which confirmed that sequenced products were murine PAR2 (data not shown). PAR2 was primarily expressed in the plasma membrane, with some punctate immunoreactivity in the cytoplasm (). Z-stack construction of optical sections of cell monolayers revealed PAR2 expression on the apical and basolateral membranes, although the majority of immunoreactivity appeared to be basolateral (). Immunoreactivity for ZO-1 showed that the CMT-93 monolayers were polarized and had formed tight junctions (). Isotype and secondary antibody controls did not show significant immunofluorescence indicating the absence of nonspecific reactivity of primary and secondary antibodies.
Figure 1. CMT-93 cells express protease-activated receptor 2 (PAR2). A: RT-PCR for PAR2 mRNA expression in CMT-93 cells. Reactions lacking reverse transcriptase enzyme (RT) and template control lacking cDNA were performed as controls. DNA ladder and the sizes (bp) are shown on the right of image, with expected product size being 167 bp (n = 3). B: Confocal immunocytochemistry for PAR2 in CMT-93 cell monolayers (n = 2). Green shows positive immunoreactivity for PAR2 and blue shows DAPI staining for nuclei. C: z stacks of optical sections through cell monolayers. Scale bar represents 10 μm.
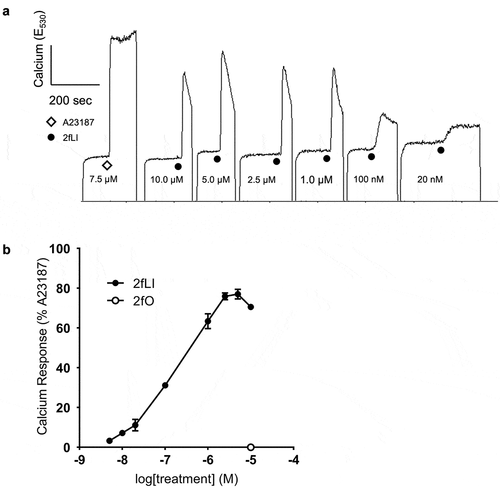
Activation of PAR2 mobilizes Ca2+ from the endoplasmic reticulum into the cytoplasm, which increases intracellular Ca2+.Citation37 Hence, we used increases in intracellular Ca2+ upon activation of PAR2 with the specific activating peptide, 2-furoyl-LIGRLO-NH2 (2fLI)Citation33 as a method of verification of PAR2 expression, activation and signaling in CMT-93 cells (). The reverse sequence peptide, 2-furoyl-OLRGIL (10 μM), which has no activity at PAR2,Citation38 served as a control. Calcium responses to PAR2 activation were compared to that elicited by the calcium ionophore, A23187 (7.5 μM), the response to which was considered 100% (). Concentrations of 10 nM to 10 µM 2fLI were tested in calcium assay and activation of PAR2 by 2fLI resulted in a concentration-dependent increase in intracellular Ca2+ (). EC50 was calculated to be 0.5 μM based on the concentration-response curve of 2fLI. The maximal PAR2-mediated Ca2+ response occurred at 5.0 µM 2fLI (77% of the response to the ionophore control). The inactive reverse-sequence peptide, 2fO (10.0 µM) elicited no response ().
Figure 2. PAR2-activating peptide 2fLI induces calcium signaling. A: Fluorescence responses resulted from the addition of varying concentrations of PAR2-activating peptide 2fLI were standardized relative to the peak fluorescence elicited by the addition of calcium ionophore A23187 (7.5 µM). The addition inactive reverse-sequence peptide 2fO (10 µM) did not elicit a response. B: 2fLI concentration response curve (n = 5).
PAR2 activation enhances intestinal epithelial wound healing
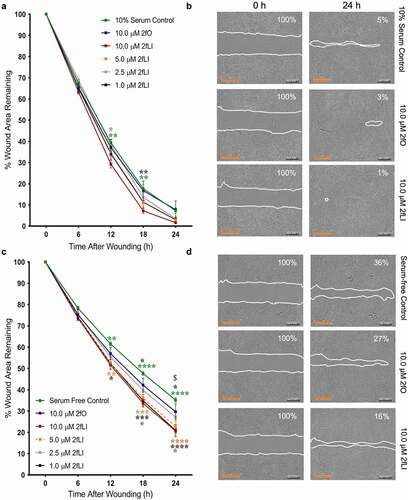
In order to test the hypothesis that PAR2 activation induces intestinal epithelial wound healing, we first assessed the capacity of 2fLI to induce cell migration using a scratch wound assay. These assays were conducted in both 10% serum and serum-free conditions at concentrations of 1.0, 2.5, 5.0 and 10.0 µM 2fLI. Wound area was measured over 24 h. Wound healing was observed in both 10% serum and serum-free conditions, reaching a maximum of ~8% and ~35% of the original wound area, respectively (, c). Exposure to 2fLI immediately after wounding of the monolayers enhanced the degree of wound healing compared with control and the inactive reverse-sequence peptide 2fO, in both 10% serum () and serum-free () conditions. All 2fLI concentrations showed significant increase in wound healing compared to control in serum-free condition, while only the maximum tested concentration of 2fLI (10 µM) significantly enhanced wound healing compared to controls in 10% serum conditions ().
Figure 3. PAR2 activation induces wound healing. CMT-93 cells were grown to post-confluence and scratch wounds were made in monolayers. Cells were treated with varying concentrations of 2fLI, 2fO or left untreated in 10% serum (A, n = 6–7) and serum-free media (C, n = 5–6). Representative images are provided at 0 h and 24 h for 10% serum (b) and serum-free (d) conditions. Data were analyzed using two-way analysis of variance with Tukey multiple comparison test. In fig A and C: green asterisks represent the p values of comparison of control with 10.0 µM 2fLI, blue asterisks represent the p values of comparison of 2fO with 10.0 µM 2fLI, and gray asterisks represent the p values of comparison of 2.5 µM 2fLI with 10.0 µM 2fLI. In Fig C: orange asterisks represent the p values of comparison of serum-free control with 5.0 µM 2fLI, black asterisks represent the p values of comparison of serum-free control with 1.0 µM 2fLI and $ represents the comparison of 1.0 µM 2fLI with 2fO (p < .01). (* p < .05; ** p < .01; *** p < .001; **** p < .0001). Scale bar represents 700 µM.
PAR2 activation does not induce COX-2 and COX enzymes do not affect PAR2-induced wound healing
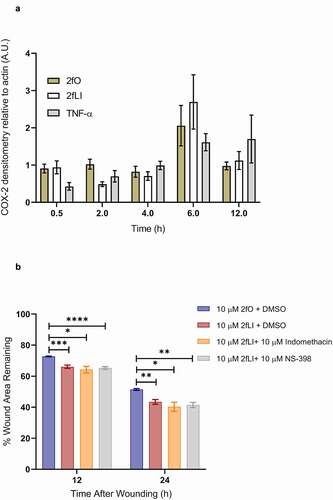
We previously observed that PAR2 activation induced COX-2 expression in Caco2 colonic epithelial cells.Citation27 Hence, we sought to characterize the CMT-93 cell line for its ability to induce COX-2 upon PAR2 activation. CMT-93 cells were grown to 5-days post-confluence and switched to serum-free medium 1 h before experiments and treated with either 2fLI (10 µM), 2fO (10 µM), or TNF-α (10 ng/ml) as a positive control.Citation39 Samples were collected from 30 min to 12 h post-treatment for western blot and subsequent densitometry. CMT-93 cells expressed COX-2 protein constitutively, however, contrary to our hypothesis, neither PAR2 activation nor TNF-α induced further COX-2 expression when compared to the 2fO control (). Because we detected basal COX-2 expression, we sought to determine if constitutively expressed COX-1 and COX-2-derived lipid mediators were involved in PAR2-induced wound healing. The effect of COX enzymes was tested by performing scratch wound assays in the presence of the nonselective COX inhibitor, indomethacin (10 µM), and the COX-2 selective inhibitor, NS-398 (10 µM). Neither indomethacin nor NS-398 altered PAR2-induced wound healing (). These results indicate that cyclooxygenase enzymes and their products are not involved in PAR2-induced wound healing.
Figure 4. PAR2 activation does not induce COX-2 expression and inhibition of COX enzymes do not affect PAR2-induced wound healing. A: CMT-93 cells were grown to post-confluence and switched to serum-free media 1 h before treatments; all treatments were performed in serum-free media. Cells were left untreated (control) or treated with 10 µM 2fLI, 10 µM 2fO or 10 ng/ml TNF-α. Cell lysates were immunoblotted for COX-2 and β-Actin. Pixel density was measured, expressed as a ratio of COX-2/β-Actin and normalized to levels observed in control cells at each time point (n = 4). Data were analyzed using Kruskal-Wallis test with Dunn’s multiple comparison test. B: The effect on PAR-2 induced wound healing was tested in the presence of nonselective COX inhibitor indomethacin (10 µM) and the COX-2 selective inhibitor NS-398 (10 µM) (n = 7–9). Data were analyzed using two-way analysis of variance with Tukey multiple comparison test. (* p < .05; ** p < .01; *** p < .001; **** p < .0001).
EGFR activity is required for PAR2-induced wound healing
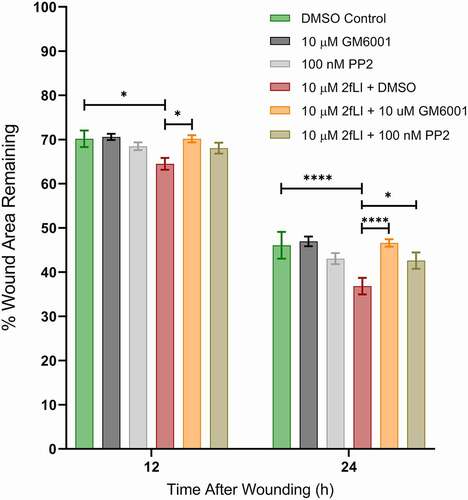
EGFR activation has long been known to be a major regulator of intestinal epithelial cell migration.Citation40 PAR2 activation is known to transactivate EGFRCitation27,Citation41,Citation42 via intracellular and extracellular pathways; intracellularly through the intracellular nonreceptor tyrosine kinase, Src, and extracellularly via matrix metalloproteinase (MMP)-mediated cleavage of membrane-bound growth factors that act as ligands to subsequently activate EGFR.Citation27 The dependency of EGFR transactivation in PAR2-induced wound healing was tested in scratch wound assays using the EGFR tyrosine kinase inhibitor, PD153035 (10 nM). EGF was used as the positive control. Wound healing capacity of two concentrations of EGF (5 and 10 ng/ml) were tested against 2fLI (10 µM). 2fLI at 10 μM and EGF at concentrations of 5 and 10 ng/ml showed similar wound healing capacity (). PD153035 on its own significantly slowed wound healing in CMT-93 cells, suggesting the involvement of an endogenous EGFR ligand in baseline cell migration. Treatment of cells with PD153035 prevented both PAR2- and EGF-induced wound healing (). These results suggest that PAR2-induced wound healing may require EGFR transactivation. Hence, we sought to determine the role of Src tyrosine kinase and MMP activity in PAR2-induced wound healing in scratch wound. Treatment with the broad spectrum MMP inhibitor, GM6001, significantly diminished PAR2-induced wound healing (). However, the Src tyrosine kinase inhibitor, PP2, had no greater effect on PAR2-induced wound healing than it did on its own ().
Figure 5. EGFR activity is required for PAR2-induced wound healing. A: CMT-93 cells were grown to post-confluence. The wound healing capacities of 2fLI (10 µM) and EGF (5 and 10 ng/ml) were tested in scratch wound healing assays (n = 5–7). B: The effect on wound healing was tested in the presence of EGFR tyrosine kinase inhibitor PD153035 (10 nM) (n = 8–4). All treatments were performed in serum-free conditions. Data were analyzed using two-way analysis of variance with Tukey multiple comparison test. (* p < .05; ** p < .01; *** p < .001).
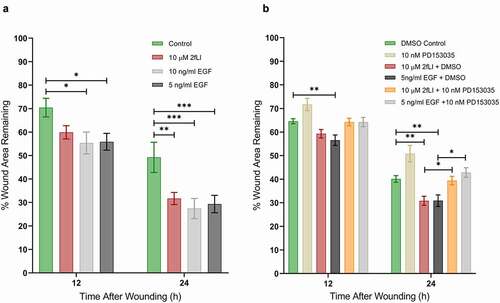
Figure 6. A: PAR2-induced wound healing was inhibited in the presence of the broad-spectrum-MMP inhibitor GM6001 and the Src tyrosine kinase inhibitor PP2. CMT-93 cells were grown to post-confluence and the effects of GM6001 and PP2 on wound healing were test in scratch wound healing assays. All treatments were performed in serum-free media. Data were analyzed using two-way analysis of variance with Tukey multiple comparison test (n = 4–8). (* p < .05; ** p < .01; *** p < .001; **** p < .0001).
Discussion
Epithelial regeneration and repair are key elements of mucosal healing, now recognized as the gold standard in assessing efficacy of treatment in IBD. The inflammatory mucosal microenvironment in which this repair must take place contains numerous host and microbial serine proteases,Citation43 some of which could be endogenous activators of PARs. We sought to determine if PAR2 activation could drive intestinal epithelial cell migration, an important facet of epithelial regeneration. We showed that PAR2 expression and its activation induce wound healing in intestinal epithelial cell monolayers via MMP-dependent EGFR transactivation. Contrary to our initial hypothesis, PAR2 activation did not induce COX-2 expression and PAR2-induced wound healing was independent of COX activity.
PAR2 is expressed by intestinal epithelial cells and cell lines, but its expression by the mouse rectal tumor cell line, CMT-93, has not been reported, so we first needed to demonstrate that CMT-93 cells express functional PAR2. Using RT-PCR and immunofluorescence confocal microscopy, we showed that CMT-93 cells express PAR2 mRNA and protein. Confocal microscopy revealed that PAR2 was localized to both the apical and basolateral membranes, with expression primarily localized basolaterally, as well as some punctate immunoreactivity in the cytoplasm. Similar PAR2 expression patterns have been observed in human, other murine and canine intestinal epithelial cell lines. Whether PAR2 is expressed and activated apically or basolaterally has important ramifications in terms of function.Citation44 For example, we previously showed in the canine intestinal epithelial cell line, SCBN, that basolateral PAR2 activation resulted in substantially higher chloride (Cl−) secretion than did apical PAR2 stimulation.Citation42 This has implications for the current study where PAR2 on the apical surface was activated on cell monolayers grown in 96 well plastic plates.
PARs can be activated without proteolytic cleavage by peptide agonists which mimic the tethered ligand sequence of the N terminal. In this study, we used 2-furoyl-LIGRLO-NH2 (2fLI), a potent and selective activator of PAR2.Citation33 Activation of PAR2 mobilizes Ca2+ from the endoplasmic reticulum into the cytoplasm via a Gq/11-mediated process which will increase intracellular Ca2+.Citation19,Citation37 Therefore, the measurement of intracellular Ca2+ upon activation by receptor agonists is widely used to assess the presence and activation of specific PARs. We showed that 2fLI triggered a rapid, concentration-dependent increase in intracellular Ca2+, thus demonstrating that the PAR2 expressed on CMT-93 cells was functionally coupled to intracellular signaling processes.
The main finding of our study is that PAR2 activation enhances wound healing capacity of post-confluent CMT-93 colonic epithelial cells. Others have shown that PAR2 activation increases wound healing in intestinal epithelial cell lines. This appears to be cell line dependent, however, as we recently showed that PAR2 decreases the rate of wound healing in Caco2 cell monolayers.Citation26 Despite previous reports of PAR2-induced wound healing, the mechanism whereby PAR2 drives epithelial cell migration is unclear. We have previously shown that PAR2 activation induces COX-2 expression and activity in intestinal epithelialCitation27 and airwayCitation45 cell lines. However, PAR2 activation failed to induce COX2 expression in CMT-93 cells. Similarly, treatment with TNF-α which is a known activator of COX2 in the intestinal epithelium, also failed to induce COX-2 expression. Nevertheless, we observed constitutive expression of COX-2 in CMT-93 cells by western blot (data not shown). While it is often reported that COX-1 is the constitutive isoform whereas COX-2 is considered inducible, it has been shown that COX-2 is also constitutively expressed in various tissues including the gastrointestinal tract, kidney, brain, and thymus.Citation46,Citation47 Furthermore, expression of COX-2 has been reported in colorectal cancers,Citation48 and since CMT-93 cells are derived from tumors of colon and rectum in C57BL/6 mice,Citation49 it is not unlikely that they constitutively express COX-2. Therefore, lack of COX-2 induction in CMT-93 cells upon PAR2 activation may relate to high basal level expression of COX-2. To investigate the role of constitutive COX isoforms, we investigated PAR2-induced wound healing in the presence of the nonselective COX inhibitor indomethacin and the COX-2 selective inhibitor NS-398. Since COX inhibition did not alter PAR2-induced wound healing, we conclude PAR2 effects on wound healing occurred independently of COX-2 activity.
It is now widely accepted that EGFR transactivation can occur in response to the activation of certain GPCRs,Citation50 including PAR2 activation in intestinal epithelial cells.Citation27,Citation42 EGFR activation elicits important signaling pathways that mediate cell migration and proliferation.Citation41,Citation51,Citation52 The involvement of EGFR transactivation was investigated to further characterize PAR2-induced wound healing. Exposure of wounded CMT-93 monolayers to 5 ng/ml EGF resulted in a wound healing response that was comparable to that of 10 μM 2fLI (10 µM). Furthermore, the EGFR tyrosine kinase inhibitor, PD153035, inhibited both EGF- and 2fLI-induced wound healing, confirming our hypothesis that EGFR transactivation is required for PAR2-induced wound healing.
PAR2 activation induces EGFR transactivation via intracellular and extracellular pathways; intracellularly through the nonreceptor tyrosine kinase, Src, and extracellularly via matrix metalloproteinase (MMP) mediated cleavage of membrane-bound growth factors subsequently activating EGFR.Citation27,Citation41,Citation42 Basolateral PAR2 activation in SCBN cells induced epithelial Cl− secretion in colonic epithelial cells via EGFR transactivation that was mediated by Src tyrosine kinase.Citation42 In addition, PAR2 activation increased the concentrations of the EGFR ligand TGF-α to activate EGFR; the release of TGF-α was abrogated by treatment with broad-spectrum MMP inhibitors.Citation41 We tested the effects of inhibition of MMP and Src tyrosine kinase in the CMT-93 wounding model using the broad-spectrum MMP inhibitor, GM6001, and Src family kinase inhibitor, PP2. MMP inhibition reduced 2fLI-induced wound healing as early as 14 h post-treatment, whereas Src inhibition did not reduce 2fLI-induced wound healing until 22 h post-treatment. These results imply that the MMP-dependent EGFR transactivation pathway plays a predominant role compared to Src kinase in transactivating EGFR. Together, these data suggest that PAR2 activation drives wound healing in CMT-93 cells possibly via EGFR transactivation, and that MMP and Src kinase may be involved in this process. Nevertheless, our results do not rule out the possibility that MMP- and Src-mediated signaling may also act through other pathways. This possibility awaits further study.
Activation of PAR2 could have other effects not investigated in our study. Alterations in the structure of the apical junctional complex (tight junction and adherens junction) often accompany epithelial cell migration, such as that which occurs during wound healing.Citation53 The expression of claudins, which are key proteins involved in the barrier established by the tight junction, can be decreased by PAR2 activation. For example, PAR2 is associated with degradation of epithelial claudin-1 in models of airway injuryCitation54,Citation55 and with downregulation of claudin-5 expression in brain endothelium and gut epithelium.Citation56,Citation57 Whether PAR2 activation drives intestinal epithelial wound healing through alteration of claudins or other apical junctional proteins remains to be determined.
Our results provide evidence to suggest that PAR2 activation may have importance in driving epithelial wound healing during gastrointestinal diseases, such as IBD, where mucosal levels of serine proteases are elevated. This is interesting when also considering that PAR2 activation can drive inflammation in mouse models of colitis.Citation58–60 However, these seemingly contradictory results support the growing hypothesis that mediators that drive acute inflammation may also serve as drivers of resolution later in the inflammatory response.Citation17 Clearly, consideration of the tissue microenvironment and the timing of inducing PAR2 activation are key factors when targeting PAR2 as a therapeutic intervention in diseases such as IBD.
Author contributions
M.B. and W.K.M. conceived and designed the research; M.B performed experiments and analyzed data; M.B and W.K.M. interpreted results of experiments; M.B. prepared figures and drafted the manuscript; M.B. and W.K.M. edited and revised the manuscript; M.B and W.K.M approved the final version of the manuscript.
Supplemental Material
Download Zip (658.8 KB)Acknowledgments
This work was supported by a Project Grant (PJT-153290) from the Canadian Institutes of Health Research. The authors gratefully acknowledge the support of Drs Pina Colarusso and Rima-Marie Wazen of the Live Cell Imaging Resource Laboratory at the University of Calgary. The authors thank Dr Morley Hollenberg for assistance with the calcium assay to measure PAR2 activity.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
References
- Luissint AC, Parkos CA, Nusrat A. Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology. 2016;151(4):1–15. doi:https://doi.org/10.1053/j.gastro.2016.07.008.
- Sumagin R, Brazil JC, Nava P, Nishio H, Alam A, Luissint AC, Weber DA, Neish AS, Nusrat A, Parkos CA. Neutrophil interactions with epithelial-expressed ICAM-1 enhances intestinal mucosal wound healing. Mucosal Immunol. 2016;9(5):1151–1162. doi:https://doi.org/10.1038/mi.2015.135.
- Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2017;4(1):33–46. doi:https://doi.org/10.1016/j.jcmgh.2017.03.007.
- Coward S, Clement F, Benchimol EI, Bernstein CN, Avina-Zubieta JA, Bitton A, Carroll MW, Hazlewood G, Jacobson K, Jelinski S, et al. Past and future burden of inflammatory bowel diseases based on modeling of population-based data. Gastroenterology. 2019;156(5):1345–1353 e4. doi:https://doi.org/10.1053/j.gastro.2019.01.002.
- Hoivik ML, Moum B, Solberg IC, Cvancarova M, Hoie O, Vatn MH, Bernklev T, Group IS. Health-related quality of life in patients with ulcerative colitis after a 10-year disease course: results from the IBSEN study. Inflamm Bowel Dis. 2012;18(8):1540–1549. doi:https://doi.org/10.1002/ibd.21863.
- van der Have M, van der Aalst KS, Kaptein AA, Leenders M, Siersema PD, Oldenburg B, Fidder HH. Determinants of health-related quality of life in Crohn’s disease: a systematic review and meta-analysis. J Crohns Colitis. 2014;8(2):93–106. doi:https://doi.org/10.1016/j.crohns.2013.04.007.
- Peyrin-Biroulet L, Bressenot A, Kampman W. Histologic remission: the ultimate therapeutic goal in ulcerative colitis? Clin Gastroenterol Hepatol. 2014;12(6):929–34 e2. doi:https://doi.org/10.1016/j.cgh.2013.07.022.
- Picco MF, Farraye FA. Targeting mucosal healing in Crohn’s disease. Gastroenterol Hepatol (N Y). 2019;15:529–538.
- Pineton de Chambrun G, Blanc P, Peyrin-Biroulet L. Current evidence supporting mucosal healing and deep remission as important treatment goals for inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2016;10:915–927. doi:https://doi.org/10.1586/17474124.2016.1174064.
- van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71(1):241–260. doi:https://doi.org/10.1146/annurev.physiol.010908.163145.
- Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol. 2008;14(3):348–353. doi:https://doi.org/10.3748/wjg.14.348.
- Miyoshi H, VanDussen KL, Malvin NP, Ryu SH, Wang Y, Sonnek NM, Lai CW, Stappenbeck TS. Prostaglandin E2 promotes intestinal repair through an adaptive cellular response of the epithelium. EMBO J. 2017;36(1):5–24. doi:https://doi.org/10.15252/embj.201694660.
- Seno H, Miyoshi H, Brown SL, Geske MJ, Colonna M, Stappenbeck TS. Efficient colonic mucosal wound repair requires Trem2 signaling. Proc Natl Acad Sci U S A. 2009;106(1):256–261. doi:https://doi.org/10.1073/pnas.0803343106.
- McCormack SA, Viar MJ, Johnson LR. Migration of IEC-6 cells: a model for mucosal healing. Am J Physiol. 1992;263:G426–35. doi:https://doi.org/10.1152/ajpgi.1992.263.3.G426.
- Iizuka M, Konno S. Wound healing of intestinal epithelial cells. World J Gastroenterol. 2011;17(17):2161–2171. doi:https://doi.org/10.3748/wjg.v17.i17.2161.
- Gordon MH, Chauvin A, Boisvert FM, MacNaughton WK. Proteolytic processing of the epithelial adherens junction molecule E-cadherin by neutrophil elastase generates short peptides with novel wound-healing bioactivity. Cell Mol Gastroenterol Hepatol. 2019;7(2):483–486 e8. doi:https://doi.org/10.1016/j.jcmgh.2018.10.012.
- Quiros M, Nusrat A. Contribution of wound-associated cells and mediators in orchestrating gastrointestinal mucosal wound repair. Annu Rev Physiol. 2019;81(1):189–209. doi:https://doi.org/10.1146/annurev-physiol-020518-114504.
- Antalis TM, Shea-Donohue T, Vogel SN, Sears C, Fasano A. Mechanisms of disease: protease functions in intestinal mucosal pathobiology. Nat Clin Pr Gastroenterol Hepatol. 2007;4(7):393–402. doi:https://doi.org/10.1038/ncpgasthep0846.
- Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol Rev. 2002;54(2):203–217. doi:https://doi.org/10.1124/pr.54.2.203.
- Kawabata A, Matsunami M, Sekiguchi F. Gastrointestinal roles for proteinase-activated receptors in health and disease. Br J Pharmacol. 2008;153(Suppl):S230–40. doi:https://doi.org/10.1038/sj.bjp.0707491.
- Raithel M, Winterkamp S, Pacurar A, Ulrich P, Hochberger J, Hahn EG. Release of mast cell tryptase from human colorectal mucosa in inflammatory bowel disease. Scand J Gastroenterol. 2001;36(2):174–179. doi:https://doi.org/10.1080/003655201750065933.
- Kim JA, Choi SC, Yun KJ, Kim DK, Han MK, Seo GS, Yeom JJ, Kim TH, Nah YH, Lee YM. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm Bowel Dis. 2003;9(4):224–229. doi:https://doi.org/10.1097/00054725-200307000-00002.
- Darmoul D, Marie JC, Devaud H, Gratio V, Laburthe M. Initiation of human colon cancer cell proliferation by trypsin acting at protease-activated receptor-2. Br J Cancer. 2001;85(5):772–779. doi:https://doi.org/10.1054/bjoc.2001.1976.
- Jiang Y, Yau MK, Kok WM, Lim J, Wu KC, Liu L, Hill TA, Suen JY, Fairlie DP. Biased signaling by agonists of protease activated receptor 2. ACS Chem Biol. 2017;12(5):1217–1226. doi:https://doi.org/10.1021/acschembio.6b01088.
- Zhou B, Zhou H, Ling S, Guo D, Yan Y, Zhou F, Wu Y. Activation of PAR2 or/and TLR4 promotes SW620 cell proliferation and migration via phosphorylation of ERK1/2. Oncol Rep. 2011;25(2):503–511. doi:https://doi.org/10.3892/or.2010.1077.
- Fernando EH, Gordon MH, Beck PL, MacNaughton WK. Inhibition of intestinal epithelial wound healing through protease-activated receptor-2 activation in Caco2 cells. J Pharmacol Exp Ther. 2018;367(2):382–392. doi:https://doi.org/10.1124/jpet.118.249524.
- Hirota CL, Moreau F, Iablokov V, Dicay M, Renaux B, Hollenberg MD, MacNaughton WK. Epidermal growth factor receptor transactivation is required for proteinase-activated receptor-2-induced COX-2 expression in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2012;303(1):G111–9. doi:https://doi.org/10.1152/ajpgi.00358.2011.
- Singer II, Kawka DW, Schloemann S, Tessner T, Riehl T, Stenson WF. Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology. 1998;115(2):297–306. doi:https://doi.org/10.1016/S0016-5085(98)70196-9.
- Ahrenstedt HR, Knutson L, KNUTSON L. Jejunal release of prostaglandin E2 in Crohn’s disease: relation to disease activity and first-degree relatives. J Gastroenterol Hepatol. 1994;9(6):539–543. doi:https://doi.org/10.1111/j.1440-1746.1994.tb01557.x.
- Rampton DS, Sladen GE, Youlten LJF. Rectal mucosal prostaglandin E2 release and its relation to disease activity, electrical potential difference, and treatment in ulcerative colitis. Gut. 1980;21(7):591–596. doi:https://doi.org/10.1136/gut.21.7.591.
- Manieri NA, Drylewicz MR, Miyoshi H, Stappenbeck TS. Igf2bp1 is required for full induction of Ptgs2 mRNA in colonic mesenchymal stem cells in mice. Gastroenterology. 2012;71:3831–3840. doi:https://doi.org/10.1158/0008-5472.CAN-10-4002.BONE.
- Mizuno H, Sakamoto C, Matsuda K, Wada K, Uchida T, Noguchi H, Akamatsu T, Kasuga M. Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology. 1997;112(2):387–397. doi:https://doi.org/10.1053/gast.1997.v112.pm9024292.
- McGuire JJ, Saifeddine M, Triggle CR, Sun K, Hollenberg MD. 2-Furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. J Pharmacol Exp Ther. 2004;309(3):1124–1131. doi:https://doi.org/10.1124/jpet.103.064584.
- Hollenberg MD, Renaux B, Hyun E, Houle S, Vergnolle N, Saifeddine M, Ramachandran R. Derivatized 2-furoyl-LIGRLO-amide, a versatile and selective probe for proteinase-activated receptor 2: binding and visualization. J Pharmacol Exp Ther. 2008;326(2):453–462. doi:https://doi.org/10.1124/jpet.108.136432.
- Bohm SK, Khitin LM, Grady EF, Aponte G, Payan DG, Bunnett NW. Mechanisms of desensitization and resensitization of protease-activated receptor-2. Biochemistry. 1996;271:22003–22016.
- Al-Ani B, Saifeddine M, Kawabata A, Hollenberg MD. Proteinase activated receptor 2: role of extracellular loop 2 for ligand-mediated activation. Br J Pharmacol. 1999;128(5):1105–1113. doi:https://doi.org/10.1038/sj.bjp.0702834.
- Bushell TJ, Plevin R, Cobb S, Irving AJ. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology. 2006;50(6):714–725. doi:https://doi.org/10.1016/j.neuropharm.2005.11.024.
- Stoermer MJ, Flanagan B, Beyer RL, Madala PK, Fairlie DP. Structures of peptide agonists for human protease activated receptor 2. Bioorg Med Chem Lett. 2012;22(2):916–919. doi:https://doi.org/10.1016/j.bmcl.2011.12.029.
- Hobbs SS, Goettel JA, Liang D, Yan F, Edelblum KL, Frey MR, Mullane MT, Polk DB. TNF transactivation of EGFR stimulates cytoprotective COX-2 expression in gastrointestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301(2):G220–9. doi:https://doi.org/10.1152/ajpgi.00383.2010.
- Polk DB, Tong W. Epidermal and hepatocyte growth factors stimulate chemotaxis in an intestinal epithelial cell line. Am J Physiol - Cell Physiol. 1999;277(6):C1149–C1159. doi:https://doi.org/10.1152/ajpcell.1999.277.6.c1149.
- Darmoul D, Gratio V, Devaud H, Laburthe M. Devaud H le`ne, Laburthe M. Protease-activated receptor 2 in colon cancer. J Biol Chem. 2004;279(20):20927–20934. doi:https://doi.org/10.1074/jbc.M401430200.
- van der Merwe JQ, Hollenberg MD, MacNaughton WK. EGF receptor transactivation and MAP kinase mediate proteinase-activated receptor-2-induced chloride secretion in intestinal epithelial cells. Am J Physiol Liver Physiol. 2007;294:G441–G451. doi:https://doi.org/10.1152/ajpgi.00303.2007.
- Kriaa A, Jablaoui A, Mkaouar H, Akermi N, Maguin E, Rhimi M. Serine proteases at the cutting edge of IBD: focus on gastrointestinal inflammation. FASEB J. 2020;34(6):7270–7282. doi:https://doi.org/10.1096/fj.202000031RR.
- Lau C, Lytle C, Straus DS, DeFea KA. Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am J Physiol - Cell Physiol. 2010;300(1):113–123. doi:https://doi.org/10.1152/ajpcell.00162.2010.
- Wang H, Wen S, Bunnett NW, Leduc R, Hollenberg MD, MacNaughton WK. Proteinase-activated receptor-2 induces cyclooxygenase-2 expression through beta-catenin and cyclic AMP-response element-binding protein. J Biol Chem. 2008;283(2):809–815. doi:https://doi.org/10.1074/jbc.M703021200.
- Kirkby NS, Chan MV, Zaiss AK, Garcia-Vaz E, Jiao J, Berglund LM, Verdu EF, Ahmetaj-Shala B, Wallace JL, Herschman HR, et al. Systematic study of constitutive cyclooxygenase-2 expression: role of NF-kappaB and NFAT transcriptional pathways. Proc Natl Acad Sci U S A. 2016;113(2):434–439. doi:https://doi.org/10.1073/pnas.1517642113.
- MacNaughton WK, Cushing K. Role of constitutive cyclooxygenase-2 in prostaglandin-dependent secretion in mouse colon in vitro. J Pharmacol Exp Ther. 2000;293:539–544.
- Dubois RN. Role of inflammation and inflammatory mediators in colorectal cancer. Trans Am Clin Climatol Assoc. 2014;125:358–372.
- Franks LM, Hemmings VJ. A cell line from an induced carcinoma of mouse rectum. J Pathol. 1978;124(1):35–38. doi:https://doi.org/10.1002/path.1711240108.
- Wang Z. Transactivation of epidermal growth factor receptor by G protein-coupled receptors: recent progress, challenges and future research. Int J Mol Sci. 2016:17. doi:https://doi.org/10.3390/ijms17010095.
- Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G276–85. doi:https://doi.org/10.1152/ajpgi.00340.2007.
- Yamaoka T, Frey MR, Dise RS, Bernard JK, Polk DB. Specific epidermal growth factor receptor autophosphorylation sites promote mouse colon epithelial cell chemotaxis and restitution. Am J Physiol Gastrointest Liver Physiol. 2011;301(2):G368–76. doi:https://doi.org/10.1152/ajpgi.00327.2010.
- Campbell K, Casanova J. A common framework for EMT and collective cell migration. Development. 2016;143(23):4291–4300. doi:https://doi.org/10.1242/dev.139071.
- Kim H-J, Lee S-H, Jeong S, Hong S-J. Protease-activated receptors 2-antagonist suppresses asthma by inhibiting reactive oxygen species-thymic stromal lymphopoietin inflammation and epithelial tight junction degradation. Allergy Asthma Immunol Res. 2019;11(4):560. doi:https://doi.org/10.4168/AAIR.2019.11.4.560.
- Wang J, Kang X, Huang Z, Shen L, Luo Q, Li M, Luo L, Tu J, Han M, Ye J. Protease-activated receptor-2 decreased zonula occludens-1 and claudin-1 expression and induced epithelial barrier dysfunction in allergic rhinitis. Am J Rhinol Allergy. 2021;35(1):26–35. doi:https://doi.org/10.1177/1945892420932486.
- Groschwitz K, Wu D, Osterfeld H, Ahrens R, Hogan S. Chymase-mediated intestinal epithelial permeability is regulated by a protease-activating receptor/matrix metalloproteinase-2-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2013;304(5):G479–G489. doi:https://doi.org/10.1152/AJPGI.00186.2012.
- Zhou Q, Wang Y-W, Ni P-F, Chen Y-N, Dong H-Q, Qian Y-N. Effect of tryptase on mouse brain microvascular endothelial cells via protease-activated receptor 2. J Neuroinflammation. 2018;15(1). doi:https://doi.org/10.1186/s12974-018-1287-1.
- Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, Wallace JL, Hollenberg MD, Bunnett NW, Garcia-Villar R, et al. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. 2002;161(5):1903–1915. doi:https://doi.org/10.1016/S0002-9440(10)64466-5.
- Hyun E, Andrade-Gordon P, Steinhoff M, Vergnolle N. Protease-activated receptor-2 activation: a major actor in intestinal inflammation. Gut. 2008;57(9):1222–1229. doi:https://doi.org/10.1136/gut.2008.150722.
- Lohman RJ, Cotterell AJ, Suen J, Liu L, Do AT, Vesey DA, Fairlie DP. Antagonism of protease-activated receptor 2 protects against experimental colitis. J Pharmacol Exp Ther. 2012;340(2):256–265. doi:https://doi.org/10.1124/jpet.111.187062.