
ABSTRACT
Maintaining epithelial homeostasis is crucial to allow embryo development but also the protective barrier which is ensured by the epidermis. This homeostasis is regulated through the expression of several molecules among which EGFR and E-cadherin which are of major importance. Indeed, defects in the regulation of these proteins lead to abnormalities in cell adhesion, proliferation, differentiation, and migration. Hence, regulation of these two proteins is of the utmost importance as they are involved in numerous skin pathologies and cancers. In the last decades it has been described several pathways of regulation of these two proteins and notably several mechanisms of cross-regulation between these partners. In this review, we aimed to describe the current understanding of the regulation of EGFR and interactions between EGFR and E-cadherin and, in particular, the implication of these cross-regulations in epithelium homeostasis. We pay particular attention to PTP1B, a phosphatase involved in the regulation of EGFR.
Introduction
Epithelia are specific tissues that support the structure of embryos and many organs but also act as a protective barrier against environmental insults and pathogens. Their structure is of graded complexities from monolayers as in the gut to multilayers as in the epidermis which is a stratified squamous epithelium composed of several cell types but mainly of keratinocytes. It is organized into discrete layers of basal, replicative cells, and suprabasal layers that undergo a stringent regulated program of terminal differentiation as cells move outward to the stratum corneum.Citation1 Epidermal keratinocytes hence exhibit growth control mechanisms that coordinate proliferation and differentiation. In human epidermis it takes about 4 weeks for cells to exit the basal layer and be extruded from the skin surface as squames. Indeed, pluristratified epithelia are polarized: basal keratinocytes are those that renew the tissue population while apical keratinocytes are highly differentiated maintaining a squamous cornified layer of dead keratinocytes endowed with highly resistant properties in contact to environmental media. The epidermis hence replenishes itself through a process during which the number of cell divisions in the basal layer compensates for the squamous dead cell lost.Citation2 Various proteins control epidermis’ homeostasis by affecting regenerative capacity and differentiation such as EGFR (Epidermal Growth Factor Receptor). The main cohesive key is the intercellular junctions that are built from cadherin as keystones of the epithelial structure. Epidermal adherens junctions are structured from transmembrane E-cadherin (). E-cadherin is of major importance for the maintenance of skin organization as it has been shown that deletion of E-cadherin causes hyperproliferation, defective differentiation and impaired barrier formation.Citation3–5
Figure 1. A Schematic representation of human epidermis. At the outermost side of the skin, the epidermis is composed of several layers of cells: basal, replicative cells, and suprabasal layers that undergo a regulated program of terminal differentiation as cells move outward to the stratum corneum (dead cells). On the dermis interface, in the basal layer of keratinocytes, EGFR signaling is highly activated and cells are proliferative. From the base and up in the epidermis, cells reduce their proliferation and start to differentiate and form more cell-cell junctions, such as tight, adherens and gap junctions; a precise regulation and balance between EGFR and E-Cadherin (E-Cad) are keystone in maintaining structure and integrity of the epidermis. B Regulation of EGFR intracellular trafficking and activation. EGFR activated in the presence of various concentrations of EGF is driven in two distinct endocytic pathways. 1: At low concentrations of EGF, the receptor undergoes the “recycling pathway”, through early and recycling endosomes, before being recycled back to the plasma membrane. 2: At high concentrations of EGF, it undergoes the “degradation pathway”. Signaling is down-regulated by two major mechanisms: PTP1B-dependent dephosphorylation at the RE membrane contact, and degradation inside the endolysosomes upon control by ubiquitination. C Overview of EGFR, E-Cadherin and PTP1B interactions, and their roles in the maintenance of AJ and intracellular signaling. 1: By dephosphorylating beta-catenin, PTP1B promotes the proper assembly of AJ. Their integrity is also conserved by the direct interaction of PTP1B with E-Cadherin (still at AJ location). 2: When PTP1B is not phosphorylated, it cannot dephosphorylate beta-catenin, and fails to maintain AJ integrity. 3: EGFR promotes proliferation, differentiation, cell growth and survival by activating several signaling pathways such as MAPK, STAT, PI3K/Akt and Erk. The latters are also regulated by E-Cadherin. EGFR interaction with E-Cadherin through galectin-7 partially impairs EGFR phosphorylation, contributing to the E-Cadherin/EGFR balance. 4: p120 direct interaction with EGFR enhances Akt and Erk signaling. 5: PTP1B inhibits EGFR by dephosphorylation, both at the plasma membrane and near the RE (Figure 1b), therefore down-regulating the corresponding signaling pathways. 6: Under tension, vinculin binds to unfolded alpha-catenin, after phosphorylation by Abl. This complex binds myosin II and F-actin, forming an organized contractile actin network.
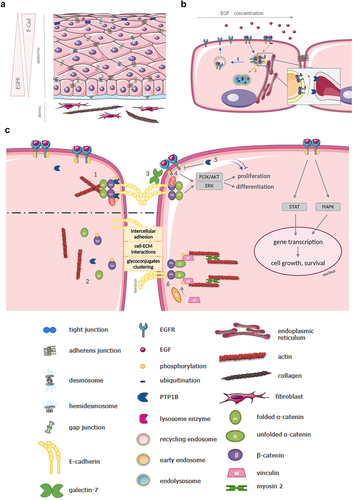
Over the last decade, numerous studies have shed light on the importance of the regulation of EGFR and interactions between EGFR and E-cadherin and, in particular, the implication of these cross-regulations in epithelium homeostasis.Citation6 We propose through this review to point out the latest advances in this area.
EGFR
EGFR, a tyrosine kinase receptor, has been identified as one of the major actors of skin homeostasis. It belongs to the ErbB family of receptors, which consists of 4 closely related receptor tyrosine kinases: EGFR (ErbB-1), HER2/c-neu (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4).Citation7 EGFR has been described to mediate cell proliferation, to modulate terminal differentiation and to control overall epidermal homeostasis.Citation8 Overexpression of EGFR is correlated with progression of numerous human cancers and therefore EGFR is a major target in many cancer therapies.Citation9,Citation10 EGFR is regulated by at least seven different activating ligands in humans: EGF itself, transforming growth factor α (TGF-α), amphiregulin (ARG), epiregulin (EPR), betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF), and epigen (EGN).Citation11 Ligand binding induces a conformational change in the extracellular domain of the receptor resulting in receptor asymmetrical dimerization and autophosphorylation.Citation12 Depending on the position and degree of its phosphorylation EGFR recruits specific signaling complexes and thus has the potential to initiate a wide variety of downstream signaling cascades associated with EGF-dependent responses.Citation10,Citation13 EGFR signaling is involved in many embryonic cellular processes such as development, tissue homeostasis and wound healing. Indeed, it activates the MAPK pathway (cell proliferation and angiogenesis), as well as the STAT pathway (cell proliferation, migration, cell adhesion, and angiogenesis) or even the PI3K/Akt pathway (metabolism, cell survival, inhibition of apoptosis and proliferation).Citation14 Moreover, EGFR overexpression and/or hyperactivation through genetic alterations have been linked to malignant transformation.Citation15
Following EGF-induced dimerization, activated EGFRs are internalized. Signaling from the endocytosed EGFR is subject to down-regulation by at least two mechanisms: (a) ESCRT (endosomal sorting complex required for transport)-mediated sequestration into the ILVs (intraluminal vesicles) of endosomes before lysosomal delivery and subsequent degradation and (b) dephosphorylation by protein tyrosine phosphatases, such as PTP1B (protein tyrosine phosphatase 1B)Citation16 ().
PTP1B
PTP1B is a ubiquitous and abundant intracellular protein Tyrosine Phosphatase (PTP) that interacts with numerous phosphotyrosine proteins present in the cytoplasm, plasma membrane, vesicles, nucleus and intercellular junctions.Citation16,Citation17 Specialized membrane contact sites may be involved in some of these interactions.Citation18 It is the first described PTP among a hundred of the protein tyrosine phosphatase (PTP) family.Citation19,Citation20 The PTP activity of PTP1B relies on the presence of a conserved and nucleophilic catalytic cysteine residue in the P loop of the enzyme.
More generally, PTP1B is involved in the regulation of several cellular pathways in the cell. Indeed, it has been shown to impact other receptors, like the insulin receptor (IR) .Citation21,Citation22 This receptor, being also a member of the tyrosine kinase family, is modulated by PTP1B-dependent dephosphorylation.
A critical role of PTP1B is its regulation of EGFR functions. Indeed, by dephosphorylating the receptor, PTP1B down regulates EGFR and several of its downstream signaling componentsCitation23 (). As mentioned above, EGFR signaling is involved in many cellular processesCitation23 and when dysregulated is involved in cancer development;Citation24 its precise regulation is therefore important in maintaining normal cellular function.Citation7 However, PTP1B regulates several other cell components, among which some may antagonize EGFR signaling. It has therefore been shown that PTP1B can display both tumor promoting and suppressing effects depending on cellular conditions.Citation25,Citation26
E-Cadherin
E-cadherin is a transmembrane glycoprotein that binds epithelial cells together via a homotypic calcium-dependent interaction.Citation27 E-cadherin contains five extracellular domains and a cytoplasmic tail that binds to several effectors to transduce physical and biochemical signals to the cell.Citation28 The five extracellular domains exhibit calcium-binding sites and are involved both in trans- and cis-homotypic interactions constituting multimolecular complexes seeding the intercellular adhesive function.Citation29 The intracellular domain of E-cadherin associates with many different cytoplasmic proteins, and notably the alpha, beta- and p120-catenins (). These E-cadherin binding intracellular partners mediate and regulate the activity of E-cadherin and in particular its association with the actin-myosin cytoskeleton, alongside with its intracellular trafficking and recycling.Citation30 E-cadherin hence forms the core of a mechano-transduction and signaling hub called the adherens junctions (AJ). Its function is critical for the induction and maintenance of tissue barriers by controlling cell adhesion, permeability, polarity, and differentiation in vitro and in vivo. Studies in mammalian cell cultures have identified a central role for E-cadherin in coordinating the establishment of apico–basolateral polarity with the formation of AJ but also desmosomes and tight junctions (TJ).Citation31 The loss of E-cadherin alters the morphology of the skin but does not affect its integrity. Dysfunctional adherens junctions are linked to various pathologies but most relevant to cancerCitation32,Citation33 and inflammatoryCitation34 diseases. As such, several studies have described E-cadherin as a tumor suppressor even if recent studies have shown it may also promote cell migration, invasion and even tumor progression.Citation33 Indeed, the cytoplasmic domains of classic cadherins interact with various signaling-competent partners.Citation35 Thus, homophilic binding of E-cadherin can down-regulates expression levels of receptor tyrosine kinase (RTK) and tyrosine kinase Src and can suppress activation of Wnt/beta-catenin pathway and RTK/PI3K pathways.Citation36
Interaction between EGFR and E-Cadherin and their consequences
Molecular interactions
Various biochemical and microscopy evidence, such as immunoprecipitation and immunofluorescence studies, demonstrated that a fraction of EGFR is detected at sites of cell–cell contactsCitation37 and is present in its phosphorylated active form, thus strongly supporting the concept that E-cadherin can transactivate EGFR.Citation38,Citation39
Qian et al. showed in MDCK cells that E-cadherin could inhibit cellular responses to EGFR stimulation documenting that mitogenic responsiveness to EGF decreased as cells grew to confluence.Citation40 This desensitization could be overcome by adding antibodies that block E-cadherin function. They also showed that reduced expression of E-cadherin increased both EGFR autophosphorylation and EGF-induced DNA synthesis. These results have been reinforced in head and neck cancer cells with the observation that downregulation of E-cadherin upregulated EGFR at the cellular membrane and at the mRNA level. This upregulation was due to the increase of EGFR mRNA stability, but not to changes in protein stability.Citation26 However, in epithelial ovarian cancer cells E-cadherin silencing was associated with a decrease of EGFR protein levels.Citation37 Notably in these cell lines EGF stimulation did not affect E-cadherin expression in control siRNA-treated and untreated cells.Citation37 Although it has not been shown for EGFR, it has been reported that the overexpression of another member of the family, ERBB2 in human mammary epithelial cells induces inhibition of the transcription of E-cadherin.Citation41
Beyond these crosstalks physical interactions between EGFR and E-cadherin have also been demonstrated as well as with some components of AJ complex such as catenin. This has been confirmed in several cell lines such as for example A431 (human epidermoid carcinoma cells),Citation42 MDCKCitation39 (kidney), EOC (epithelial ovarian carcinoma)Citation37 or HaCaT (human keratinocyte cells).Citation43
The crosstalk between both molecules has firstly been described through interactions via intracellular partners belonging to the AJ complex. As early as the 1990s, it was demonstrated using recombinant proteins that beta-catenin directly interacts with EGFR. This interaction seems to be mediated by the central region of beta-catenin. This region having about 80% identity at the amino acid level with plakoglobin and armadillo protein suggests possible interactions of EGFR with these molecules.Citation42 More recently, it has been shown that EGFR also interacts with another catenin of the AJ complex: δ-catenin. This direct interaction has been demonstrated in bosc23 (human kidney cell line) and CWR22Rv-1 cells (Human Prostate cancer cell line) cells. Moreover, the authors noted that this interaction was decreased in the presence of EGF without affecting the localization of δ-catenin.
Beyond the catenins, other partners of the AJ complex have been shown to be crucial in the interaction between EGFR and E-cadherin. PLEKHA7, a recently demonstrated new component of the AJ, is known to stabilize the molecular complex of E-cadherin by linking it to the minus ends of non-centrosomal microtubules.Citation29 However, it also appears that this molecule obliterates the interaction between EGFR and E-cadherin since upon PLEKHA7 overexpression in epithelial ovarian cancer, the association of E-cadherin with EGFR is lost and subsequent EGFR activation inhibited.Citation37,Citation44 This type of interaction has also been illustrated for the neurofibromatosis type-2 (NF2) tumor suppressor and Merlin (also known as schwannomin). Merlin has been described to be recruited at the cadherin/catenin complex and to regulate cell adhesion.Citation45,Citation46 A few years later it has been demonstrated that it could also binds to and regulates EGFR trafficking through NHE-FR1Citation47 and hence by this interaction with both E-cadherin and EGFR coordinate membrane receptor signaling and adhesion in mammalian cells.Citation48
However, there are other possible interactions through post-translational modifications such as glycosylation. Indeed, three canonical N-glycosylation sites in its extracellular domain were reported on EGFR in the UniProt database and several evidence that EGFR functions are linked to the N-glycosylation status of EGFR have been published.Citation44 These results and the importance of EGFR glycosylation and E-cadherin glycosylation have been nicely illustrated recently by Manwar-Hussain and colleagues.Citation49 Interestingly, we recently identified a galectin as a new partner of EGFR. Galectins are a family of proteins characterized by a common affinity for β-galactoside containing carbohydrates and an evolutionary conserved Carbohydrate Recognition Domain (CRD).Citation39 Among them, galectin-7, which is restricted to the pluristratified epithelia, has newly been shown to impair EGFR phosphorylation through a direct interaction with the carbohydrates of EGFR extracellular domain.Citation43 Moreover, galectin-7 was already known to regulate cell adhesion through the direct regulation of E-cadherin.Citation38 Evidence has now been established by protein recombinant experiments and in silico data that this molecule establishes a bridge between E-cadherin and EGFR regulating the proliferation/differentiation balance notably in the skinCitation43 ().
The interactions between these two molecules can be even more indirect involving additional partners in the regulation of their membrane stability or intracellular trafficking. Hence, reggie proteins (also known as flotillins) favor EGFR internalization by enhancing its phosphorylation and stabilize cell adhesion and AJ formation and dynamics. The defect in AJ formation and dynamics which results in the case of the down-regulation of reggie proteins is caused by an imbalanced rate of the macropinocytic uptake and redelivery of E-cadherin to AJ.Citation50 Thus, it appears that the regulation between E-cadherin and EGFR can also involve many other proteins, particularly at the level of intracellular trafficking and signaling. It is to note that in spite it is not the main subject of this review E-cadherin and EGFR can also impact tight junctions (TJ) through the regulation of claudins, a family of 24 proteins in humans.Citation51 For example, it has been shown that EGF induces expression of claudin −1, −3 and −4 cells but also claudin-2 and −18 in several cell types and cancersCitation52–56
Interactions between PTP1B and cadherins have also been described. The enzyme interacts directly with cadherins leading to a PTP1B/cadherins/catenins complex.Citation57,Citation58 PTP1B direct association with the N-cadherin cytoplasmic domain, especially in neural cells, has been well described.Citation58 More recently, its direct interaction with E-cadherin was also reported in intestinal epithelial cells (CaCo2).Citation59 This binding has been shown to be essential for AJ integrity and cell–cell adhesion in general. Indeed, dephosphorylation of beta-catenin tyrosine residues by PTP1B is needed for its binding to cadherinsCitation58 (). It has been shown that the absence or inhibition of PTP1B prevents those bindings and disrupts adhesion.Citation60 PTP1B is therefore an up-regulator of cell–cell adhesion in several cell types. However, this mechanism also depends on PTP1B phosphorylated state. Indeed, the enzyme is only capable of binding cadherins and dephosphorylating catenins when itself phosphorylated on a tyrosine residue.Citation60 This underscores the importance of PTP1B regulation itself and suggests the presence of a more complex mechanism involving kinases as well as phosphatases.
Intracellular trafficking and signaling
Several findings suggest that E-cadherin may exert an extrinsic control on outside-in signal transducing pathways through interaction with tyrosine kinase receptors such as EGFR. Indeed, during the formation of AJ in keratinocytes, clustering of E-cadherins can induce a ligand-independent activation of the EGFR kinase which seems to involve the formation of higher order molecular complexes between E-cadherin and EGFR.Citation39 This aggregation of E-cadherin into assembling AJ has also been demonstrated in mammary epithelial cells where it induces tyrosine phosphorylation of specific substrates within sites of cell-cell contact.Citation61 Furthermore Fedor-Chaiken and colleagues have demonstrated that EGFR phosphorylation requires the extracellular domain of E-cadherin. However, and interestingly this activation does not impact mammary epithelial cell motility governed by E-cadherin.Citation62 Perrais et al. further demonstrated that E-cadherin homophilic ligation inhibited serum-stimulated cell proliferation by preventing E-cadherin binding to beta-catenin.Citation63
At the same time, it was observed that downregulation of E-cadherin upregulated EGFR expression compared with control siRNA-transfected cells after 72 hours. Cellular membrane localization of EGFR was also increased and downstream activated molecules of the EGFR signaling pathway, p-AKT, and p-ERK, were thus increased.Citation64 Current knowledge of E‐cadherin/catenin–EGFR cross‐talk hence suggests that loss of E‐cadherin function activates PI3K, mTOR, EGFR, or FAK signaling.Citation65
Furthermore, E-cadherin promotes EGFR-mediated cell differentiation of cultured human airway epithelial cells via a pathway involving protein tyrosine phosphatase-dependent EGFR dephosphorylation.Citation66
In a recent study Sehgal P. et al. revealed a key role for EGFR in E-cadherin force sensitive mechanism in A431 (human epidermoid carcinoma cells) and MCF7 (human epithelial breast carcinoma).Citation67 The tension dependent junctional E-cadherin response was shown to involve EGFR mediated Abl activated vinculin via stressed E-cadherin activation of integrin (). This complex cascade of activation further recruits vinculin to junctional domain linking cytoskeletal reorganization and force-modified E-cadherin.
Of note soluble E-Cadherin has been shown to act as a survival factor and to signal through EGFR to mediate its anti-apoptotic effects hence suggesting that soluble E-Cadherin present in patients’ sera might contribute to cancer progression by inhibiting apoptosis in cancer cells.Citation68
Conclusion
Current knowledge underscores that there is a strong link between adhesion molecules such as E-cadherin and growth factor receptors such as EGFR in the regulation of epithelial homeostasis. These interactions involve direct contact but also other partners, such as PTP1B, which regulate both activation pathways. Recent findings provide evidence that E-cadherin can differentially and concurrently regulate specific growth-related signaling pathways in a context-specific fashion, with direct functional consequences for cell proliferation and population growth. Several observations not only reveal a novel, complex role for E-cadherin in normal epithelial cell homeostasis and tissue regeneration, but also provide the basis for a more complete understanding of the consequences of E-cadherin loss on malignant transformation.Citation69 Furthermore, EGFR is also able to regulate differentiation by modulating the expression of adhesion molecules, notably E-cadherin which itself integrates the various concomitant signals.Citation70
Mutations of PTP1B have been documented in various types of cancer.Citation71,Citation72 More precisely, frequent somatic mutations have been identified in primary mediastinal B cell lymphoma and Hodgkin lymphoma.Citation73,Citation74 Most of them (i.e. 60%) are missense mutations located in the catalytic domain (i.e. 83%), inactivating the catalytic activity. Other frequent mutations affect glutamine residues (Q) of the first exon, leading to premature inactivation of the protein. In this case, but also in colon and thyroid cancersCitation75 and leukemia,Citation76 PTP1B mutations are associated with tumor development, as reduced phosphatase activity induces higher phosphorylation of several pathways, such as EGFR, JAK-STAT, Src or some downstream oncogenic targets.Citation77 Refined analysis of deletion mutants of PTP1B demonstrated enhanced proteolysis of WT enzyme linked to higher affinity dimer formation with the exon 6 deleted mutant.Citation78 On the other hand, mutants deleted from exon 2–4 did not impair coexpressed WT PTP1B suggesting additional mechanisms such as competition with ligands and substrates, interaction with novel molecular partners or interference with diverse signaling pathways.Citation79 Interestingly, combinatorial mutations in JAK/STAT effectors and PTP1B have been reported, adding further complexity.Citation73
However, PTP1B can also be a tumor promoter, as it has been described to activate the c-src pathway in colon and breast cancers, the enzyme being frequently overexpressed, alongside with increasing the formation of invadopodia.Citation26 This role has also been shown in prostate cancer cells, where an elevated PTP1B expression correlates with neuroendocrine differentiation.Citation80
Even if mutations have been described outside of the catalytic domain, for example, in the proline-rich motifs thus preventing the enzyme to interact with p130Cas;Citation81 most of them are localized in the catalytic domain and have been identified in several cancers. However, its wide range of substrates and interactions broaden its implication in cellular processes.
PTP1B, by regulating E-cadherin and EGFR via its phosphatase activity, probably allows a very precise control of this balance which opens new therapeutic perspectives. Therefore, the development of PTP1B modulators is a very promising point that has been considered in recent years.Citation82 Moreover, it has been demonstrated that environmental xenobiotics such as naphthoquinones are potent inhibitors of PTP1B and increase EGFR signaling. Furthermore, Li et al., showed that menadione can reduce the secondary effects of treatment with EGFR inhibitors in epithelial tumors. Indeed, the toxicity of EGFR inhibitors on the skin has long been reported, and, if not life-threatening, skin toxicity may cause significant physical and psycho-social discomfort. In their study the authors showed that the use of menadione topical cream can be efficient in the treatment or prevention of EGFR inhibitor-induced skin toxicity.Citation83 In parallel, using mice expressing HER2/Neu and lacking PTP1B specifically in the mammary epithelium, it was also shown that PTP1B delays HER2/Neu-induced mammary cancer.Citation84
In conclusion, the EGFR/E-cadherin balance is a major component of epithelial regulation and homeostasis and its regulation by the phosphatase PTP1B is a major and promising element.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Kulukian A, Fuchs E. Spindle orientation and epidermal morphogenesis. Philos Trans R Soc Lond B Biol Sci. 2013;368(1629):211. doi:10.1098/rstb.2013.0016.
- Fuchs E. Epithelial skin biology: three decades of developmental biology, a hundred questions answered and a thousand new ones to address. Curr Top Dev Biol. 2016;116:357–221.
- Tinkle CL, Lechler T, Pasolli HA, Fuchs E. Conditional targeting of E-cadherin in skin: insights into hyperproliferative and degenerative responses. Proc Natl Acad Sci U S A. 2004;101(2):552–557. doi:10.1073/pnas.0307437100.
- Tunggal JA, Helfrich I, Schmitz A, Schwarz H, Günzel D, Fromm M, Kemler R, Krieg T, Niessen CM. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 2005;24(6):1146–1156. doi:10.1038/sj.emboj.7600605.
- Young P, Boussadia O, Halfter H, Grose R, Berger P, Leone DP, Robenek H, Charnay P, Kemler R, Suter U. E-cadherin controls adherens junctions in the epidermis and the renewal of hair follicles. EMBO J. 2003;22(21):5723–5733. doi:10.1093/emboj/cdg560.
- Andl CD, Rustgi AK. No one-way street: cross-talk between e-cadherin and receptor tyrosine kinase (RTK) signaling: a mechanism to regulate RTK activity. Cancer Biol Ther. 2005;4(1):28–31. doi:10.4161/cbt.4.1.1431.
- Herbst RS. Review of epidermal growth factor receptor biology. Int J Radiat Oncol Biol Phys. 2004;59(2):21–26. doi:10.1016/j.ijrobp.2003.11.041.
- Peus D, Hamacher L, Pittelkow MR. EGF-receptor tyrosine kinase inhibition induces keratinocyte growth arrest and terminal differentiation. J Invest Dermatol. 1997;109(6):751–756. doi:10.1111/1523-1747.ep12340759.
- Roskoski R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi:10.1016/j.phrs.2013.11.002.
- Roskoski R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res. 2019;139:395–411. doi:10.1016/j.phrs.2018.11.014.
- Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2014;6(4):a020768. doi:10.1101/cshperspect.a020768.
- Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24:26–34.
- Holcmann M, Sibilia M. Mechanisms underlying skin disorders induced by EGFR inhibitors. Mol Cell Oncol. 2015;2(4):e1004969. doi:10.1080/23723556.2015.1004969.
- Di Domenico M, Giordano A. Signal transduction growth factors: the effective governance of transcription and cellular adhesion in cancer invasion. Oncotarget. 2017;8(22):36869–36884. doi:10.18632/oncotarget.16300.
- Baumdick M, Brüggemann Y, Schmick M, Xouri G, Sabet O, Davis L, Chin JW, Bastiaens PIH. EGF-dependent re-routing of vesicular recycling switches spontaneous phosphorylation suppression to EGFR signaling. Elife. 2015;4:e12223. doi:10.7554/eLife.12223.
- Mertins P, Eberl HC, Renkawitz J, Olsen JV, Tremblay ML, Mann M, Ullrich A, Daub H. Investigation of protein-tyrosine phosphatase 1B function by quantitative proteomics. Mol Cell Proteomics. 2008;7(9):1763–1777. doi:10.1074/mcp.M800196-MCP200.
- Feldhammer M, Uetani N, Miranda-Saavedra D, Tremblay ML. PTP1B: a simple enzyme for a complex world. Crit Rev Biochem Mol Biol. 2013;48(5):430–445. doi:10.3109/10409238.2013.819830.
- Wong LH, Eden ER, Futter CE. Roles for ER:endosome membrane contact sites in ligand-stimulated intraluminal vesicle formation. Biochem Soc Trans. 2018;46(5):1055–1062. doi:10.1042/BST20170432.
- Tonks NK, Diltz CD, Fischer EH. Characterization of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. 1988;263(14):6731–6737. doi:10.1016/S0021-9258(18)68703-4.
- Tonks NK, Diltz CD, Fischer EH. Purification of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. 1988;263(14):6722–6730. doi:10.1016/S0021-9258(18)68702-2.
- Shi K, Egawa K, Maegawa H, Nakamura T, Ugi S, Nishio Y, Kashiwagi A. Protein-tyrosine phosphatase 1B associates with insulin receptor and negatively regulates insulin signaling without receptor internalization. J Biochem. 2004;136(1):89–96. doi:10.1093/jb/mvh094.
- Galic S, Hauser C, Kahn BB, Haj FG, Neel BG, Tonks NK, Tiganis T. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol. 2005;25(2):819–829. doi:10.1128/MCB.25.2.819-829.2005.
- Haj FG, Markova B, Klaman LD, Bohmer FD, Neel BG. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B. J Biol Chem. 2003;278(2):739–744. doi:10.1074/jbc.M210194200.
- Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12(1):3–20. doi:10.1002/1878-0261.12155.
- Bentires-Alj M, Neel BG. Protein-tyrosine phosphatase 1B is required for HER2/ Neu –Induced breast cancer. Cancer Res. 2007;67(6):2420–2424. doi:10.1158/0008-5472.CAN-06-4610.
- Lessard L, Stuible M, Tremblay ML. The two faces of PTP1B in cancer. Biochim Biophys Acta. 2010;1804(3):613–619. doi:10.1016/j.bbapap.2009.09.018.
- Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6(8):622–634. doi:10.1038/nrm1699.
- Lecuit T, Yap AS. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat Cell Biol. 2015;17(5):533–539. doi:10.1038/ncb3136.
- Niessen CM, Leckband D, Yap AS. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev. 2011;91(2):691–731. doi:10.1152/physrev.00004.2010.
- Coopman P, Djiane A. Adherens junction and E-Cadherin complex regulation by epithelial polarity. Cell Mol Life Sci. 2016;73(18):3535–3553. doi:10.1007/s00018-016-2260-8.
- Watabe M, Nagafuchi A, Tsukita S, Takeichi M. Induction of polarized cell-cell association and retardation of growth by activation of the E-cadherin-catenin adhesion system in a dispersed carcinoma line. J Cell Biol. 1994;127(1):247–256. doi:10.1083/jcb.127.1.247.
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, Ewald AJ. E-cadherin is required for metastasis in multiple models of breast cancer. Nature. 2019;573(7774):439–444. doi:10.1038/s41586-019-1526-3.
- Kaszak I, Witkowska-Piłaszewicz O, Niewiadomska Z, Dworecka-Kaszak B, Ngosa Toka F, Jurka P. Role of cadherins in cancer-A review. Int J Mol Sci. 2020;21(20):E7624. doi:10.3390/ijms21207624.
- Bandyopadhyay C, Schecterson L, Gumbiner BM. E-cadherin activating antibodies limit barrier dysfunction and inflammation in mouse inflammatory bowel disease. Tissue Barriers. 2021;9(4):1940741. doi:10.1080/21688370.2021.1940741.
- van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65(23):3756–3788. doi:10.1007/s00018-008-8281-1.
- Loh C-Y, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, Chong PP, Looi CY. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: signaling, therapeutic implications, and challenges. Cells. 2019;8(10):E1118. doi:10.3390/cells8101118.
- Rea K, Roggiani F, De Cecco L, Raspagliesi F, Carcangiu ML, Nair-Menon J, Bagnoli M, Bortolomai I, Mezzanzanica D, Canevari S, et al. Simultaneous E-cadherin and PLEKHA7 expression negatively affects E-cadherin/EGFR mediated ovarian cancer cell growth. J Exp Clin Cancer Res. 2018;37(1):146. doi:10.1186/s13046-018-0796-1.
- Advedissian T, Proux-Gillardeaux V, Nkosi R, Peyret G, Nguyen T, Poirier F, Viguier M, Deshayes F. E-cadherin dynamics is regulated by galectin-7 at epithelial cell surface. Sci Rep. 2017;7(1):17086. doi:10.1038/s41598-017-17332-y.
- Pece S, Gutkind JS. Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem. 2000;275(52):41227–41233. doi:10.1074/jbc.M006578200.
- Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23(8):1739–1748. doi:10.1038/sj.emboj.7600136.
- D’souza B, Taylor-Papadimitriou J. Overexpression of ERBB2 in human mammary epithelial cells signals inhibition of transcription of the E-cadherin gene. Proc Natl Acad Sci U S A. 1994;91(15):7202–7206. doi:10.1073/pnas.91.15.7202.
- Hoschuetzky H, Aberle H, Kemler R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127(5):1375–1380. doi:10.1083/jcb.127.5.1375.
- Proux-Gillardeaux V, Advedissian T, Perin C, Gelly J-C, Viguier M, Deshayes F. Identification of a new regulation pathway of EGFR and E-cadherin dynamics. Sci Rep. 2021;11(1):22705. doi:10.1038/s41598-021-02042-3.
- Ling Y-H, Li T, Perez-Soler R, Haigentz M. Activation of ER stress and inhibition of EGFR N-glycosylation by tunicamycin enhances susceptibility of human non-small cell lung cancer cells to erlotinib. Cancer Chemother Pharmacol. 2009;64(3):539–548. doi:10.1007/s00280-008-0902-8.
- Curto M, McClatchey AI. Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. Br J Cancer. 2008;98(2):256–262. doi:10.1038/sj.bjc.6604002.
- Lallemand D, Curto M, Saotome I, Giovannini M, McClatchey AI. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003;17(9):1090–1100. doi:10.1101/gad.1054603.
- Lazar CS, Cresson CM, Lauffenburger DA, Gill GN. The Na + /H + exchanger regulatory factor stabilizes epidermal growth factor receptors at the cell surface. Mol Biol Cell. 2004;15(12):5470–5480. doi:10.1091/mbc.e04-03-0239.
- Curto M, Cole BK, Lallemand D, Liu C-H, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177(5):893–903. doi:10.1083/jcb.200703010.
- Manwar Hussain MR, Iqbal Z, Qazi WM, Hoessli DC. Charge and polarity preferences for N-glycosylation: a genome-wide in silico study and its implications regarding constitutive proliferation and adhesion of carcinoma cells. Front Oncol. 2018;8:29. doi:10.3389/fonc.2018.00029.
- Solis GP, Schrock Y, Hülsbusch N, Wiechers M, Plattner H, Stuermer CAO, Nusrat A. Reggies/flotillins regulate E-cadherin-mediated cell contact formation by affecting EGFR trafficking. Mol Biol Cell. 2012;23(10):1812–1825. doi:10.1091/mbc.e11-12-1006.
- Zhang Y, Wu S, Xia Y, Sun J, Denning PW. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PLoS One. 2013;8(3):e58606. doi:10.1371/journal.pone.0058606.
- de Souza WF, Fortunato-Miranda N, Robbs BK, de Araujo WM, de-Freitas-Junior JC, Bastos LG, Viola JPB, Morgado-Díaz JA, Lee JW. Claudin-3 overexpression increases the malignant potential of colorectal cancer cells: roles of ERK1/2 and PI3K-Akt as modulators of EGFR signaling. PLoS One. 2013;8(9):e74994. doi:10.1371/journal.pone.0074994.
- Peter Y, Comellas A, Levantini E, Ingenito EP, Shapiro SD. Epidermal growth factor receptor and claudin-2 participate in A549 permeability and remodeling: implications for non-small cell lung cancer tumor colonization. Mol Carcinog. 2009;48(6):488–497. doi:10.1002/mc.20485.
- Singh AB, Harris RC. Epidermal growth factor receptor activation differentially regulates claudin expression and enhances transepithelial resistance in Madin-Darby canine kidney cells. J Biol Chem. 2004;279(5):3543–3552. doi:10.1074/jbc.M308682200.
- Takasawa K, Takasawa A, Osanai M, Aoyama T, Ono Y, Kono T, Hirohashi Y, Murata M, Sawada N. Claudin-18 coupled with EGFR/ERK signaling contributes to the malignant potentials of bile duct cancer. Cancer Lett. 2017;403:66–73. doi:10.1016/j.canlet.2017.05.033.
- Zhang L, Wang Y, Zhang B, Zhang H, Zhou M, Wei M, Dong Q, Xu Y, Wang Z, Gao L, et al. Claudin-3 expression increases the malignant potential of lung adenocarcinoma cells: role of epidermal growth factor receptor activation. Oncotarget. 2017;8(14):23033–23047. doi:10.18632/oncotarget.14974.
- Balsamo J, Arregui C, Leung T, Lilien J. The nonreceptor protein tyrosine phosphatase PTP1B binds to the cytoplasmic domain of N-cadherin and regulates the cadherin-actin linkage. J Cell Biol. 1998;143(2):523–532. doi:10.1083/jcb.143.2.523.
- Xu G, Arregui C, Lilien J, Balsamo J. PTP1B modulates the association of beta-catenin with N-cadherin through binding to an adjacent and partially overlapping target site. J Biol Chem. 2002;277(51):49989–49997. doi:10.1074/jbc.M206454200.
- Sheth P, Seth A, Atkinson KJ, Gheyi T, Kale G, Giorgianni F, Desiderio DM, Li C, Naren A, Rao R. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem J. 2007;402(2):291–300. doi:10.1042/BJ20060665.
- Balsamo J, Leung T, Ernst H, Zanin MK, Hoffman S, Lilien J. Regulated binding of PTP1B-like phosphatase to N-cadherin: control of cadherin-mediated adhesion by dephosphorylation of beta-catenin. J Cell Biol. 1996;134(3):801–813. doi:10.1083/jcb.134.3.801.
- Kinch MS, Petch L, Zhong C, Burridge K. E-cadherin engagement stimulates tyrosine phosphorylation. Cell Adhes Commun. 1997;4(6):425–437. doi:10.3109/15419069709004459.
- Fedor-Chaiken M, Hein PW, Stewart JC, Brackenbury R, Kinch MS. E-cadherin binding modulates EGF receptor activation. Cell Commun Adhes. 2003;10(2):105–118. doi:10.1080/cac.10.2.105.118.
- Perrais M, Chen X, Perez-Moreno M, Gumbiner BM, Schwarzbauer J. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol Biol Cell. 2007;18(6):2013–2025. doi:10.1091/mbc.e06-04-0348.
- Wang D, Su L, Huang D, Zhang H, Shin DM, Chen ZG. Downregulation of E-Cadherin enhances proliferation of head and neck cancer through transcriptional regulation of EGFR. Mol Cancer. 2011;10(1):116. doi:10.1186/1476-4598-10-116.
- Li D, Lo W, Rudloff U. Merging perspectives: genotype-directed molecular therapy for hereditary diffuse gastric cancer (HDGC) and E-cadherin-EGFR crosstalk. Clin Transl Med. 2018;7(1):7. doi:10.1186/s40169-018-0184-7.
- Kim S, Schein AJ, Nadel JA. E-cadherin promotes EGFR-mediated cell differentiation and MUC5AC mucin expression in cultured human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289(6):L1049–1060. doi:10.1152/ajplung.00388.2004.
- Sehgal P, Kong X, Wu J, Sunyer R, Trepat X, Leckband D. Epidermal growth factor receptor and integrins control force-dependent vinculin recruitment to E-cadherin junctions. J Cell Sci. 2018;131:jcs206656. doi:10.1242/jcs.206656.
- Inge LJ, Barwe SP, D’Ambrosio J, Gopal J, Lu K, Ryazantsev S, Rajasekaran SA, Rajasekaran AK. Soluble E-cadherin promotes cell survival by activating epidermal growth factor receptor. Exp Cell Res. 2011;317(6):838–848. doi:10.1016/j.yexcr.2010.12.025.
- Georgopoulos NT, Kirkwood LA, Walker DC, Southgate J, Anderson K. Differential regulation of growth-promoting signalling pathways by E-cadherin. PLoS One. 2010;5(10):e13621. doi:10.1371/journal.pone.0013621.
- Rübsam M, Mertz AF, Kubo A, Marg S, Jüngst C, Goranci-Buzhala G, Schauss AC, Horsley V, Dufresne ER, Moser M, et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat Commun. 2017;8(1):1250. doi:10.1038/s41467-017-01170-7.
- Hendriks WJAJ, Pulido R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim Biophys Acta. 2013;1832(10):1673–1696. doi:10.1016/j.bbadis.2013.05.022.
- Villamar-Cruz O, Loza-Mejía MA, Arias-Romero LE, Camacho-Arroyo I. Recent advances in PTP1B signaling in metabolism and cancer. Biosci Rep. 2021;41(11):BSR20211994. doi:10.1042/BSR20211994.
- Gunawardana J, Chan FC, Telenius A, Woolcock B, Kridel R, Tan KL, Ben-Neriah S, Mottok A, Lim RS, Boyle M, et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet. 2014;46(4):329–335. doi:10.1038/ng.2900.
- Liu R, Sun Y, Berthelet J, Bui L-C, Xu X, Viguier M, Dupret J-M, Deshayes F, Rodrigues Lima F. Biochemical, enzymatic, and computational characterization of recurrent somatic mutations of the human protein tyrosine phosphatase PTP1B in primary mediastinal B cell lymphoma. IJMS. 2022;23(13):7060. doi:10.3390/ijms23137060.
- Mei W, Wang K, Huang J, Zheng X, Maki CG. Cell transformation by PTP1B truncated mutants found in human colon and thyroid tumors. PLoS One. 2016;11(11):e0166538. doi:10.1371/journal.pone.0166538.
- Le Sommer S, Morrice N, Pesaresi M, Thompson D, Vickers MA, Murray GI, Mody N, Neel BG, Bence KK, Wilson HM, et al. Deficiency in protein tyrosine phosphatase PTP1B shortens lifespan and leads to development of acute leukemia. Cancer Res. 2018;78(1):75–87. doi:10.1158/0008-5472.CAN-17-0946.
- Stuible M, Doody KM, Tremblay ML. PTP1B and TC-PTP: regulators of transformation and tumorigenesis. Cancer Metastasis Rev. 2008;27(2):215–230. doi:10.1007/s10555-008-9115-1.
- Zahn M, Marienfeld R, Melzner I, Heinrich J, Renner B, Wegener S, Mießner A, Barth TFE, Dorsch K, Brüderlein S, et al. A novel PTPN1 splice variant upregulates JAK/STAT activity in classical Hodgkin lymphoma cells. Blood. 2017;129(11):1480–1490. doi:10.1182/blood-2016-06-720516.
- Zahn M, Kaluszniak B, Möller P, Marienfeld R. The PTP1B mutant PTP1B∆2-4 is a positive regulator of the JAK/STAT signalling pathway in Hodgkin lymphoma. Carcinogenesis. 2021;42(4):517–527. doi:10.1093/carcin/bgaa144.
- Wu C, Zhang L, Bourne PA, Reeder JE, Di Sant’agnese PA, Yao JL, Na Y, Huang J. Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differentiation of prostate cancer. Prostate. 2006;66(11):1125–1135. doi:10.1002/pros.20412.
- Liu F, Sells MA, Chernoff J. Transformation suppression by protein tyrosine phosphatase 1B requires a functional SH3 ligand. Mol Cell Biol. 1998;18(1):250–259. doi:10.1128/MCB.18.1.250.
- Sharma B, Xie L, Yang F, Wang W, Zhou Q, Xiang M, Zhou S, Lv W, Jia Y, Pokhrel L, et al. Recent advance on PTP1B inhibitors and their biomedical applications. Eur J Med Chem. 2020;199:112376. doi:10.1016/j.ejmech.2020.112376.
- Li T, Perez-Soler R. Skin toxicities associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009;4(2):107–119. doi:10.1007/s11523-009-0114-0.
- Balavenkatraman KK, Aceto N, Britschgi A, Mueller U, Bence KK, Neel BG, Bentires-Alj M. Epithelial protein-tyrosine phosphatase 1B contributes to the induction of mammary tumors by HER2/Neu but is not essential for tumor maintenance. Mol Cancer Res. 2011;9(10):1377–1384. doi:10.1158/1541-7786.MCR-11-0198.