
Abstract
The aim of the present work is to formulate heparin-modified-polycaprolactone (HEP) core shell nanoparticles (NPs) of 5-fluorouracil (5-FU). These NPs were characterized for various in vitro parameters like particle size, zeta potential, etc. HEP NPs were found to maintain comparatively slower drug release pattern (98.9% in 96 h) than PCL NPs. Cytotoxicity studies demonstrated a massive cytotoxic potential of 5-FU-loaded HEP NPs in A549, MDA-MD-435, and SK-OV-3 cancer cell lines. Pharmacokinetic parameters were also determined in blood after IV administration of HEP NPs: AUC, Cmax, MRT, and Tmax values are 6096.075 ± 5.90 μg h/mL, 144.38 ± 1.52 μg/L, 58.71 ± 0.25 h, 96 ± 0.50 h, respectively and 117.92 ± 1.78, 45.35 ± 3.00, 1.2 ± 0.25, 0.5 ± 0.02 in plain 5-FU solution.
Introduction
Cancer has been recognized as one of the most lethal diseases worldwide, for which the new cases are promptly rising every year (Boyle and Levin Citation2008). In spite of hasty advancements in investigative procedures and therapies, the global survival rate from cancer has not been upgraded significantly over the previous 30 years (Jemal et al. Citation2010). Cancer is undoubtedly acknowledged as one of the most prominent basis of mortality in national as well as global scene. It is well known that interaction among the various molecules expressed by cancer cells is affected by effectors/stimuli that are squirted by stromal cells within the tumor microenvironment (Theocharis et al. Citation2010). Presently there is a requisite to exploit the innovative approaches for the accurate recognition of initial stage of cancer and also for the targeted therapies centered on the cancer-specific markers, which could result in individualized medicine. Latest innovations in nanotechnology have explored passive and active targeting approaches for enriching the intratumoral drug concentrations while restricting the unsolicited toxicity to healthy tissue (Allen Citation2002). The targeted delivery of nanomaterials can prevail over many hurdles associated with traditional anticancer drugs, i.e., insolubility under aqueous conditions, rapid clearance, and a lack of selectivity, causing nonspecific toxicity toward normal cells and thereby lessening the dose of drugs to be liberated on the cancer cells (Ashley et al. Citation2011). Heparin (HE) belongs to a group of polyanionic polysaccharides known as glycosaminoglycans (GAGs) having repeated disaccharide sequences of alternating uronic acid and amino sugar residues. Heparin is a polydisperse composite with a molecular weight ranging from 3000 to 30,000 Da (mean weight – approximately 15,000 Da). By deactivating thrombin, HE not only prevents fibrin formation but also inhibits thrombin-induced triggering of platelets and factors V and VIII. In addition to bind to anti-thrombin, HE also unites with a wide variety of other proteins by means of electrostatic interactions (Hirsh Citation1991).
5-Fluorouracil (5-FU) is a pyrimidine analogue. 5-FU is a white crystalline powder that is soluble in water (10 mg/mL). 5-FU is air sensitive and should be stored at 0–5 °C. 5-FU is light sensitive, combustible, incompatible with strong oxidizing agents and strong bases. It is suggested for the treatment of proliferative skin diseases, for example superficial basal cell carcinoma (Ghoshal and Jacob Citation1997). 5-FU has been shown to be effective against a range of solid tumors of colon, breast, cervix, and rectum (Ansifield et al. Citation1962). 5-FU is poorly absorbed when given via oral route, with a remarkable variations in bioavailability found between 0% and 80% while after parenteral administration of 5-FU, the prompt elimination of the 5-FU was obtained with possible half-life of approximately 8–20 min (Diasio and Harris Citation1989). Various research groups explored various drug delivery systems for selective delivery of 5-FU (Alanazi et al. Citation2014, Citation2015, Shakeel et al. Citation2014, Citation2015).
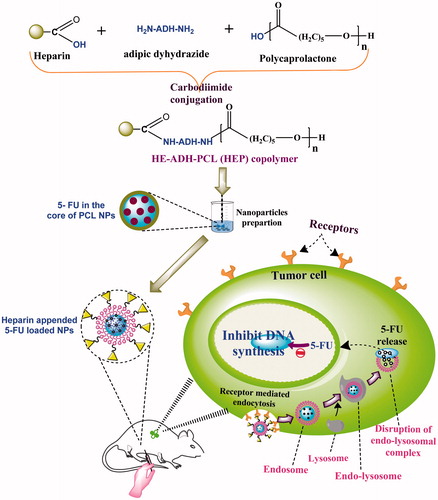
The aim of the current research is to evaluate the efficiency of HE conjugated PCL nanoparticles (NPs) designed for the drug delivery to tumor tissues. In the recent investigation, HE conjugated PCL NPs were synthesized using adipic dihydrazide as linkers and verified by in vitro study. These HE-modified-polycaprolactone (HEP)-conjugated NPs were meticulously investigated as vehicles for antitumor specific delivery using 5-FU as a prototype drug against MDA-MB-435, SK-OV-3, and A-549 cell lines ().
Scheme 1. Project Scheme
Materials and methods
Materials
5-Fluorouracil was obtained from Neon Laboratories Ltd., Mumbai, India. ɛ-Caprolactone, pluronic F-68 and dialysis membranes, adipic dyhydrazide (ADH), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC), N-hydroxysuccinimide (NHS) were purchased from Himedia, Mumbai, India. Heparin was purchased from Sigma-Aldrich, St. Louis, MO. Acetonitrile, isopropyl alcohol, and acetone were purchased from Spectrochem, Mumbai, India. All other chemicals used were of AR grade and were used as received.
Synthesis of HE–ADH–PCL copolymer (HEP)
HE (50 mg) was dissolved in 10 mL of distilled water and then fivefold excess ADH of 250 mg was added with continuous stirring followed by 250 mg of EDAC. The reaction was performed at room temperature for 12 h. HE–ADH dispersion was added into the activated PCL dispersion and the reaction mixture was taken in a round-bottom flask kept on reflux at 110 °C for 5 h. The reaction mixture was extensively dialyzed (MWCO 12–14 kDa) to separate HE–ADH–PCL from unreacted HE, PCL, and HE–ADH. Mixture was subjected to gel filtration on a column of Sephadex G-100 and then unreacted PCL and HE molecules were quantified by a titration method (Demoré et al. Citation1998). The obtained copolymer named HEP was dried under vacuum. For synthesis of PCL, ɛ-caprolactone (250 mg) was taken in the round-bottom flask and 22 mg of stannous octoate and 4 mL of toluene were added on it and refluxed at 110 °C for 5 h. Then, the PCL was dried and dissolved in 25 mL of dichloromethane. Synthesis of HEP was confirmed by IR and NMR spectroscopy. IR spectra of HEP and PCL were recorded with a FT-IR spectrophotometer (Cary-89 630 FTIR, Agilent Technologies, Santa Clara, CA). Nuclear Magnetic Resonance (NMR) spectroscopy of HEP and PCL were performed at 300 MHz, after dissolving in DMSO (Bruker DRX, Billerica, MA).
Preparation of HE–ADH–PCL nanoparticles (HEP NPs)
HEP (10 mg) was dissolved in 20 mL of dichloromethane:ethanol (1:1) and 5-FU (10 mg) was added into HEP solution. Pluronic solution (0.05%) was formed by dissolving 3.75 mg pluronic F-68 in 75 mL of acetone. HEP solution was added drop wise in pluronic solution under constant stirring on magnetic stirrer for 2 h. Nanoparticles were formed on solvent interface. Then, the solvent was evaporated at room temperature. The resulting suspension of NPs was filtered through 0.45 μm membrane filter (Millipore, Billerica, MA) and centrifuged for 40 min at 10,000 rpm (Remi, Mumbai, India). The supernatant was discarded and HEP NPs were lyophilized and kept for further use. Moisture content of lyophilized NPs was measured by Karl-Fischer Titrator (Dolphin Instruments, Mumbai, India).
Characterization of HEP NPs
Morphology
The shape and surface morphology of HEP NPs were observed by Atomic Force Microscopy (AFM) or Scanning Probe Microscopy, AIST-NT Smart SPM 1000. AFM of the NPs was carried out at glass substrate in AC mode. AFM measurements were performed in air and at room temperature, with a nominal tip radius of 10 nm and resonant frequency of about 220 kHz. The image processing was carried out using SPMLab software. The determination of the particle height was carried out with external processing software. The height was determined as a difference between maximum height in the inner part of annulus and the average height in the area of annulus. Particles with noise spikes and obvious agglomerates were excluded from the analysis.
Particle size and zeta potential
A properly diluted suspension of NPs (HEP and PCL NPs) was crammed in the chamber of a laser diffraction particle size analyzer (DTS Ver. 4.10, Malvern Instruments, Malvern, UK) and the average particle size and PDI were determined. The zeta potential of the NPs was determined by laser Doppler anemometry using a Malvern Zetasizer (DTS Ver. 4.10; Malvern Instruments).
Differential scanning calorimetry
Differential scanning calorimetry (DSC) analysis of samples was done using DSC 60 instruments (Shimadzu, Tokyo, Japan). The physical state of 5-FU inside the NPs was characterized by the analysis of the DSC curves. The DSC of 5-FU, CAP, ADH, HE, HE–ADH–CAP NPs (without 5-FU), and 5-FU-loaded HEP NPs was performed. The samples (2 g) were weighted into aluminum pan and closed with a pin-holed lid. Thermograms were recorded under nitrogen atmosphere from ambient to 300 °C at a heating rate of 10 °C per min.
X-ray diffraction
Powder X-ray diffraction (XRD) analysis was carried out in order to characterize crystalline nature of polymer and drug. Powder XRD patterns of samples were obtained using power X-ray diffractometer (Bruker, Munich, Germany). X-ray diffraction analysis of 5-FU, CAP, ADH, HE, HE-ADH-CAP NPs (Without 5-FU), and 5-FU-loaded HEP NPs was performed using a nickel-filtered Cu-Ka radiation (at a voltage of 40 kV and a current of 20 mA). The scanning rate was 2θ/min over the range of 0–40° and with an interval of 0.02°.
Entrapment efficiency
Ten milligrams of HEP NPs was dissolved in acetone/isopropyl alcohol (9:1). The mixture was centrifuged and the supernatant was thinned with methanol–PBS (pH 7.4) mixture. Further, the quantity of entrapped drug was analyzed by HPLC (Agilent Technologies, 1220 infinity LS, UK).
The HPLC system (Agilent Technologies, 1220 infinity LS, UK) consisted of a variable wavelength detector, and a zorbax 5 μ C18 column (250 × 4.60 mm) was used for the analysis of the drug. The mobile phase was composed of acetonitrile/acetate buffer pH 4.4 (ratio 15:85). At a flow rate of 0.8 mL/min, 5-FU was detected at 260 nm, with a detection limit of ∼20 ng. Similarly entrapment efficiency of PCL NPs was determined using the following formula (de Mattos et al. Citation2013):
In vitro release study
Ten milligrams of NPs (HEP) was suspended in 2 mL of PBS (pH 7.4) and subsequently put into a dialysis tube (MWCO 2000 Da). The dialysis tube was positioned into a 50 mL of aqueous recipient medium of PBS (pH 7.4). The release media were agitated at 100 rpm at 37 ± 2 °C. A whole-media change method was used to circumvent drug saturation in the drug release study. At specific time intervals, the whole medium (50 mL) was taken and replaced with the same volume of fresh PBS (pH 7.4) (50 mL). The samples were then analyzed by HPLC. Similarly in vitro release of 5-FU from PCL NPs were determined.
Hemolytic toxicity
Whole human blood was collected and stored in HiAnticlot blood collection vials as mentioned in our previous paper (Garg et al. Citation2014, Yadav et al. Citation2010). The human blood was centrifuged and then red blood cells were alienated and resuspended in normal saline solution (10% hematocrit). One mL of the red blood cell suspension was separately incubated with 5 mL of distilled water (taken as 100% hemolytic standard) and 5 mL of normal saline (taken as blank for spectrophotometric estimation). Further, 1 mL of adequately diluted HEP NPs and PCL NPs (negative control), 5-FU-loaded HEP NPs, 5-FU-loaded PCL NPs, and plain 5-FU were added to 5.0 mL of normal saline and interacted with RBC suspension. The suspensions were centrifuged for 10 min at 2000 rpm, and the absorbance of supernatants was measured at 540 nm, which was used to estimate the percentage hemolysis using distilled water as 100% hemolytic standard.
In vitro cellular cytotoxicity
Cellular cytotoxicity was assessed by tetrazolium dye-based MTT assay following a previously reported procedure (Garg et al. Citation2014, Kolhe et al. Citation2003). MDA-MB-435, SKOV-3, and A-549 cells were maintained in RPMI-1640 medium supplemented with 10% heat inactivated fetal bovine serum and antibiotics at 37 °C in a humidified incubator containing 5% CO2. The cells were treated with drug-loaded NPs in various concentrations (10, 20, 40, and 80 μg/mL) for 24 h. The amount of formulation needed to prepare molar equivalents of 5-FU was calculated, based on the drug content in the formulation. Control was taken without any drug treatment. Subsequently, MTT was added and plates were then incubated for another 3 h, the medium was pippetted off and 250 μL DMSO was added. The absorbance of individual wells was noted at 570 nm via an ELISA plate reader at 25 °C. Average values from triplicate were subtracted from average value of control and the survival fraction of cells was calculated by the formula.
Pharmacokinetic study
Pharmacokinetic parameters were also determined after IV administration of 5-FU-loaded HEP NPs and plain 5-FU in albino rats. The protocol was approved by the Institutional Animal Ethics Committee of the Guru Ramdas Khalsa Institute of Science and Technology, Pharmacy, Jabalpur, India with approval number GRKIST/406/02/IAEC/16A dated 16/01/2013 (Registration Number 1471/PO/a/11/CPCSEA, India). Healthy male albino rats having uniform body weight (100 ± 20 g) with no prior drug treatment were used for the study. The rats were stabilized with a standard diet and water. The rats were divided into three groups having six animals in each group. 5-FU-loaded HEP NPs and plain 5-FU solution (30 mg/kg) were administered to the first group and second group of the rats, respectively, from i.v. route, and third group were kept as a control. Blood samples (0.1 mL) were withdrawn from retro-orbital plexus in every 1, 2, 4, 6, 8 h for up to 24 h. The blood samples were centrifuged at 2000 rpm for 15 min and serum was collected and deproteinized with acetonitrile (1 mL/mL of serum). Then, the obtained samples were centrifuged and the supernatants were analyzed for drug content by HPLC (Agilent Technologies, 1220 infinity LS, UK) (Alsarra and Alarifi Citation2004).
Statistical analysis
Experimental results were expressed as mean ± SD (number of experiments). Statistical comparisons were made by t-test. p < 0.05 was considered significant. All experiments were repeated three times unless otherwise indicated.
Results and discussion
The primary aim of present research is to study the delivery of 5-FU from HE appended PCL NPs prepared using HE–ADH–PCL copolymers. NPs of triblock copolymer, i.e., HA–PEG–PLGA, HA–PEG–PCL, and DEX–PEG–CAP copolymers containing anticancer drugs have been reported previously by our research groups (Garg et al. Citation2014, Yadav et al. Citation2008, Citation2010). In the present work, we explored PCL using a variety of targeting ligands, a number of research groups attempted to increase the tissue specificity of the drug carriers by coupling targeting agents, such as hyaluronic acid, peptide, biotin, folic acid for the delivery of anticancer drug.
FTIR
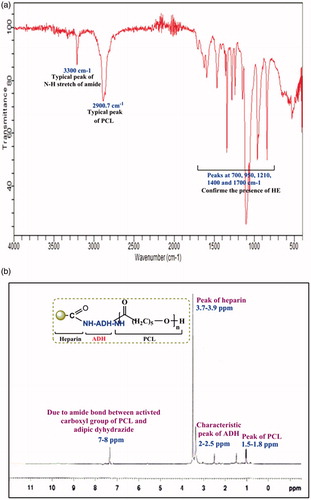
Coupling of HE on PCL NPs was confirmed by FTIR spectroscopy. Synthesis of HEP copolymer was confirmed by FTIR and NMR spectroscopy as shown in . In the FTIR spectra of HEP copolymer, peaks at 700, 950, 1210, 1400, and 1700 cm−1 confirmed the presence of HE. Typical peaks at 600, 700, 990, and 2800 cm−1 showed the presence of ADH. PCL showed typical peaks at 750, 950, 1103.3, 1422.14, 1700, and 2900.7 cm−1. A typical peak of N–H stretch of amide at 3300 cm−1 and C=O stretch at 1650 cm−1 was used to confirm the formation of HEP copolymer.
Figure 1. (a) FTIR of HEP copolymer. (b) 1H NMR spectra of HEP copolymer.
NMR analysis
The presence of HE, PCL, and ADH in NPs was verified by the characteristic peaks appearing in the 1H NMR spectra (). The peak of HE was observed at 3.7–3.9 and N-acetyl group of HE at 2.05 ppm, the characteristic peak of ADH shown in 2–2.5 and 2.10 ppm and the peak of PCL was observed at 1.5, 1.6–1.8 ppm, which confirmed the presence of HE, ADH, and PCL in HEP NPs formulation.
Morphology
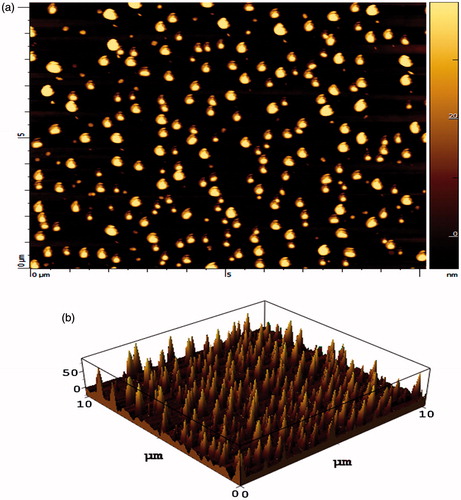
The shape of NPs (HEP NPs) was visualized under AFM and photomicrographs were obtained. HEP NPs were found to be spherical and of nanometric-size range (). Using the AFM, HEP NPs was visualized and the height was found to be relatively constant, which revealed the size of NPs. Unlike other microscopic techniques, the AFM offers visualization in three dimensions. gives the tapping mode atomic force microscopic images of freshly prepared HEP. Results are in good agreement with the previous report (Garg et al. Citation2014). The prime advantage of AFM is its ability to image non-conducting samples without any specific treatment, thus allowing imaging of delicate biological and polymeric nano- and microstructures (Shi et al. Citation2003). AFM provides the most accurate description of size and size distribution and requires no mathematical treatment. Moreover, particle size obtained by AFM technique provides real picture which helps us to understand the effect of various biological conditions (Polakovic et al. Citation1999).
Figure 2. AFM image of HEP NPs. (a) One dimension and (b) three dimension.
Particle size and zeta potential determination
The average particle size of HEP NPs was found to be 82 ± 3.50 nm and particles size distribution of HEP NPs was found to be 81.20% of 83.2 nm, 14.90% of 78.20 nm and 3.9% of 73.5 nm and zeta potential of HEP NPs was found as −5.13 mV. The obtained particle size distribution ranges from 82 to 210 nm with either larger particles or agglomerates up to 210 nm. The effect of surfactant concentration on size distribution has a unique significance in HEP NPs. Thermodynamic effect of surfactant addition is primordial in emulsion formation. Besides, the use of small glassware in the lab scale is commonly associated to strong wall effects, which may cause the multimodal size distributions (Dieckmann et al. Citation2009).
As results shown in , the drug entrapment efficiency and particle size of HEP NPs were mainly influenced by polymer concentration and concentration of surfactant. With increasing concentration of polymer (10–30 mg), particle size was increased from 82 ± 3.50 to 210 ± 1.40 nm and drug encapsulation efficiency of HEP NPs was decreased from 95.45 ± 1.65% to 72.50 ± 1.10%. It may be attributed that increase in viscosity of dispersed phase negatively impacts dispersibility of the HEP solution into the aqueous phase (Xie and Hwa Wang Citation2004). PCL NPs have shown similar pattern in the drug entrapment efficiency and particle size. PCL NPs were influenced by polymer concentration and concentration of surfactant as similar to HEP NPs. With increasing concentration of polymer (10 to 30 mg), particle size was increased from 140 ± 1.5 nm to 285 ± 1.2 nm and drug encapsulation efficiency of NPs was decreased from 81.28 ± 1.60% to 49.20 ± 1.80% (Xie and Hwa Wang Citation2004).
Table 1. Ingredients and concentration used in the formulation of HEP NPs.
Table 2. Ingredients and concentration used in the formulation of PCL NPs.
Pluronic F-68 concentration influences the HEP NPs characteristics, and was used in different concentrations and hence some batches were prepared. When the concentration of surfactant was increased (1–4%), the size of NPs was decreased (185 ± 1.20 to 110 ± 1.10 nm) and entrapment efficiency was increased (67.50 ± 0.50% to 87.22 ± 1.78%) when concentration of surfactant further increased (1 and 4%). In case of PCL NPs, particles size was decreased from 279 ± 1.5 nm to 183 ± 1.10 nm and entrapment efficiency was increased from 52.44 ± 1.54% to 74.22 ± 1.45% by increasing concentration of surfactant from 1 to 4% ().
The higher concentration of surfactant reduces the size of the NPs. When the amount of Pluronic F-68 was increased it was found that the granulometric distribution became narrower. This phenomenon can be expected from the stabilizing function of a surfactant up to a definite concentration, further increase in amount of surfactant exerts little or no effect on particle and entrapment efficiency. It was also observed that when a small concentration of surfactant is used, it causes aggregation of the particles and the size of particles may increase (Yadav et al. Citation2007).
Polydispersity index of all HEP NPs, prepared during the optimization of process variable, was between 0.074 and 0.092 and in case of all PCL NPs it was between 0.085 and 0.120. The entrapment efficiency of optimized HEP (HEP 1) and PCL NPs (PCL 1) was found to be 95.45 ± 1.65% and 81.28 ± 1.60% and the particle size was found to be 82 ± 3.50 nm and 140 ± 1.5 nm, respectively, as shown in > (Nidhin et al. Citation2008). Particles size distribution of PCL NPs (PCL 1) was found to be 75.4% of 176.4 nm, 24.6% of 50.60 nm and zeta potential of PCL NPs (PCL 1) was found to be −5.13 mV.
Differential scanning calorimetry
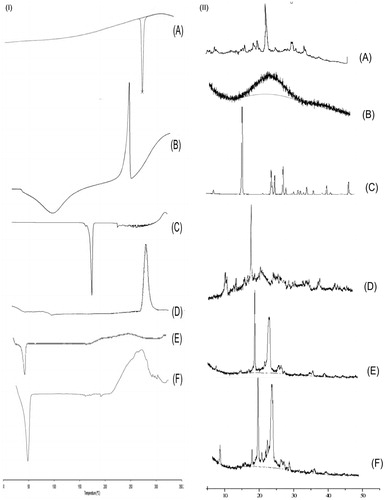
DSC thermogram of 5-FU, HE, ADH, PCL HEP NPs (without drug), and HEP NPs (5-FU-loaded) was performed and the thermogram is shown in . In case of 5-FU, an endothermic peak was observed at 280 °C (). In case of HE, a broad endothermic peak was observed at 100 °C (), which is a characteristic peak of HE and a broad exothermic peak on 240 °C (Martins et al. Citation2011). In the case of ADH, an endothermic peak was observed at 170–180 °C () (Tomuta et al. Citation2012). In the case of PCL, endothermic peak at 60 °C and exothermic degradation peak at 300 °C were found (). In case of HEP NPs, endothermic peak of HE was observed at 80 °C, an exothermic peak of HE-ADH at 270 °C indicated the crystalline structure of the copolymers (). In case of 5-FU-loaded HEP NPs, endothermic peak of HE at 50 °C, exothermic peak of HE-ADH at 270–280 °C indicated the crystalline structure of the copolymers, endothermic peak of 5-FU at 290 °C, and degradation endothermic peak at 300 °C (). However, the melting endotherm of 5-FU is observed at 280 °C in the DSC thermogram of 5-FU-loaded HEP NPs, revealing that the drug is present in crystalline form inside the NPs (). Similar results were obtained in our earlier studies (Pinon-Segundo et al. Citation2005).
Figure 3. (I) DSC thermograph of (A) 5-FU, (B) HE, (C) ADH, (D) PCL, (E) HEP, and (F) 5-FU loaded HEP NPs (HEP 1). (II) XRD of (A) 5-FU, (B) HE, (C) ADH, (D) PCL, (E) HEP, and (F) 5-FU loaded HEP NPs (HEP 1).
X-ray diffraction
The XRD of 5-FU, HE, PCL, ADH, HEP NPs (without 5-FU), and 5-FU-loaded HEP NPs are shown in . 5-FU has shown characteristic intense peaks between 2θ values of 14 θ, 18 θ, 19 θ, 21 θ, 30 θ, and 35 θ which confirmed its crystalline nature (). XRD diffractogram of HE has a peak at 24 θ which indicates the crystalline nature of HE (). XRD diffractogram of ADH has a peak at 15, 22, 23 25, 26, 31, 35, and 42 θ which indicates the crystalline nature of ADH (). However, the diffractograms of HEP (with 5-FU) has a peak at 8, 19, 20, 22, 23, 24, 27, 29, 36, 40 θ () which indicates an increase in crystallinity and 5-FU associated with the crystalline form, it is suggested that the 5-FU-loaded HEP NPs are suitable for sustained and prolonged release.
In vitro drug release
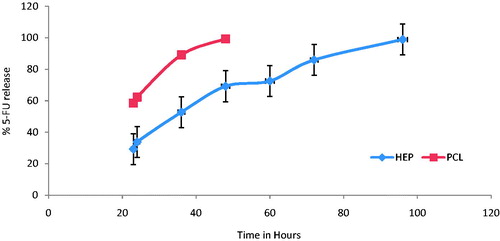
The release of the 5-FU from the NPs shows that the formulation has sustained release characteristics (). The results showed that there was a pronounced time prolongation of drug release from NPs system. HEP NPs were able to sustain 5-FU more than 96 h (98.9% in 96 h) where PCL NPs release only up to 48 h (). This may be due to high negative charge of the NPs, indicating that HE is forming a shell around the NPs, electrostatically stabilizing them in aqueous solution and it may also be due to the hydrophilic nature of HE leading to make NPs with long-circulating nature (Hu et al. Citation2004). Shakeel et al. also reported the LDE nanoemulsions of cholesteryl-maleoyl-5-fluorouracil (5-FU conjugate) and the researcher reported 97% 5-FU release after 24 h.
Figure 4. In vitro 5-FU release from HEP and PCL NPs from PBS, pH 7.4.
In vitro cellular cytotoxicity
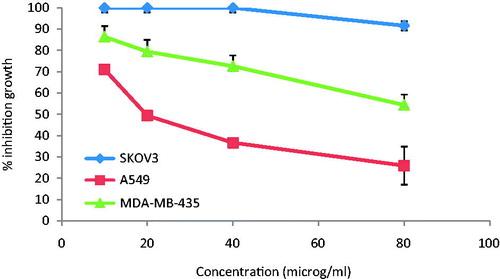
The growth inhibition of NPs on MDA-MB-435, SK-OV-3, and A-549 cells was investigated by MTT assay. The results clearly suggested a dose-dependent cytotoxicity, i.e. reduced cellular viability upon increasing the concentration of 5-FU. The survival fraction of cells upon incubation of plain 5-FU and NPs formulation in varying concentration is shown in . After incubation, 5-FU-loaded HEP NPs formulation shows inhibitory effect on cell growth, the % control growth was reduced with the increasing the concentration of 5-FU. Further, the cell viability decreased when the concentration of 5-FU either in free form or inside the NPs was increased. In the concentration range of 10–80 μg/mL, NPs were cytotoxic to a greater degree in comparison to plain 5-FU. This is concordant with carbohydrate receptor-binding capacity exhibited by NPs. These results demonstrated that HEP NPs cytotoxicity induced by 5-FU to the tumor cells were dose dependent and had a greater cytotoxic effect on the tumor cells than did plain 5-FU. This is attributed because HE has excellent dispersibility in aqueous solution and endows the NPs with the “stealth” property. Specifically, HE is of interest for use as a drug delivery system for treatment of cancers because it has been shown to inhibit angiogenesis and metastasis, particularly nonanticoagulant HP (Hu et al. Citation2004, Ono et al. Citation2002, Yoshitomi et al. Citation2004). Park et al. (Citation2004) developed self-assembled HP–deoxycholic acid NPs to be used as anticancer drug delivery. Heparin as specific ligand for Anaplastic Lymphoma Kinase (ALK) reveals a potential mechanism for the regulation of ALK activity in vivo and suggests an approach for developing ALK-targeted therapies for cancer (Murray et al. Citation2015).
Figure 5. In vitro cytotoxicity assay from HEP NPs in SKOV3, A549, and MDA-MB-435.
Pharmacokinetic study
Blood level studies of nanoparticulate formulations were finally performed on albino rats to determine the feasibility of delivering 5-FU into blood through intravenous route. It is attributed from that HEP NPs (HEP 1) contributed remarkably towards 5-FU concentration in comparison to 5-FU plain solution (control). In the pharmacokinetic study, AUC, Cmax, MRT, and Tmax values of HEP NPs were observed as 6096.075 ± 5.90 μg h/mL, 144.38 ± 1.52 μg/L, 58.71 ± 0.25 h, 96 ± 0.50 h, respectively, and 117.92 ± 1.78 μg h/mL, 45.35 ± 3.00 μg/L, 1.2 ± 0.25 h, 0.5 ± 0.02 h in case of plain 5-FU solution. This may be attributed because hydrophilic HE coating on PCL NPs lead to enhance the penetration of drug. These results are in good correlation with earlier reported studies (Garg et al. Citation2014).
Conclusion
The HEP NPs (HEP 1) loaded with 5-FU were obtained successfully by nanoprecipitation method with high 5-FU entrapment efficiency and low particle size. HEP NPs (HEP 1) were sustained by 5-FU for a period of up to 96 h and also found more hemocompatable. The results of cytotoxicity study showed that the cytotoxicity of 5-FU-loaded HEP NPs (HEP 1) seems to be higher than on A549 compared to MDA-MB 435 and SK-OV-03. Based on these studies, the formulations fabricated in this work could be promising for in vivo 5-FU drug delivery systems. These observations suggest that the present NPs offer an exciting mode of target delivery for potent 5-FU.
Funding information
The present research work was financially supported by MPCST, Bhopal (MP), India (Grant Number: 1766/CST/R&D/Bio, Proj/2013).
Acknowledgements
The author acknowledges SAIF-CDRI (Lucknow, India) for NMR facility and Indian Institute of Technology (Indore, India) for providing AFM facility. The author expresses his sincere thanks to Indian Institute of Technology (Ropar, India) for providing XRD facility. The author also expresses his sincere thanks to SAIF-RGPV (Bhopal, India) for providing particle size and zeta potential facilities, and also expresses his sincere thanks to ACTREC (Mumbai, India) for providing in vitro cytotoxicity assay facility.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Alanazi F, Haq N, Radwan AA, Alsarra IA, Shakeel F. 2015. Formulation and evaluation of cholesterol-rich nanoemulsion (LDE) for drug delivery potential of cholesteryl-maleoyl-5-fluorouracil. Pharm Dev Technol. 20:266–270.
- Alanazi FK, Haq N, Radwan AA, Alsarra IA, Shakeel F. 2014. Cholesterol-rich nanoemulsions (LDE) for drug targeting of cholesteryl-succinyl-5-fluorouracil conjugate. Curr Nanosci. 10:287–229.
- Alanazi FK, Haq N, Radwan AA, Alsarra IA, Shakeel F. 2014. Potential of lipid nanoemulsions for drug delivery of cholesteryl-hexahydrophthaloyl-5-fluorouracil. J Drug Deliv Sci Technol. 24:459–463.
- Allen TM. 2002. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2:705–763.
- Alsarra IA, Alarifi MN. 2004. Validated liquid chromatographic determination of 5-fluorouracil in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 804:435–439.
- Ansifield FJ, Schroeder JM, Curreri AR. 1962. Five years clinical experience with 5-fluorouracil. JAMA. 181:295–299.
- Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, et al. 2011. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 10:389–397.
- Boyle P, Levin B. 2008. World Cancer Report. World Health Organization Press.
- de Mattos AC, Khalil NM, Mainardes RM. 2013. Development and validation of an HPLC method for the determination of fluorouracil in polymeric nanoparticles. Braz J Pharma Sci. 49:117–126.
- Demoré B, Benoit E, Maincent P, Hoffman M, Bessière J. 1998. Determination of heparin in aqueous solutions. J Clin Pharm Ther. 23:381–384.
- Diasio RB, Harris BE. 1989. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 16:215–237.
- Dieckmann Y, Collfen H, Hofmann H, Fink AP. 2009. Particle size distribution measurements of manganese-doped ZnS nanoparticles. Anal Chem. 81:3889–3895.
- Garg A, Rai G, Lodhi S, Jain AP, Yadav AK. 2014. In-vitro and in-vivo assessment of dextran-appended cellulose acetate phthalate nanoparticles for transdermal delivery of 5-fluorouracil. Drug Deliv. 24:1–11.
- Ghoshal K, Jacob ST. 1997. An alternative molecular mechanism of action of 5-fluorouracil, a potent anticancer drug. Biochem Pharmacol. 53:1569–1575.
- Hirsh J. 1991. Heparin. N Engl J Med. 324:1565–1574.
- Hu Z, Xia X, Tang L. 2004. Process for synthesizing oil and surfactant-free hyaluronic acid nanoparticles and microparticles. US Patent App. 20,060/040,892.
- Jemal A, Siegel R, Xu J, Ward E. 2010. Cancer statistics, 2010. CA Cancer J Clin. 60:277–300.
- Kolhe P, Misra E, Kannan RM, Kanna S, Lieh-Lai M. 2003. Drug complexation, in vitro release and cellular entry of dendrimers and hyperbranched polymers. Int J Pharm. 259:143–160.
- Martins AF, Pereira AGB, Fajardo AR, Rubira AF, Muniz EC. 2011. Characterization of polyelectrolytes complexes based on N, N, and N-trimethyl chitosan/heparin prepared at different pH conditions. Carbohydr Polym. 86:1266–1272.
- Murray PB, Lax I, Reshetnyak A, Ligon GF, Lillquist JS, Natoli EJ, Jr, et al. 2015. Heparin is an activating ligand of the orphan receptor tyrosine kinase ALK. Sci Signal. 20:ra6.
- Nidhin M, Indumathy R, Sreeram KJ, Nair BU. 2008. Synthesis of iron oxide nanoparticles of narrow size distribution on polysaccharide templates. Bull Mater Sci. 31:93–96.
- Ono K, Ishihara M, Ishikawa K, Ozeki Y, Deguchi H, Sato M, et al. 2002. Periodate-treated, non-anticoagulant heparin carrying polystyrene (NAC-HCPS) affects angiogenesis and inhibits subcutaneous induced tumour growth and metastasis to the lung. Br J Cancer. 86:1803–1812.
- Park K, Kim K, Kwon IC, Kim SK, Lee S, Lee DY, Byun Y. 2004. Preparation and characterization of self-assembled nanoparticles of heparin-deoxycholic acid conjugates. Langmuir. 20:11726–11731.
- Pinon-Segundo E, Ganem-Quintanar A, Alonso-Perez V, Quintanar-Guerrero D. 2005. Pharmaceutical nanotechnology preparation and characterization of triclosan nanoparticles for periodontal treatment. Int J Pharm. 294:217–232.
- Polakovic M, Gorner T, Gref R, Dellacherie E. 1999. Lidocaine loaded biodegradable nanospheres. II. Modelling of drug release. J Control Release. 60:169–177.
- Shakeel F, Alanazi FK, Raish M, Haq N, Radwan AA, Alsarra IA. 2015. Pharmacokinetic and in vitro cytotoxic evaluation of cholesterol-rich nanoemulsion of cholesteryl-succinyl-5-fluorouracil. J Mol Liq. 211:164–168.
- Shakeel F, Haq N, Aldhfyan A, Alanazi FK, Alsarra IA. 2014. Double w/o/w nanoemulsion of 5-fluorouracil for self-nanoemulsifying drug delivery system. J Mol Liq. 200:183–190.
- Shakeel F, Haq N, Aldhfyan A, Alanazi FK, Alsarra IA. 2015. Chemoprevention of skin cancer using low HLB surfactant nanoemulsion of 5-fluorouracil: a preliminary study. Drug Deliv. 22:573–580.
- Shi L, Farber JN, Michaels KC, Dickey A, Thompson KC, Shelukar SD, et al. 2003. Characterization of crystalline drug nanoparticles using atomic force microscopy and complementary techniques. Pharm Res. 20:479–484.
- Theocharis AD, Skandalis SS, Tzanakakis GN, Karamanos NK. 2010. Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 277:3904–3923.
- Tomuta AM, Ramis X, Ferrandoc F, Serra A. 2012. The use of dihydrazides as latent curing agents in diglycidyl ether of bisphenol a coatings. Progress Org Coat. 74:59–66.
- Xie J, Hwa Wang C. 2004. Paclitaxel-loaded biodegradable nanoparticles developed by direct dialysis and electrodydrodynamic atomization methods. AIChE Annual Meeting, Texas; 7–12.
- Yadav AK, Agarwal A, Jain S, Mishra AK, Bid H, Rai G, Agrawal H, Agrawal GP. 2010. Chondroitin sulphate decorated nanoparticulate carriers of 5-fluorouracil: development and in vitro characterization. J Biomed Nanotechnol. 6:1–11.
- Yadav AK, Mishra P, Jain S, Mishra P, Mishra AK, Agrawal GP. 2008. Preparation and characterization of HA–PEG–PCL intelligent core–corona nanoparticles for delivery of doxorubicin. J Drug Target. 6:464–478.
- Yadav AK, Mishra P, Mishra AK, Mishra P, Jain S, Agrawal GP. 2007. Development and characterization of hyaluronic acid-anchored PLGA nanoparticulate carriers of doxorubicin. Nanomedicine. 3:246–257.
- Yoshitomi Y, Nakanishi H, Kusano Y, Munesue S, Oguri K, Tatematsu M. 2004. Inhibition of experimental lung metastases of Lewis lung carcinoma cells by chemically modified heparin with reduced anticoagulant activity. Cancer Lett. 207:165–174.