
ABSTRACT
Previous data provided evidence for a critical role of desmosomes to stabilize intestinal epithelial barrier (IEB) function. These studies suggest that desmosomes not only contribute to intercellular adhesion but also play a role as signaling hubs. The contribution of desmosomal plaque proteins plakophilins (PKP) in the intestinal epithelium remains unexplored. The intestinal expression of PKP2 and PKP3 was verified in human gut specimens, human intestinal organoids as well as in Caco2 cells whereas PKP1 was not detected. Knock-down of PKP2 using siRNA in Caco2 cells resulted in loss of intercellular adhesion and attenuated epithelial barrier. This was paralleled by changes of the whole desmosomal complex, including loss of desmoglein2, desmocollin2, plakoglobin and desmoplakin. In addition, tight junction proteins claudin1 and claudin4 were reduced following the loss of PKP2. Interestingly, siRNA-induced loss of PKP3 did not change intercellular adhesion and barrier function in Caco2 cells, while siRNA-induced loss of both PKP2 and PKP3 augmented the changes observed for reduced PKP2 alone. Moreover, loss of PKP2 and PKP2/3, but not PKP3, resulted in reduced activity levels of protein kinase C (PKC). Restoration of PKC activity using Phorbol 12-myristate 13-acetate (PMA) rescued loss of intestinal barrier function and attenuated the reduced expression patterns of claudin1 and claudin4. Immunostaining, proximity ligation assays and co-immunoprecipitation revealed a direct interaction between PKP2 and PKC. In summary, our in vitro data suggest that PKP2 plays a critical role for intestinal barrier function by providing a signaling hub for PKC-mediated expression of tight junction proteins claudin1 and claudin4.
Introduction
The gastrointestinal tract is lined by a single layer of epithelial cells that provide a critical interface between the inner part of the body and the gut lumen which contains nutrients, microbiota and pathogens.Citation1,Citation2 One of the pivotal tasks of the intestinal epithelium is its selective barrier function to prevent bacterial and pathogen translocation.Citation3
The paracellular space is sealed by the apical junctional complex.Citation1,Citation2 Within this complex, tight junctions (TJ) regulate permeability by the presence of different transmembrane proteins, known as claudins and occludin, in the cell lateral membrane.Citation1,Citation4 Furthermore, adherens junctions (AJ) containing the transmembrane protein E-cadherin contribute to the mechanical stability of the intestinal barrier. A comparable role has been attributed to the desmosomes. Previous data indicate that the integrity of desmosomes is crucial to maintain intestinal barrier function, both under basal and inflammatory states, as observed in IBD.Citation5–8 Interestingly, there is increasing evidence that desmosomes not only stabilize intercellular adhesion but provide signaling hubs to regulate cellular behavior and TJs.Citation9
Desmosomes in the intestine consist of the adhesion molecules desmoglein 2 (DSG2) and desmocollin 2 (DSC2). They bind in either homo- or heterophilic manner in the intercellular space and mediate intercellular adhesion between neighboring enterocytes. At their intracellular domains, DSG2 and DSC2 are linked to plaque proteins known as plakoglobin (PG), plakophilins (PKP) and desmoplakin (DP), which, in turn, tethers the desmosome complex to the intermediate filament system.Citation1
There is evidence especially in keratinocytes and cardiomyocytes, that plakophilins are critical to regulate desmosome function and cell adhesion.Citation10–12 Plakophilins belong to the armadillo protein family which are characterized by a repetitive series of 45 amino acids, named arm repeats. This conserved domain is flanked by a carboxyl-terminal tail and an amino-terminal head that interacts with numerous binding partners.Citation13 As a result of alternative splicing, PKP1 and PKP2 exist in a long a and a shorter b isoform whereas there were no reports on isoforms for PKP3. In the heart, the genetic disease arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVC) is partly caused by PKP2 mutations leading to vulnerable cell contacts and ultimately to heart failure especially in young adults and athletes.Citation14 In lung carcinomas, PKP3 overexpression was correlated with poor prognosis, suggesting that PKP3 functions as an oncogene.Citation15 Moreover, reduced levels of PKP3 lead to the stimulation of neoplastic progression and metastasis in poorly differentiated tumors, underlining the essential roles of PKPs in cancer.Citation16 All these findings clearly indicate that the role of PKPs is strongly dependent on the cellular context. However, when focusing on the gut, the specific role of PKPs remains almost entirely unexplored.
Therefore, the aim of the present study was to characterize the presence of PKPs in the human intestine and in different in vitro models to assess the basic contribution of PKPs in the context of intestinal epithelial barrier regulation.
Methods
Test reagents
For PKC activation, we used 100 nM Phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, Munich, Germany) as has been reported previously.Citation17
Human tissue samples
Control human tissue samples were obtained from patients diagnosed with colon carcinoma in which the surgical procedure involved resection from a healthy part of the intestine. Prior to surgery, a written informed consent form to be included in the study, was signed by all patients. The ethical approval was given by the ethical board of the University of Wuerzburg (proposal numbers 113/13, 46/11, 42/16).
Human intestinal organoids
Organoids derived from healthy human small intestines were obtained from gut resections as described previously.Citation18 In brief, the villi were extracted, the residual tissue was washed with HBSS solution (Sigma-Aldrich) and incubated with 20 mL 2 mM EDTA/HBSS solution (Sigma-Aldrich) on a shaker for 30 minutes at 4°C. Thereafter, the tissue was washed with HBSS once more and shaken manually to capture cell fractions. These were controlled under the microscope to ensure the proper quantity of crypts. Subsequently, the supernatants containing the crypts were centrifuged at 300 g for 3 minutes. The pellet was then resuspended in 10 mL basal medium, which consists of DMEM-F12 Advanced (Invitrogen) with N2, B27, antibiotic-antimycotics, Hepes, 2 mM GlutaMAX (all Invitrogen, Carlsbad, USA), 1 mM N-acetylcysteine (Sigma-Aldrich, Munich, Germany). The prepared mixture was finally centrifuged at 300 g for 3 minutes and the supernatant was extracted. The pellet was resuspended in 5000 crypts/ml cold embedding mixture, consisting of 50% Matrigel (Corning, New York, USA) and 50% basal medium. Three droplets at 10 µl per well of the mixture was seeded in a 24-well plate and incubated for ca. 30 minutes until solidified. The following growth factors and inhibitors were added to the culture medium: 500 ng/ml hR-Spondin 1, 100 ng/ml mNoggin, 50 ng/ml mEGF (all from PeproTech, Hamburg, Germany), 10 μM Y-27632 (ROCK inhibitor; Tocris Bioscience, R&D Systems), 10 nM Gastrin ([Leu15]-Gastrin I; Sigma-Aldrich), 10 mM Nicotinamide (Sigma-Aldrich), 500 nM A83-01 (Tocris Bioscience, R&D Systems), 10 μM SB202190 (Sigma-Aldrich), and 500 nM LY2157299 (Axon MedChem, Groningen, Niederlande). Intestinal epithelial cells were expanded as organoids for 3–4 weeks by adding 300 μl of this medium per well. The medium was changed every third day. Organoids for this study were chosen from an existing cryo cell bank, defrosted, cultured, and thereafter used for this study up to passage 30.Citation7
Cell culture
The well-established human CaCo2 cell line (ATCC), which mimics barrier properties represented in human intestine, was cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher) containing 10% FCS, 5% pyruvate and 50 U/ml penicillin-G, 50 μg streptomycin (Biochrom). Experiments were carried out after cells were grown to a confluent monolayer.Citation7
siRNA-mediated silencing of PKP2/3
After grown in 6-well plates overnight, cells were transfected with human PKP2-, PKP3-specific siRNA or with a mix of both siRNAs (Thermo Fisher Scientific, Waltham, USA) and nontarget (n.t.) siRNA as controls (Abcam; Cambridge, UK). Transfection was carried out when cells had reached 80% of confluence.
Transfection was performed using Lipofectamine Transfection LTX Reagent (Thermo Fisher Scientific; Waltham, USA) according to the manufacturer’s protocol. According to preliminary experiments sufficient knockdown of PKP2, PKP3 or both was achieved after 48 h, so that all experiments using knockdown conditions were started after 48 h.
Overexpression of PKP2 and PKP3
PKP2-wt-pCMV5a-Flag and PKP3-wt-pCMV5 plasmids were kindly supplied by Mechthild Hatzfeld (Institute for Molecular Medicine, University of Halle; Germany). First, the plasmids were amplified using MaxiPrep protocol according to manufacturer’s recommendation (Macherey-Nagel, Düren, Germany; Cat.No.740414). Thereafter, 2.5 µg of the plasmids were used for transfection into Caco2 grown to 80% confluency using Lipofectamine LTX Transfection Reagent (Thermo Fisher Scientific; Waltham, USA) according to the manufacturer’s protocol. Experiments were carried out after 24–48 h following transfection. For co-transfection experiments of cells with PKP2 siRNA and PKP3-wtpCMV5, cells were incubated for the same time. Overexpression or knockdown was verified by Western blotting.
Western blot
Cells were grown in 6-well plates and harvested with lysis buffer containing 25 mM HEPES, 2 mM EDTA, 2 mM NaF, and 1% SDS after knockdown was achieved, the lysates were then homogenized and total amount of protein was measured using a BCA assay kit (Thermo Fisher Scientific). After SDS gel electrophoresis, proteins were transferred onto nitrocellulose membrane and incubated with primary antibodies () overnight. The secondary antibodies were then incubated for 1 h at room temperature (RT), signals were detected using chemiluminescent HRP substrate. The measurement of optical density and normalization to ß-actin were performed using ImageLab (ChemiDoc Touch Bio-Rad Laboratories).
Table 1. Primary and secondary antibodies used for this study are shown including the concentrations in Western blot (WB) experiments and in immunostaining (IS).
Immunostaining
The human specimens were embedded in paraffin and sectioned as 1-µm slices. The organoids were digested from Matrigel with Cell Recovery Solution (Thermo Fisher, Waltham, USA) for 1 h on ice, washed with phosphate-buffered saline (PBS) (Sigma Aldrich) and fixed with 4% formaldehyde solution in PBD prepared from PFA. After embedding in histogel (Thermo Fisher) the organoids were also embedded in paraffin. Immunostaining was performed after removal of paraffin as described for epithelial monolayers and human specimens previously.Citation7,Citation18 For Caco2 immunostaining, the medium of confluent monolayers cells grown on coated coverslips were removed with several washing steps using PBS. After fixing cells with 4% paraformaldehyde and permeabilizing them using 0.1% Triton immunostaining was carried out.
Monolayers of Caco2 cells and tissue slides were incubated at 4°C overnight using the primary antibodies outlined in . As secondary antibodies, we used Cy3- or Alexa Fluor 488 labeled goat anti-mouse or goat anti-rabbit, respectively (all diluted 1:600; Dianova, Hamburg, Germany). Coverslips and filters were mounted on glass slides with Vector Shield Mounting Medium as anti-fading compound, which included 4′,6-diamidin-2-phenylindol (DAPI) to stain cell nuclei additionally (Vector Laboratories; Peterborough, UK).
Representative experiments were documented using a confocal microscope (Leica LSM 780; Zeiss, Oberkochen, Germany).
TER-measurements
Cells were grown on 8-well electrode arrays overnight and transfected with anti Plakophilin2/3 siRNA and nontarget (nt) siRNA as a control. Transepithelial electrical resistance (TER) measurements were carried out immediately after transfection at 400 Hz (Applied Biophysics, New York USA; Ibidi, Gräfelfing, Germany). For measuring the effects of PMA activation, cells were treated with PMA 6 h post transfection. Resistance was then monitored throughout the following 24 h.
Protein kinase C activity measurements
After knockdown of plakophilin, cells were treated with PMA for 6 h and harvested with lysis buffer as described above. Protein Kinase C activity was measured using the PKC Kinase Activity Assay Kit (Abcam 139437, Cambridge, UK) according to the manufacturer’s recommendation. The assay is based on a solid phase enzyme-linked immunosorbent assay (ELISA) that utilizes a specific synthetic peptide as a substrate for PKC and a polyclonal antibody that recognizes the phosphorylated form of the substrate. Optical density was measured at 450 nm and relative Protein Kinase C activity was calculated after subtracting the background activity.
Cell viability assay
The amount of viable cells were obtained with CellTiter-Glo 2.0 assay (Promega; catalog G9241, Mannheim, Germany). Cells were grown in 96well opaque-walled multiwall plates and transfected with siRNA as described previously.Citation7 CellTiter-Glo® 2.0 Reagent was added and luminescence signal, which is proportional to the present amount of ATP, was measured. After subtracting background signal, the present amount of ATP is directly proportional to the number of viable cells.
Dispase-based enterocyte dissociation assay
As described beforeCitation9 dispase-based enterocyte dissociation assays were carried to test for changes in intercellular adhesion under the different experimental conditions. Cells were seeded in 12-well plates and after reaching confluence, cells were washed with PBS and incubated with Dispase-II (Sigma-Aldrich) at 36°C for 30 minutes to release the monolayer from the well bottom. Afterward, the cell sheet was exposed to shear stress by constant pipetting 10 times each well. Four pictures with a 4x magnification in brightfield with BZ-9000 (BioRevo, Keyence, Osaka) were taken and the number of fragments/area were counted using with ImageJ.Citation19
FITC–dextran flux measurements across monolayers of cultured epithelial cells
Caco2 cells were seeded on top of transwell filter chambers on 12-well plates (0.4 μm pore size; Falcon, Heidelberg, Germany) as described previously.Citation19 After reaching confluence, cells were rinsed with PBS and incubated with fresh DMEM without phenol red (Sigma) containing 10 mg/ml FITC-dextran (4 kDa). Paracellular flux was assessed by taking 100 μl aliquots from the outer chamber over 2 h of incubation. Fluorescence was measured using a Tecan GENios Microplate Reader (MTX Lab systems, Bradenton, USA) with excitation and emission at 485 and 535 nm, respectively. For all experimental conditions, permeability coefficients (PE) were calculated by the following formula: PE = (ΔCA/Δt* VA)/S* CL where PE = diffusive permeability (cm/s), ΔCA = change of FITC- Dextran concentration, Δt = change of time, VA = volume of the abluminal medium, S = surface area, and CL = constant luminal concentration.
Quantitative (q)RT-PCR
RNA from CaCo2 cells was isolated using TRIZOL and cDNA was synthesized with iScriptTM cDNA Synthesis Kit (Biorad, Munich, Germany). (q)RT-PCR was performed using MESA GREEN qPCR MasterMix Plus for SYBR® Assay No ROX (Eurogentec, Cologne, Germany) on the CFX96 Touch Real-Time PCR Detection System (Biorad, Munich, Germany). Gene expression was analyzed via the Bio-Rad CFX Manager software with ß-Aatin as a reference gene. All reactions were done in duplicates at 60.0°C annealing temperature. Primers were applied at concentration of 5 µM. The following primer sequences were used:
PKP1 f: 5’- GAC CAG GAC AAC TCC ACG TT – 3’; PKP 1 r: 5’ – CTG CTG GTG GTC CCA TAG TT- 3’ (Biomol, Hamburg, Germany)
PKP2 f: 5’- GCC TCC AAC AAA GCA AGT AA – 3’; PKP2 r: 5’ – GAT TTT TGC AGC CGA GAA TA – 3’ (Biomol, Hamburg, Germany)
PKP3 f: 5’ – TGA CTG GCC TCA TCC GAA AC-3’; PKP3 r: 5’ – CTC ACC ACC TTC GTG GAC ATC-3’ (Eurofins)
Proximity ligation assay (PLA)
Cells were seeded on coverslips and knockdown of PKP2 and PKP3 was established as described earlier in the text. After blocking of unspecific binding sites, slides were incubated with primary antibody PKP2 (BD biosciences, Heidelberg, Germany, CatNo: BD610788), and PKC (Abcam, Berlin, Germany, CatNo: ab18746) or PKP3 (thermos Fisher Scientific, Germany, CatNO: 35–7600) and PKC (Abcam, Berlin, Germany, CatNo: ab18746). Next, a pair of oligonucleotide-labeled secondary antibodies (PLUS and MINUS Probes) (Merck, Taufkirchen, Germany) which bind to the primary antibody were applied. When the PLA probes are in close proximity connector oligos join the PLA probes and become ligated by addition of ligase at a dilution of 1:40. As a consequence, a closed circle DNA template is formed and acts as a primer for a DNA polymerase. Finally, labeled oligos hybridize to the complementary sequences within the amplicon, which are then visualized as discrete spots (PLA signals) by microscopy analysis. As negative controls, the same procedure was carried without application of primary antibodies as recommended by the manufacturer.Citation9
Co-immunoprecipitation assay
Cells were seeded on 6-well plates. Monolayer cells at confluency were harvested in RIPA-buffer. Samples were steamed for 1 min and centrifuged for 15 min at 15.000 g and 4°C. Total protein concentration was determined by measuring absorbance at 280 nm.
The Co-IP experiments were done using the immuno- precipitation Starter Pack (GE Healthcare, Germany). The amount of 300–600 μg protein was used. After an initial pre- clearing step of one h at 4°C (500 μl of whole cell lysate with respectively 25 μl protein G/A sepharose beads), antigens were coupled overnight at 4°C to 2.5 μg purified antibody rabbit anti- PKC (Abcam, Cambridge, UK), anti-PKP2, anti-PKP3, or IgG antibodies (all from BD Transduction Laboratories, Heidelberg, Germany) as negative controls, respectively. Protein-antibody complexes were precipitated with a mix of 25 μl protein A and G sepharose beads for 1 h at 4°C. The beads were washed three times with isotonic salt buffer (RIPA-buffer), once with wash-buffer (50 mM TRIS, pH 8) and suspended in 50 μl Laemmli buffer. After denaturation for 5 min at 95°C and a following centrifugation step, the supernatant was analyzed by Western Blot analyses as described above. Detection was performed with mouse anti-PKC diluted 1:1000 (Invitrogen, Carlsbad, CA, USA).
Statistical analysis
Statistical analysis was performed using Prism (GraphPad Software, La Jolla, CA, USA). Data are presented as means ± SE. Statistical significance was assumed for p < .05. Paired Student’s t-test was performed for two-sample group analysis after checking for a Gaussian distribution. Analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test and Bonferroni correction was used for multiple sample groups.
Results
Plakophilin2 and plakophilin3 are expressed in human intestinal tissue
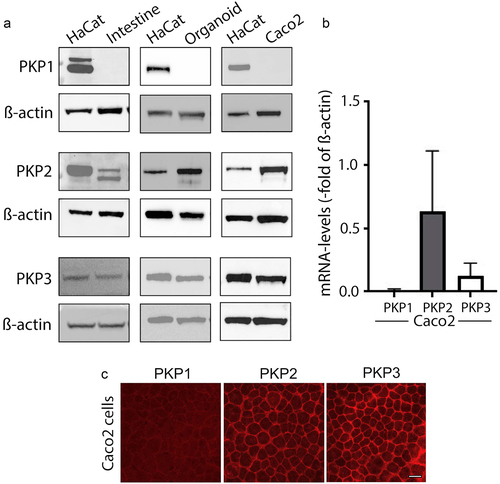
First, we verified which of the plakophilins are expressed in human intestinal tissue, human intestinal organoids and differentiated Caco2 cells. In Western Blot analyses, PKP1 was detected in none of the samples, whereas PKP2 and PKP3 were both present in human intestinal tissue, organoids and Caco2 cells (). Quantitative real-time-PCR confirmed the presence of mRNA for PKP2 and PKP3, but not PKP1 in Caco2 cells (). In immunostaining, both PKP2 and PKP3 were present in Caco2 cells. No specific staining pattern was observed for PKP1 (). In summary, these data confirmed that in intestinal epithelia express PKP2 and PKP3 but not PKP1.
Figure 1. Plakophilin2 and Plakophilin3 are expressed in human intestinal tissue.
Plakophilin2 is required for intercellular adhesion and formation of barrier function in intestinal epithelial cells
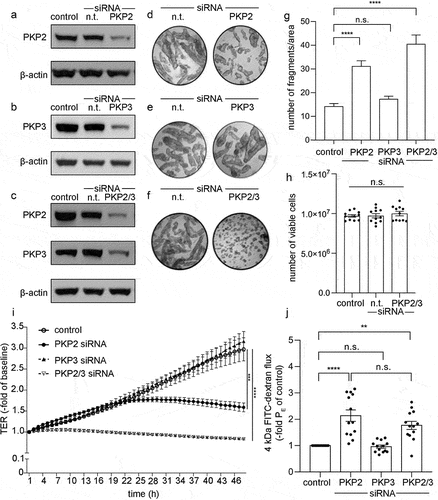
To assess the functional role for PKP2 and PKP3 in the intestinal epithelium, we used differentiated Caco2 cells following knockdown of PKP2, PKP3 or both using a siRNA approach. Sufficient knockdown to 0.31 ± 0.03-fold of controls for PKP2, to 0.19 ± 0.03-fold of controls for PKP3, and of 0.25 ± 0.01-fold/0.18 ± 0.05-fold of controls for PKP2/3 was induced after 48 h of transfection with the various siRNAs (). In dispase-based enterocyte dissociation assays we found that loss of PKP2 induced an increased fragmentation of Caco2 monolayers whereas loss of PKP3 did not affect intercellular adhesion. Simultaneous knockdown of PKP2 and PKP3 resulted in even more pronounced fragmentation than loss of PKP2 alone (). Importantly, control experiments using an MTT-based cell viability assay demonstrated that the number of viable cells was not affected by the knockdown of PKP2, PKP3 or both (). In measurements of TER, Caco2 cell transfected with non-target siRNA (control) showed a continuous increase to 2.97 ± 0.28-fold of baseline within 48 h, indicating a normal differentiation of epithelial barrier function (). In contrast, in cells transfected with siRNA for PKP2, TER augmented only within the first hours of measurements and then remained at a level reaching 1.59 ± 0.11-fold of baseline after 48 h. Caco2 monolayers transfected with siRNA for PKP3 revealed an increase of TER to 3.16 ± 0.24-fold of baseline comparable to the monolayers transfected with nontarget siRNA. Knock down of PKP2/3 even reduced TER to 0.84 ± 0.02-fold of baseline during the 48 h of measurement. Next, permeability measurements of 4kDa FITC-dextran flux across Caco2 monolayers were performed (). In line with the measurements of TER, knockdown of PKP2 and PKP2/3, but not PKP3 alone, resulted in augmented permeability to 1.98 ± 0.21-fold and 1.86 ± 0.13-fold compared to controls. In summary, these experiments pointed to a predominant role of PKP2 for intercellular adhesion and epithelial barrier formation in intestinal epithelial cells.
Figure 2. Effects following knockdown of PKP2, 3 or PKP2/3.
PKP2 and PKP2/3 knockdown led to a reduction of the desmosome complex and tight junction proteins
Western blot analyses were performed to evaluate potential changes on the junctional complex and especially of desmosomes following loss of PKP2, PKP3 or both (). Loss of PKP2 resulted in reduced levels of desmosomal proteins DSG2 (0.76 ± 0.06-fold of controls), DSC2 (0.75 ± 0.05-fold of controls), Plakoglobin (PG) (0.46 ± 0.07-fold of controls). In contrast, desmoplakin (DP (1.09 ± 0.12-fold of controls) and E-cadherin (0.92 ± 0.08-fold of controls) levels were not affected. Comparable to the observations for PKP2, simultaneous knockdown of PKP2/3 resulted in a reduction of DSG2 and DSC2 and PG (0.78 ± 0.04-fold, 0.75 ± 0.03-fold and 0.62 ± 0.04-fold of controls, respectively). In addition, DP was reduced (0.50 ± 0.10-fold of controls) and revealed an elevated cytoplasmic localization (). Again, changes of E-cadherin were absent (0.95 ± 0.09 of controls) following the loss of PKP2/3. In line with this, immunostaining showed a reduction of the staining patterns at the cell borders for the desmosomal proteins DSG2, DSC2, DP and PG following knock down of PKP2 and PKP2/3, whereas controls showed a regular staining pattern of all of these proteins along the cell borders ().
Figure 3. Effects of PKP knockdown on desmosomal proteins in Caco2 cells
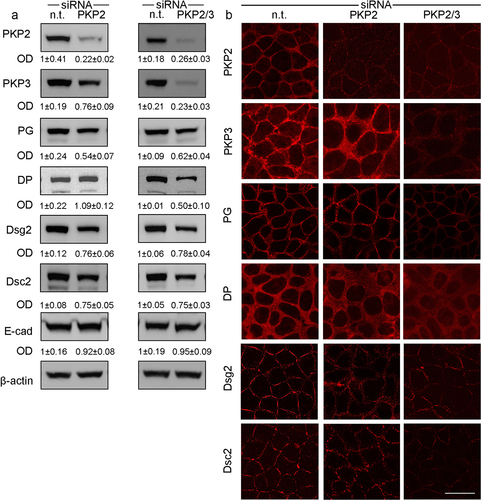
Interestingly, levels of barrier-sealing proteins Claudin1 (0.81 ± 0.05-fold of controls) and Claudin4 (0.84 ± 0.07-fold of controls) were also reduced in response to PKP2 knockdown. This was also observed in the PKP2/3 knockdown (claudin 1 0.89 ± 0.03-fold of controls; claudin4 (0.88 ± 0.05-fold of controls; ). In contrast, changes of Claudin1 (0.93 ± 0.05-fold of controls) and Claudin4 (1.02 ± 0.04-fold of controls) were not observed following knockdown of PKP3 (not shown).
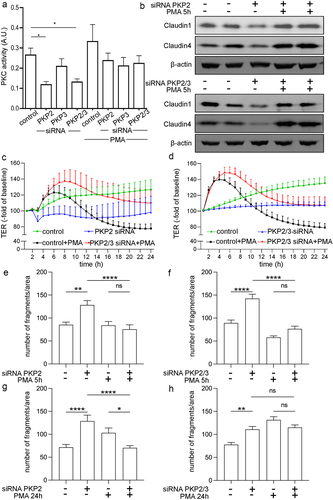
Figure 4. Protein kinase C is involved in barrier-compromising effects by PKP2.
PKC activity is reduced following loss of PKP2 and PKP2/3 in Caco2 cells
A previous study in keratinocytes and squamous carcinoma cells has pointed out the potential role of PKP2 as a scaffold to regulate PKC-dependent desmosome formation.Citation20 Therefore, we hypothesized that beside its structural role, PKP2 may affect PKC-dependent signaling in intestinal epithelial cells. First, we performed measurements of PKC activity using an ELISA-based system that utilizes a specific synthetic peptide as a substrate for PKC and a polyclonal antibody that recognizes the phosphorylated form of the substrate. Using this assay, we found a reduction of PKC activity to 0.47 ± 0.06-fold of controls following knockdown of PKP2 which was also found in Caco2 cells with PKP2/3 knockdown to 0.52 ± 0.06-fold (). The knockdown of PKP3, however, did not alter PKC activity compared to controls. Using the PKC activator, Phorbol 12-myristate 13-acetate (PMA) at 100 nM restored PKC activity in Caco2 with PKP2 and PKP2/3 knockdown to control levels. However, PMA did not augment PKC activity in untreated controls, nontarget siRNA or PKP3 siRNA treated Caco2 cells. This indicates that PMA was not used at supraphysiological doses under these experimental conditions.
Based on these observations, we tested next whether restoration of PKC activity using PMA would result in a restoration of the changes we had observed in response to PKP2 and PKP2/3 knockdown. On a protein level, as revealed by Western blot analyses, loss of Claudin1 and Claudin4 were restored in Caco2 cells with PKP2 and PKP2/3 knockdown when incubated with PMA for 6 h to control levels (). Similarly, loss of DSG2 as observed following knockdown of PKP2 and PKP2/3 was attenuated when cells were treated with PMA (data not shown). In functional measurements, application of PMA to Caco2 monolayers transfected with PKP2 or PKP2/PKP3 siRNA resulted in a rapid increase of TER to 1.25 ± 0.08-fold of baseline in PKP2 knockdown or 1.42 ± 0.06-fold of baseline in PKP2/3 knockdown Caco2 cells after PMA treatment which was maintained for 8 h before TER declined to baseline levels ( c and d). Similarly, cells transfected with nontarget siRNA showed increased TER to 1.38 ± 0.06-fold of baseline for 8 h and 0.78 ± 0.05-fold of baseline of baseline following application of PMA before TER returned to baseline levels. Untreated controls transfected with nontarget siRNA showed a gradual increase of TER to 1.07 ± 0.02-fold of baseline and 1.33 ± 0.09-fold of baseline after 24 h whereas TER remained at baseline levels in cells transfected with PKP2 siRNA or PKP2/PKP3 siRNA, respectively. It has to be pointed out that measurements started 48 h after transfection, which is different to the conditions shown in .
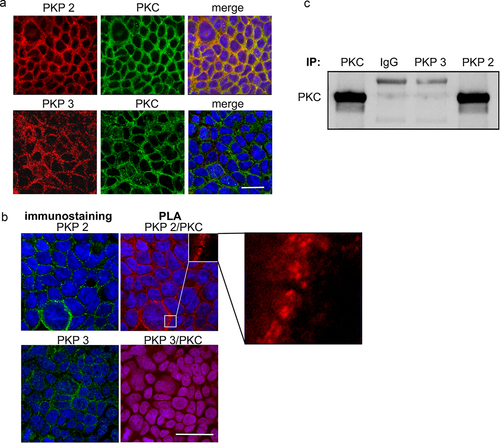
Figure 5. PKP2 interacts with protein kinase C.
Application of PMA for 5–8 h restored compromised intercellular adhesion following PKP2- and PKP2/3 knockdown as revealed by dispase-based enterocyte dissociation assays application of PMA (). Comparable to the observation made in TER measurements, the beneficial effect of PMA was not evident after long-term incubation with PMA for 24 h in cells transfected with PKP2 siRNA or PKP2/3 siRNA ().
PKP2/PKC association is essential for barrier integrity
In order to test for a structural or spatial interaction of PKP2 and PKC, we performed immunostaining for both proteins. Under basal conditions, PKP2 and PKC were both present as co-localized proteins in form of a dominant ring in the cell periphery at the cell borders (). Using PLAs to test for a direct interaction demonstrated the presence of red spots along the cell borders when PKP2/PKC antibodies were applied, indicating a direct interaction between these proteins (). Co-staining with PKP2 after PLA confirmed the presence of PKP2 at sites where the red PLA spots were present. In contrast no red spots were visible when PKP3/PKC antibodies were applied following PLA although PKP3 was clearly present at the cell borders in immunostaining (). In line with these observations, co-immunoprecipitation of PKC with PKP2 revealed a direct interaction between both proteins, suggesting that PKP2 may serve as a scaffold to control PKC activity under basal conditions. This was not observed when co-immunoprecipitation was carried out for PKC and PKP3 ().
Overexpression of PKP2 and PKP3 does not affect barrier integrity in Caco2 cells
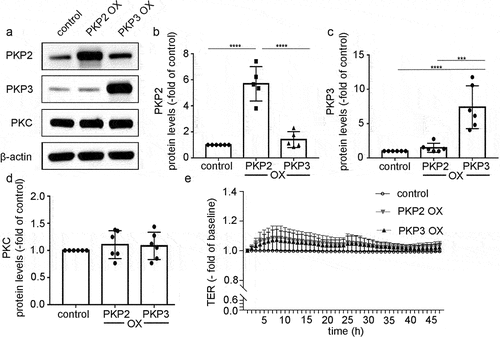
Finally, we tested whether overexpression (OX) of PKP2 or PKP3 would affect barrier function in intestinal epithelial cells. Caco2 cells were transfected with plasmids expressing PKP2 or PKP3 cDNA. Transfection of the plasmids in Caco2 cells resulted in a 5.7 ± 0.6-fold overexpression of PKP2 and 7.4 ± 1.3-fold overexpression of PKP3 compared to controls (). PKC protein levels were not affected by overexpression of PKP2 and PKP3 (). Measurements of TER starting 24 h after transfection of the cells with the plasmids did not show any differences between controls and Caco2 monolayers transfected with PKP2 and PKP3 within 48 h.
Figure 6. PKP2 and PKP3 overexpression had no effect on barrier functions.
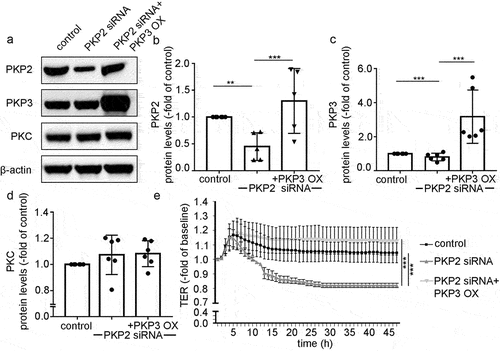
In view of the results that loss of PKP2 but not PKP3 led to reduced barrier function, we tested whether overexpression of PKP3 may compensate for the siRNA-induced loss of PKP2. PKP2 protein expression was reduced after silencing with PKP2 siRNA (0.4 ± 0.1-fold of control). Unexpectedly, simultaneous knockdown of PKP2 and PKP3 overexpression restored protein levels of PKP2 (1.3 ± 0.2-fold of control (). Under the same conditions, PKP3 overexpression was validated by a 3.2 ± 0.6-fold increase in controls (). Protein levels of PKC were neither affected by knockdown of PKP2 nor by simultaneous overexpression of PKP3 (). In TER measurements, reduced barrier function observed after knockdown of PKP2 alone was restored to control levels when cells overexpressed PKP3 simultaneously (). Taken together, this indicates that PKP3 overexpression may compensate for the loss of PKP2 to restore intestinal epithelial barrier integrity.
Figure 7. PKP3 overexpression in Caco2 cells attenuated the effect of PKP2 siRNA.
Discussion
This is the first study that specifically focuses on the functional role of plakophilins in intestinal barrier regulation. Our data confirm that PKP2 and PKP3, but not PKP1 are present in the intestinal epithelium. As revealed by siRNA-induced knockdown of PKP2, PKP3 or PKP2/3, we identified the predominant role of PKP2 in the contribution to intercellular adhesion and intestinal epithelial barrier stabilization. In contrast, PKP3 alone appears not to be critically involved in either intestinal barrier stabilization or intercellular adhesion. This is in agreement with data from keratinocytes where PKP1 and PKP3 are co-expressed. In these cells, intercellular adhesion and barrier formation critically depends on PKP1 whereas PKP3 did not strengthen adhesion but rendered desmosomes more dynamic.Citation21,Citation22
Our current data point to a role of PKP2 as a scaffold protein to modulate PKC activity, which, in turn, controls tight junction integrity. This is documented by the experiments showing that PKP2 and PKC co-localize in immunostaining and in proximity ligation assays which indicate a spatial interaction. Moreover, immunoprecipitation assays confirm that there is a direct interaction between PKP2 and PKC. Since siRNA-induced loss of PKP2 was associated with decreased PKC activity but did not affect PKC protein levels, our current data suggest that PKP2 stabilizes PKC function contributing to the regulation of its activity. Furthermore, the observation that the reduction of tight junction proteins claudin1 and claudin4 is attenuated following pharmacological activation of PKC indicates that PKP2-dependent PKC activation is required for tight junction stabilization. Overexpression of PKP2 and PKP3 did not change barrier function while PKP3 overexpression appeared to compensate for the loss of PKP2. This strengthens the current view that desmosomes, beside their role for intercellular adhesion, are important signaling hubs to mediate junctional stability in the intestinal epithelium.
The functional role of PKP2 and PKP3 for intestinal barrier function remained unexplored
Previous research from different groups provide evidence that the integrity of the desmosomes critically contributes to the stability of the intestinal epithelial barrier.Citation1,Citation6,Citation7,Citation9,Citation18,Citation19,Citation23–26 The underlying mechanism of how precisely intestinal permeability is affected by desmosomes remained unclear although it has been repeatedly reported that reduced DSG2 and DSC2 levels not only lead to loss of intercellular adhesion but also affect tight junction integrity in intestinal epithelial cells.Citation5,Citation6,Citation9,Citation19 Since plakophilins drive critical cellular functions in several cell types including keratinocytes, cardiomyocytes or cancer cells from different tissues, we have decided to test whether PKPs in intestinal epithelium are required for intestinal barrier regulation.
Data from previous studies revealed that PKP1 expression is restricted to stratified and complex epithelia, whereas PKP2 and PKP3 are found in simple epithelia and non-epithelial tissue, such as cardiomyocytes and lymphocytes.Citation27 Accordingly, our present data confirm that PKP2 and PKP3, but not PKP1 are expressed in intestinal epithelium in analysis of human tissue specimens, intestinal organoids and Caco2 cells using Western blotting. This supports previous observations where several junctional proteins including PKPs were assessed in the context of inflammation. In this study no PKP1 but PKP2 was detected in the intestinal mucosa and PKP2 was reduced in patients with inflammatory bowel diseases. However, the presence of PKP3 was not assessed.Citation28 In a study that compared the expression patterns of PKPs in different tissues, the expression of PKP3 in intestinal and colonic epithelium but not in hepatic tissue was described.Citation27,Citation29 The functional implications of the differential expression of different PKPs in intestinal tissue however remained largely unexplored since then. Similar to the situation in intestinal epithelium there are no studies available studying plakophilin expression and function in other simple epithelia such as lung or epididymis in detail. Only PKP1 was shown to be expressed in squamous cell carcinoma of the lung.Citation15,Citation30
Limitations and advantages of the Caco2 model to test PKP2/3 function
In general, organoid cultures representing primary intestinal epithelial cells would be desirable as a model for functional analysis of PKP2/3 function in intestinal epithelial cells.Citation23,Citation31 In our preliminary experiments using organoids, this remained challenging due to the heterogeneous growth patterns, insufficient transfection results following knockdown of PKPs as well as the lack to carry out reproducible functional measurements by TER or 4 kDa FITC dextran flux in this setting. Therefore, we decided to use Caco2 cells for our experiments. Although they have several disadvantages due to their cancer origin, Caco2 cells have proven to be an adequate model to assess intestinal epithelial functions in the past.Citation19 We used these cells to assess PKP function by siRNA-induced knockdown since this is a suitable approach to observe how the cells react to specific changes in a short-time without the potential compensatory changes when performing permanent knock-out cell lines.
PKP2 and PKP3 have differential roles in the intestinal epithelium
In knockdown experiments, loss of PKP2 but not PKP3 had significant effects on intestinal barrier function and maturation which indicates a specific and differential role for each of these proteins. This is supported by the observation that PKP2 and PKP2/3 knockdown led to the reduction of proteins of the whole desmosome complex and tight junction proteins, whereas loss of PKP3 alone induced only minor changes. Previously, it was shown that in MDCK cells most of the desmosomal complex remains stable while PKP2 remained highly mobile suggesting a regulatory role of PKP2 in desmosome formation.Citation32 Following knockdown in DLD1 cells, PKP3 was identified to facilitate the recruitment of desmosomes into the apical junctional complex.Citation33 In view of the minor changes following loss PKP3 in our model, it can be speculated that PKP2, at least in part, compensates for loss of PKP3. This idea is supported by the observation that knockdown of both PKP2 and PKP3 led to a phenotype with significantly more pronounced changes on a functional level and on the overall reduction of desmosomal proteins.
The observation that PKP3 is not required for the regulation of intestinal barrier function is supported by a previous study in PKP3-deficient mice: only deficiency for PKP3 in the hematopoietic system, but not in enterocytes, resulted in increased susceptibility to dextran sodium sulfate and LPS with consecutively increased intestinal permeability.Citation34 Since overexpression of PKP3 under conditions of PKP2 knockdown restored TER levels on a functional level, it could be argued that PKP3 compensates for loss of PKP2. However, when looking at PKP2 expression in cells treated with PKP2 siRNA following overexpression of PKP3 it appears as if the compensation of PKP3 for PKP2 is due to a PKP3-induced upregulation of PKP2. It has been reported before, that PKP3 affects the mRNA stability and translation of specific mRNAs including that of PKP2 and DP.Citation35 Thus, we speculate that PKP3 binding to the PKP2 mRNA may protect it from siRNA mediated degradation and increase its translation in cooperation with FXR1. Beside this, it was reported that PKP3 is lost in invasive and metastatic colon carcinoma, which leads accelerated tumor formation and decreased cell-cell adhesion followed by metastasis.Citation16
Interestingly, loss of intestinal epithelial DSC2 in mice led to reduced PKP3 expression leading to reduced activity of Rap1. This, in turn, resulted in reduced cell migration and wound closure.Citation36 Based on all observations, it may be concluded that PKP2 is predominately required for intestinal barrier formation and maturation whereas PKP3 may rather be involved in regulating intestinal cell migration.
Overexpression of PKP2 and PKP3 did not have a functional impact in epithelial barrier measurements. Since the barrier measurement includes the stabilization of tight junctions it can be assumed that junctions that are already intact do not benefit from increased PKP levels.
PKP2/PKC interaction regulates intercellular adhesion and tight junction integrity
Our present data show that in intestinal epithelial cells PKP2 and PKC interact with each other as revealed by proximity ligation assays and co-IP. The interaction between PKP2 and PKC has been observed before in skin squamous cancer.Citation20 Nonetheless, it must be emphasized that it is important to look at such interactions in a cell-type specific manner because protein interactions are dependent on the cellular context and may lead to different and even opposing functional consequences. Previous research has shown that PKC modulates the binding capacity of DP to intermediate filaments and thereby facilitates desmosomal stabilization.Citation37 This also appears to be the case in our present study since the loss of several desmosomal proteins, including DP, DSG2 and DSC2, in response to knockdown of PKP2 resulted in a reduction of intercellular adhesion as revealed by dispase-based enterocyte dissociation assays. Since total protein levels of PKC were neither affected by knockdown nor by overexpression of PKP2 and PKP3 it may be concluded that the interaction between PKC and PKP2 is required to facilitate the activation of PKP2. This is supported by the observation that the PKC activator PMA partially restored PKC activity. The exact molecular mechanism however remains to be explored. Nonetheless, there is a clear link between loss of PKP2, reduced PKC activity and reduced proteins levels of tight junction proteins claudin1 and claudin4. The observation that this could be reversed by PMA-induced activation of PKC points to an important link between the loss of desmosomal integrity and increased intestinal epithelial permeability. In general, a direct functional link between desmosomal integrity and tight junction regulation has been reported recently for the pore forming tight junction protein claudin2 which was strongly upregulated in response to reduced DSG2 levels. This was explained by the observation that DSG2 controls PI-3-kinase activation which is one of the most important regulators for claudin2 expression.Citation9 The novel observation in the present study is that PKP2 and PKC interaction appears to control the regulation of the sealing tight junction proteins claudin1 and claudin4. The critical role for PKC activity in the regulation of tight junctions has been recognized in previous studies where in kidney cells the PAR/Shank2 complex was stabilized by PKC.Citation38,Citation39 This resulted in the activation of small GTPase Rap1 and in the stabilization of tight junction-associated protein ZO1.Citation38,Citation39
In summary, our present data provide novel insights how desmosomes and especially PKPs contribute to the stability of intestinal epithelial barrier functions. Nonetheless, our data support the view that desmosomes are not confined to strengthening intercellular adhesion but provide signaling hubs to mediate tight junction stability in the intestinal epithelium. The physiological role of PKP2 in intestinal barrier function in vivo, however, remains to be tested in the future.
Author contribution
SN, YG, NS: conception and design of the whole work, performed experiments, interpreted data, drafting the manuscript.
CK, MK, BG, JW, NB: acquisition of data, analysis and interpretation, conception of parts of the study.
All authors have contributed finally approved the version to be published.
Acknowledgments
We thank Eva Riedel, Veronika Heimbach, Thekla Selig and Daniela Kraemer-Alm for skilful technical assistance. The authors are grateful to Prof. Dr. Mechthild Hatzfeld for providing plasmids (Institute for Molecular Medicine, University of Halle; Germany).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Schlegel N, Boerner K, Waschke J. Targeting desmosomal adhesion and signalling for intestinal barrier stabilization in inflammatory bowel diseases-Lessons from experimental models and patients. Acta Physiol. 2021;231(1):361. doi:10.1111/apha.13492.
- Luissint AC, Parkos CA, Nusrat A. Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology. 2016;151(4):616–379. doi:10.1053/j.gastro.2016.07.008.
- Fortea M, Albert-Bayo M, Abril-Gil M, Ganda Mall J-P, Serra-Ruiz X, Henao-Paez A, Expósito E, González-Castro AM, Guagnozzi D, Lobo B, et al. Present and Future Therapeutic Approaches to Barrier Dysfunction. Front Nutr. 2021;8:718093. doi:10.3389/fnut.2021.718093.
- Zuo L, Kuo WT, Turner JR. Tight junctions as targets and effectors of mucosal immune homeostasis. Cell Mol Gastroenterol Hepatol. 2020;10(2):327–340. doi:10.1016/j.jcmgh.2020.04.001.
- Gross A, Pack LAP, Schacht GM, Kant S, Ungewiss H, Meir M, Schlegel N, Preisinger C, Boor P, Guldiken N, et al. Desmoglein 2, but not desmocollin 2, protects intestinal epithelia from injury. Mucosal Immunol. 2018;11(6):1630–1639. doi:10.1038/s41385-018-0062-z. Epub 2018 Aug 16; PMID 30115995.
- Spindler V, Meir M, Vigh B, Flemming S, Hütz K, Germer C-T, Waschke J, Schlegel N. Loss of Desmoglein 2 Contributes to the Pathogenesis of Crohn’s Disease. Inflamm Bowel Dis. 2015;21(10):2349–2359. doi:10.1097/MIB.0000000000000486.
- Meir M, Burkard N, Ungewiß H, Diefenbacher M, Flemming S, Kannapin F, Germer C-T, Schweinlin M, Metzger M, Waschke J, et al. Neurotrophic factor GDNF regulates intestinal barrier function in inflammatory bowel disease. J Clin Invest. 2019;129(7):2824–2840. doi:10.1172/JCI120261.
- Nava P, Laukoetter MG, Hopkins AM, Laur O, Gerner-Smidt K, Green KJ, Parkos CA, Nusrat A. Desmoglein-2: a novel regulator of apoptosis in the intestinal epithelium. Mol Biol Cell. 2007;18(11):4565–4578. doi:10.1091/mbc.e07-05-0426.
- Burkard N, Meir M, Kannapin F, Otto C, Petzke M, Germer C-T, Waschke J, Schlegel N. Desmoglein2 regulates claudin2 expression by sequestering PI-3-kinase in intestinal epithelial cells. Front Immunol. 2021;12:756321. doi:10.3389/fimmu.2021.756321.
- Green KJ, Roth-Carter Q, Niessen CM, Nichols SA. Tracing the evolutionary origin of desmosomes. Curr Biol. 2020;30(10):R535–R543. doi:10.1016/j.cub.2020.03.047.
- Hofmann I. Plakophilins and their roles in diseased states. Cell Tissue Res. 2020;379(1):5–12. doi:10.1007/s00441-019-03153-0.
- Fuchs M, Foresti M, Radeva MY, Kugelmann D, Keil R, Hatzfeld M, Spindler V, Waschke J, Vielmuth F. Plakophilin 1 but not plakophilin 3 regulates desmoglein clustering. Cell Mol Life Sci. 2019;76(17):3465–3476. doi:10.1007/s00018-019-03083-8.
- Bonne S, Gilbert B, Hatzfeld M, Chen X, Green KJ, van Roy F. Defining desmosomal plakophilin-3 interactions. J Cell Biol. 2003;161(2):403–416. doi:10.1083/jcb.200303036.
- Tsatsopoulou AA, Protonotarios NI, McKenna WJ. Arrhythmogenic right ventricular dysplasia, a cell adhesion cardiomyopathy: insights into disease pathogenesis from preliminary genotype–phenotype assessment. Heart. 2006;92(12):1720–1723. doi:10.1136/hrt.2005.081679.
- Furukawa C, Daigo Y, Ishikawa N, Kato T, Ito T, Tsuchiya E, Sone S, Nakamura Y. Plakophilin 3 oncogene as prognostic marker and therapeutic target for lung cancer. Cancer Res. 2005;65(16):7102–7110. doi:10.1158/0008-5472.CAN-04-1877.
- Kundu ST, Gosavi P, Khapare N, Patel R, Hosing AS, Maru GB, Ingle A, DeCaprio JA, Dalal SN. Plakophilin3 downregulation leads to a decrease in cell adhesion and promotes metastasis. Int J Cancer. 2008;123(10):2303–2314. doi:10.1002/ijc.23797.
- Egu DT, Kugelmann D, Waschke J. Role of PKC and ERK signaling in epidermal blistering and desmosome regulation in pemphigus. Front Immunol. 2019;10:2883. doi:10.3389/fimmu.2019.02883.
- Meir M, Salm J, Fey C, Schweinlin M, Kollmann C, Kannapin F, Germer C-T, Waschke J, Beck C, Burkard N, et al. Enteroids generated from patients with severe inflammation in crohn’s disease maintain alterations of junctional proteins. J Crohn's Colitis. 2020;14(10):1473–1487. doi:10.1093/ecco-jcc/jjaa085.
- Schlegel N, Meir M, Heupel W-M, Holthöfer B, Leube RE, Waschke J. Desmoglein 2-mediated adhesion is required for intestinal epithelial barrier integrity. Am J Physiol Gastrointest Liver Physiol. 2010;298(5):G774–83. doi:10.1152/ajpgi.00239.2009.
- Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, Wahl JK, Denning MF, Green KJ. Plakophilin 2: a critical scaffold for PKCα that regulates intercellular junction assembly. J Cell Biol. 2008;181(4):605–613. doi:10.1083/jcb.200712133.
- Keil R, Rietscher K, Hatzfeld M. Antagonistic regulation of intercellular cohesion by plakophilins 1 and 3. J Invest Dermatol. 2016;136(10):2022–2029. doi:10.1016/j.jid.2016.05.124.
- Rietscher K, Wolf A, Hause G, Rother A, Keil R, Magin TM, Glass M, Niessen CM, Hatzfeld M. Growth retardation, loss of desmosomal adhesion, and impaired tight junction function identify a unique role of plakophilin 1 in vivo. J Invest Dermatol. 2016;136(7):1471–1478. doi:10.1016/j.jid.2016.03.021.
- Meir M, Kannapin F, Diefenbacher M, Ghoreishi Y, Kollmann C, Flemming S, Germer C-T, Waschke J, Leven P, Schneider R, et al. Intestinal epithelial barrier maturation by enteric glial cells is GDNF-dependent. Int J Mol Sci. 2021;22(4):1887. doi:10.3390/ijms22041887.
- Jiang K, Rankin CR, Nava P, Sumagin R, Kamekura R, Stowell SR, Feng M, Parkos CA, Nusrat A. Galectin-3 regulates desmoglein-2 and intestinal epithelial intercellular adhesion. J Biol Chem. 2014;289(15):10510–10517. doi:10.1074/jbc.M113.538538.
- Kamekura R, Kolegraff KN, Nava P, Hilgarth RS, Feng M, Parkos CA, Nusrat A. Loss of the desmosomal cadherin desmoglein-2 suppresses colon cancer cell proliferation through EGFR signaling. Oncogene. 2014;33(36):4531–4536. doi:10.1038/onc.2013.442.
- Kamekura R, Nava P, Feng M, Quiros M, Nishio H, Weber DA, Parkos CA, Nusrat A. Inflammation-induced desmoglein-2 ectodomain shedding compromises the mucosal barrier. Mol Biol Cell. 2015;26(18):3165–3177. doi:10.1091/mbc.e15-03-0147.
- Borrmann CM, Mertens C, SCHMIDT A, LANGBEIN L, Kuhn C, FRANKE WW. Molecular diversity of plaques of epithelial-adhering junctions. Ann N Y Acad Sci. 2006;915(1):144–150. doi:10.1111/j.1749-6632.2000.tb05237.x.
- Gassler N, Rohr C, Schneider A, Kartenbeck J, Bach A, Obermüller N, Otto HF, Autschbach F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281(1):G216–28. doi:10.1152/ajpgi.2001.281.1.G216.
- Schmidt A, Langbein L, Prätzel S, Rode M, Rackwitz HR, Franke WW. Plakophilin 3--a novel cell-type-specific desmosomal plaque protein. Differentiation; Research in Biological Diversity. 1999;64(5):291–306. doi:10.1046/j.1432-0436.1999.6450291.x.
- Gomez-Morales M, Cámara-Pulido M, Miranda-León MT, Sánchez-Palencia A, Boyero L, Gómez-Capilla JA, Fárez-Vidal ME. Differential immunohistochemical localization of desmosomal plaque-related proteins in non-small-cell lung cancer. Histopathology. 2013;63(1):103–113. doi:10.1111/his.12126.
- Wilson SS, Mayo M, Melim T, Knight H, Patnaude L, Wu X, Phillips L, Westmoreland S, Dunstan R, Fiebiger E, et al. Optimized culture conditions for improved growth and functional differentiation of mouse and human colon organoids. Front Immunol. 2021;11:547102. doi:10.3389/fimmu.2020.547102.
- Fulle JB, Huppert H, Liebl D, Liu J, Alves de Almeida R, Yanes B, Wright GD, Lane EB, Garrod DR, Ballestrem C, et al. Desmosome dualism – most of the junction is stable, but a plakophilin moiety is persistently dynamic. J Cell Sci. 2021;134(21). doi:10.1242/jcs.258906.
- Indra I, Troyanovsky RB, Green KJ, Troyanovsky SM. Plakophilin 3 and Par3 facilitate desmosomes’ association with the apical junctional complex. Mol Biol Cell. 2021;32(19):1824–1837. doi:10.1091/mbc.E21-01-0001.
- Sklyarova T, van Hengel J, Van Wonterghem E, Libert C, van Roy F, Vandenbroucke RE. Hematopoietic plakophilin-3 regulates acute tissue-specific and systemic inflammation in mice. Eur J Immunol. 2015;45(10):2898–2910. doi:10.1002/eji.201445440.
- Fisher-Keso R, Breuninger S, Hofmann S, Henn M, Röhrig T, Ströbel P, Stoecklin G, Hofmann I. Plakophilins 1 and 3 bind to FXR1 and thereby influence the mRNA stability of desmosomal proteins. Mol Cell Biol. 2014;34(23):4244–4256. doi:10.1128/MCB.00766-14.
- Flemming S, Luissint A-C, Kusters DHM, Raya-Sandino A, Fan S, Zhou DW, Hasegawa M, Garcia-Hernandez V, García AJ, Parkos CA, et al. Desmocollin-2 promotes intestinal mucosal repair by controlling integrin-dependent cell adhesion and migration. Mol Biol Cell. 2020;31(6):407–418. doi:10.1091/mbc.E19-12-0692.
- Broussard JA, Jaiganesh A, Zarkoob H, Conway DE, Dunn AR, Espinosa HD, Janmey PA, Green KJ. Scaling up single-cell mechanics to multicellular tissues – the role of the intermediate filament–desmosome network. J Cell Sci. 2020;133(6). doi:10.1242/jcs.228031.
- Sasaki K, Kojitani N, Hirose H, Yoshihama Y, Suzuki H, Shimada M, Takayanagi A, Yamashita A, Nakaya M-A, Hirano H, et al. Shank2 binds to aPKC and controls tight junction formation with Rap1 signaling during establishment of epithelial cell polarity. Cell Rep. 2020;31(1):107407. doi:10.1016/j.celrep.2020.02.088.
- Suzuki T, Elias BC, Seth A, Shen L, Turner JR, Giorgianni F, Desiderio D, Guntaka R, Rao R. PKCη regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci U S A. 2009;106(1):61–66. doi:10.1073/pnas.0802741106.