
Abstract
We previously synthesized some novel benzoxazole derivatives-containing sulfonamide. In this study, the compounds were investigated for their antitumor activities against the HL-60 human leukemia cells, using the MTT assay. Moreover, quantum chemical calculations using the DFT methods were applied for understanding the difference in antitumor activity. Additionally, molecular docking into active site of the DNA Topo II enzyme was performed on 3QX3. PDB file in order to find out possible mechanism of antitumor effect.
According to all obtained results showed that compounds 1b, 1c, and 1d could be potential drug candidates as new antitumor agents, and are promising for cancer therapy.
Introduction
Cancer remains a major threat to human health, representing the second leading cause of death worldwide. It is estimated by the World Health Organization that in 2030, 13.1 million people will die from cancer. Thus, the medical need for cancer treatment remains one of the most demanding areas in scientific research (Akbal et al. Citation2016, Jagani et al. Citation2016). The discovery and development of new treatments for cancer are urgently needed because of problems, such as toxicity and drug-resistance with current treatment (Krishna and Mayer Citation2000). Recently, many efforts have been made to develop safer and more effective ways of treating cancers and to search for novel chemotherapeutic agents with minimal side effects (Caleta et al. Citation2009). In this context, the major challenge is the development of more effective and less toxic new anticancer drugs (Abdelgawad et al. Citation2014).
The benzoxazole scaffold is a core structure found in a wide class of natural and synthetic compounds and has been targeted by considerable research over many years because they constitute an important class of heterocyclic compounds that exhibit substantial chemotherapeutic activity (Akbay et al. Citation2003, Ertan et al. Citation2009, Gautam et al. Citation2012, Kim et al. Citation1996, Lage et al. Citation2006, Oksuzoglu et al. Citation2008, Omar et al. Citation2012, Oren et al. Citation1997, Oren et al. Citation1998, Pinar et al. Citation2004, Rida et al. Citation2005, Sato et al. Citation2001, Shi et al. Citation1996, Varga et al. Citation2005, Xiang et al. Citation2012, Yildiz Oren et al. Citation2004a,b).
Benzoxazole and its ring derivatives structurally resemble adenine and guanine heterocyclic bases present in the nucleic acid structure. Therefore, assuming that the chemotherapeutic activities of the benzoxazole derivatives occur by inhibiting the nucleic acid synthesis, research interest in biological activities of these compounds has increased. Benzoxazole moiety is expanding their biological importance and has been studied frequently for the exploration of their biological assistance in varied pharmacological circumstances (Gautam et al. Citation2012).
Through searching the literature, it was found that benzoxazole derivatives play an important role in the design of new drugs, since they present interesting biological and pharmacological activities, including antimicrobial (Padalkar et al. Citation2012, Yildiz-Oren et al. Citation2004a,Citationb), antiviral (Plemper et al. Citation2004), antituberculosis (Klimesová et al. Citation2009), antioxidant, antihelmintic (Satyendra et al. Citation2011), and anticancer (Abdelgawad et al. Citation2014, El-Hady and Abubshait Citation2015, Kamal et al. Citation2010, Rida et al. Citation2005, Sato et al. Citation2001, Xiang et al. Citation2012) activities.
On the other hand, heterocycles possessing sulfonamido moieties have attracted obvious attention due to their role as pharmacophores (Akurathi et al. Citation2010, Chandak et al. Citation2013). Sulfonamides have been in clinical use for several decades. Recently, many novel sulfonamide derivatives have shown substantial antitumor activities (Crespo et al. Citation2010, Owa et al. Citation1999).
Recently, we reported the synthesis of some novel 2-(p-substitutedbenzyl/phenyl)-5-(p-nitro/aminophenylsulfonamido)benzoxazole derivatives and their biological screening for antimicrobial activities against human pathogenic microbes and inhibitory activities of human Glutathione-S-Transferase (hGST) P1–1 (Ertan-Bolelli et al. Citation2014, Ertan-Bolelli et al. Citation2016). Glutathione-S-transferases (GSTs) are enzymes involved in cellular detoxification by catalyzing the nucleophilic attack of glutathione (GSH) on the electrophilic center of numerous toxic compounds and xenobiotics, including chemotherapeutic drugs (Mathew et al. Citation2006). hGST P1–1 is the most prevalent isoform of the mammalian cytosolic GSTs. GST P1–1 is frequently over-expressed in rat and human tumors, including carcinoma of the colon, lung, kidney, ovary, pancreas, esophagus, and stomach. It is suggested that over-expression of hGST P1–1 by human tumor cells may play a role in resistance to anticancer drugs, such as adriamycin, cisplatin, melphalan, and etoposide (Schisselbauer et al. Citation1990, Tsuchida and Sato Citation1992, Waxman Citation1990). According to our previous study (Ertan-Bolelli et al. Citation2014, Ertan-Bolelli et al. Citation2016), it was found that among the tested sulfonamido benzoxazoles only 2-(p-chlorobenzyl)-5-(p-nitrophenylsulfonamido) benzoxazole (compound 1d) showed a hGST P1–1 enzyme inhibitor even it indicated a similar potency to the reference drug ethacrynic acid (Ertan-Bolelli et al. Citation2014). This study showed us that further studies were needed in order to find a lead compound for treating cancer cells.
For that reason, we decided to evaluate our anti-cancer candidate compound (1d) in a human leukemia cancer cell line (HL-60). In here, not only we have screened compound 1d against HL-60 cells but also we have selected the compounds with phenyl, which has a different substituent at the para position, instead of benzyl at the second position of the benzoxazole ring for testing. Thus, we have selected three more compounds in order to understand the effect of substitution on the HL-60 cells. These are; one non-substituted phenyl, one bearing CH3, which increases the electron density of the phenyl, and one fluorine atom (). In addition, the effects of tested compounds have been compared with the reference drug, etoposide.
Table 1. 2-(p-substitutedbenzyl/phenyl)-5-(nitrophenylsulfonamido)benzoxazole derivatives and their antitumor activities against HL-60 cell line.
Moreover, during the quantum chemical calculations, density functional theory (DFT) methods inside the Gaussian 09 package (Becke Citation1993, Chai and Head-Gordon Citation2008, Gaussian 09, Revision D.01, Frisch et al. Citation2009, Lee et al. Citation1988) were applied to calculate the frontier molecular orbitals (HOMO, LUMO), the magnitude of the dipole moment, and molecular electrostatic potential (MEP) distributions of geometrically optimized structures of each tested sulfonamido benzoxazoles. These theoretical studies were performed to find out the difference in anti-proliferative activity against HL-60 cells of the sulfonamide benzoxazole derivatives.
Recent molecular studies have greatly expanded the biological contexts where DNA topoisomerase II (Topo II) enzyme plays critical roles, including DNA replication, transcription, and chromosome segregation. Although the biological functions of Topo II are important for insuring genomic integrity, the ability to interfere with Topo II and generate enzyme mediated DNA damage is an effective strategy for cancer chemotherapy. Hence, Topo II has become popular targets for cancer chemotherapy treatments. It is thought that Topo II inhibitors block the ligation step of the cell cycle, generating double stranded breaks that harm the integrity of the genome. Introduction of these breaks subsequently leads to apoptosis and cell death (Nitiss Citation2009).
Due to the target of Topo II enzyme in the development of new cancer drugs, this study also aimed to determine whether they would inhibit the Topo II of compounds (1a–d). For that reason, molecular docking studies using CDocker method in Accelrys Discovery Studio version 3.5 (Wu et al. Citation2003, Discovery Studio 3.5, Citation2012; Accelrys Inc., San Diego, CA) were performed onto the active site of the DNA Topo II enzyme (PDB ID: 3QX3) (Wu et al. Citation2011) in order to understand the mechanism of antitumor activity of the tested compounds (1a–d). The main goal of this study was to discover new anticancer drug candidates having sulfonamide benzoxazole for the HL-60 cells.
Materials and methods
Chemicals and reagents
The tested 2-(p-substituted phenyl)-5-(p-nitrophenylsulfonamido)benzoxazole derivatives were previously synthesized by our group (Ertan-Bolelli et al. Citation2014, Ertan-Bolelli et al. Citation2016). The chemical structure of the compounds can be seen in . All sulfonamido benzoxazoles were dissolved in dimethylsulfoxide (DMSO) and ethanol [20% + 80% v/v] and freshly prepared just before the experiments.
Reagents for cell culture, including RPMI-1640, fetal bovine serum (FBS), Penicillin-Streptomycin, dPBS, and MTT solution were obtained from Life Tech. (Life Technologies Corp., Carlsbad, CA), DMSO and ethanol were purchased from Santa Cruz Biotechnology (Santa Cruz Biotechnology Inc., Heidelberg, Germany). All chemicals and reagents used in this study were of analytical grade.
Cell cultures, conditions, and treatment
The HL-60 (human promyelocytic leukemia) cell line used in this study was obtained from German Collection of Microorganisms and Cell Cultures (Leibniz Institute DSMZ, Braunschweig, Germany). HL-60 cells were routinely cultured in RPMI 1640 medium containing 2 mM L-glutamine, supplemented with 10% (v/v) FBS, 100 U/mL penicillin and 0.1 mg/mL streptomycin. The cells were grown in 75-cm² flasks in an incubator at 37 °C in a humidified atmosphere containing 5% CO2. Incubation was continued until a confluent growth was achieved and the cells were freshly subcultured before each experiment to be in the exponential phase of growth.
The MTT cytotoxicity assay
The MTT cell proliferation assay is a colorimetric assay for assessing cell viability. It can be used for measurement of cell proliferation and analysis of cytotoxic and cytostatic compounds, such as anticancer drugs and other pharmaceutical compounds. The assay is based on the cleavage of the tetrazolium salt MTT in the presence of an electron-coupling reagent (Mossmann Citation1983).
We carried out the assay according to the method described by D. Seenaiah et al. (Citation2014). HL-60 cells were seeded in 96-well plates with 7.5 × 10³/well in 0.1 mL of RPMI 1640 medium supplemented with 10% FBS and cultured in a humidified incubator (at 37 °C in 5% CO2) for 24 h. The tested compounds were dissolved in DMSO and ethanol and serial dilutions were made for the compounds. Different concentrations of dilutions were added to cells in 96 well plates and the cells were incubated further for 48 h.
In each well plate run, wells for solvent control (medium + cells + [DMSO and ethanol]), blank (medium + solvent [DMSO and ethanol]), and growth control (medium + cells) were also included.
MTT (3–(4,5-Dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide) solution (20 μL of 5 mg/mL) was added to each well and the incubation was continued for an additional 4 h. The medium was removed and 100 μL of DMSO was added to dissolve the formazan crystals. The dark blue formazan crystals formed within the viable cells were solubilized with DMSO and the absorbance (OD) of each well was measured using a ELISA plate reader (Molecular Devices) at 492 nm. The percent survival of the cells was calculated by using non-linear regression. Cell survival was measured as the absorbance percentage compared with the control (non-treated cells with the compounds). The proliferation inhibitory effects of the compounds on cancer cells were expressed as IC50, which is the compound concentration required to inhibit cell growth by 50%. IC50 values of the compounds were obtained by plotting the percentage of survival versus the concentrations. We used etoposide as the standard anticancer drug against HL-60 cells at our experiments.
Data analysis
The MTT cell cytotoxicity experiments were performed in triplicates; and each point on the graphs was given with standard deviation of the mean value. IC50 values of the compounds were determined by regression analysis using GraphPad Prism software version 4.0 (GraphPad Software Inc., San Diego, CA). Data are shown as mean ± SD (standard deviation) of three independent experiments.
Computational methods
All computations presented within were carried out using Gaussian 09 package (Gaussian 09, Revision D.01, Frisch et al. Citation2009). The geometries of compounds were optimized by DFT methods. For comparison, different DFT functionals were applied during the calculations of geometries and molecular orbitals. B3LYP (Becke, three-parameter, Lee-Yang-Parr) (Becke Citation1993, Lee et al. Citation1988) and ωB97X-d (Chai and Head-Gordon Citation2008) functionals with 6–31G(d) basis set were used for all the calculations. The geometry of the saddle point was confirmed by checking of the vibrational modes and frequencies.
Molecular docking studies
The crystal structure of the Topo II enzyme complexed with etoposide was retrieved from the Protein Data Bank (PDB ID: 3QX3) (Wu et al. Citation2011). AccelrysDiscoveryStudio version 3.5 software (Discovery Studio 3.5, 2012; Accelrys Inc., San Diego, CA) was used for preparation and docking processes. The water molecules were removed, hydrogens were added and their positions were optimized using the all atom CHARMm force field and the adopted basis set Newton Raphson (ABNR) method until the root mean square deviation (RMSD) gradient was <0.05 kcal/mol/Ų. The binding site was defined from the cavity finding method. The binding sphere was selected from the active site using the binding site tools (33.73, 95.77, 50.84, and 11.80). The tested compounds were sketched, all atom CHARMm forcefield parameterization were assigned and then minimized using the ABNR method as described above. Conformational searches were carried out using a simulated annealing molecular dynamics (MD) approach. Afterwards, CDocker (Wu et al. Citation2003) method was performed by using Discovery Studio 3.5. The protein was held rigid while the ligands were allowed to be flexible during refinement. Finally, all docked poses were scored by applying analyze ligand poses subprotocol and binding energies were calculated by applying calculate binding energy subprotocol in Discovery Studio 3.5 by using in situ ligand minimization step (ABNR method) and using implicit solvent model (GBMV). The lowest binding energy was taken as the best-docked conformation of the compound for the Topo II enzyme. The pictures were taken by using Discovery Studio 4.1 Visualizer (Accelrys Inc., San Diego, CA).
Results and discussion
In vitro antitumor activity of the compounds againstHL-60 cells
For the evaluation of the anti-tumor cytotoxicity of 2-(p-subtitutedbenzyl/phenyl)-5-(p-nitrophenylsulfonamid)benzoxazole derivatives, the HL-60 (human acute promyelocytic leukemia) cancer cell line was used. Leukemia is a cancer of the white blood cells. Acute promyelocytic leukemia is a subtype of leukemia that can affect people at any age. Most forms of leukemia are treated with chemotherapy. Etoposide is a potent Topo II enzyme inhibitor as an antitumor drug that was approved by the FDA for the treatment of various tumors, such as small-cell lung cancer, the germ-line cancers, leukemia, and lymphoma subtypes (Baldwin and Osheroff Citation2005). For this reason, we used etoposide as the standard anticancer drug against human promyelocytic leukemia (HL-60) cells in our experiments.
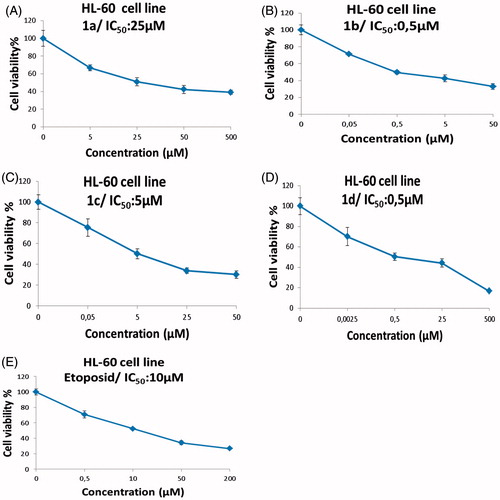
The results of antitumor activity against the HL-60 cancer cell line of the sulfonamido benzoxazole derivatives are shown in and ), respectively. Based on the data obtained, the tested compounds have anti-proliferative potentials on HL-60 cells to varying degrees. The tested derivatives are arranged, based on their antitumor potentials against the human HL-60 cancer cell line and on the obtained IC50 values, in the following order: compounds 1b (IC50 = 0.5 ± 1.2 μM) = 1d (IC50 = 0.5 ± 1.3 μM) > 1c (IC50 = 5 ± 1.5 μM) > 1a (IC50 = 25 ± 1.1 μM). Thus, compounds 1b and 1d especially seemed to have the most potent anti-proliferative effects with the IC50 values of 0.5 μM (, )). At the same time, compound 1c was found to have noteworthy effectiveness with the IC50 value of 5 ± 1.5 μM (). Moreover, the cytotoxicities of our compounds have been compared with etoposide (IC50 = 10 ± 1 μM) (), which is a very special drug for human leukemia cancer therapy as given above (Baldwin and Osheroff Citation2005).
Figure 1. The anti-proliferative activities of the sulfonamido-benzoxazoles against human HL-60 cancer cell line. (A) compound 1a, (B) compound 1b, (C) compound 1c, (D) compound 1d, and (E) standard drug etoposide showed a dose-dependent growth inhibition (cells viability % versus the concentrations) on the cells.
Structure-activity relationships indicated that compounds bearing substituent on the para position of phenyl ring at the second position of benzoxazole indicated significantly anti-proliferative activity for the HL-60 cancer cell line. Surprisingly, these potent derivatives (1b, 1d, and 1c) were found to be more active than the well-known anti-cancer drug etoposide (). Obtained data showed that benzyl or phenyl being in the second position of the benzoxazole structure did not create any major difference in activity. In here, the other considerable point is that attaching a halogen atom on the para position of phenyl ring is more important than CH3 group for increasing the anti-proliferative activity against HL-60 cells.
Theoretical analysis of the anti-tumor activity
In order to understand the difference in antitumor activity against HL-60 cells of the tested sulfonamido benzoxazole derivatives, computational studies using the DFT functional. For this reason, the frontier molecular orbitals (HOMO, LUMO), the magnitude of the dipole moment, and MEP distributions were calculated for geometrically optimized structures of each compound. Frontier molecular orbitals can present valuable information about the bonding and other chemical properties (Fleming Citation1976). The highest occupied molecular orbital (HOMO) includes the electrons that are possible to take an important role in a chemical reaction. The values of HOMO and LUMO and their energy gap are one of the indicators of chemical activity and kinetic stability of the molecules. Whilst the hard molecules possess a large HOMO–LUMO energy gap, soft molecules have a small HOMO–LUMO energy gap. To find the basis set effect, the geometry optimizations and orbitals calculation were carried out using 6–31G(d) and 6–311++G(d,p) basis sets at the B3LYP level of theory. And also, dispersion-corrected functional ωB97X-d was used with 6–31G(d) basis set. The results of molecular orbital energies show the importance of inclusion the dispersion effects in single point energy calculations (). While high level basis set was used (6–311++G(d,p)), the energies of frontier molecular orbitals, and their gaps became smaller according to 6–31G(d) basis set. The effect of long-range corrected ωB97X-d functional on orbital energies was observed as reductions in energy values. The HOMO–LUMO gap energy values are observed nearly close to each other for all compounds at the different level of calculations. Nevertheless, we may say that HOMO, LUMO, and their gap energy values can correlate the biological activity. Most active compounds (1b and 1d) have smaller HOMO and LUMO energy values in all calculations. The molecular orbitals displayed that the electron density in HOMO mostly located from p-substituted benzyl to sulfonamide part of the compound (). LUMO cover the p-nitrophenyl and sulfonamide part of the compounds. Furthermore, compound 1d has bended structure due to the methylene bridge between the two rings. This bended structure of compound 1d leads to pi–pi stacking between the phenyl and benzoxazole rings. Introducing the electron withdrawing groups, such as halogen atoms (F and Cl) in 1b and 1d compounds, lead to small dipole moment due to balancing of electron withdrawing effect of nitro group, which is located in the another part of the molecule. In our case, smaller dipole moment causes higher anti-tumor activity.
Figure 2. Plots of the frontier orbitals compounds 1a–d by ωB97X-d/6–31G(d) level of theory.
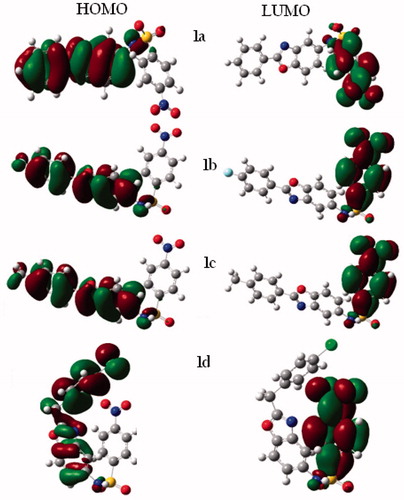
Table 2. The frontier molecular orbital energies and their gaps, and dipole moments. Energies are in eV. The unit of dipole moment is Debye.
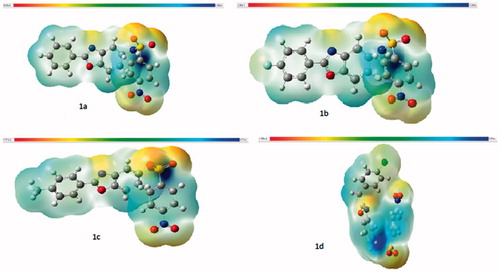
MEP displays the electronic density and is a very valuable descriptor in finding sites suitable for electrophilic attack and nucleophilic reactions plus hydrogen bonding interactions. Attraction is represented by negative values; repulsion is indicated by positive values display the electronic density. ωB97X-d/6–31G(d) level of theory was used to explore the MEP maps (). In the maps, potential increases in the order red < orange < yellow < green < blue. The positive (blue) regions of MEP correspond to electrophilic reactivity and the negative (red) regions to nucleophilic reactivity displayed in . In the MEP map of compounds 1a–d, the negative regions are mostly cover the oxygen atoms of nitro and sulphoxide groups, halogen, and nitrogen atoms, demonstrating a promising site for electrophilic attack when interacts with positively charged species. The molecules have mostly maximum positive region, which are localized on the hydrogen and carbons, especially on hydrogen, which belongs to benzoxazole nitrogen. Positive regions of the molecule can be evaluated that electrostatically attract negatively charged atoms and can act as an ideal hydrogen bond donor group.
Figure 3. Maps of molecular electrostatic potentials (MEP) of compounds 1a–d.
Consequently, we can report that the results obtained from computational studies are in good agreement with experiments for the most active compounds 1b and 1d.
Molecular docking studies
Recently, it was reported by us that 2-(p-chlorobenzyl)-5-(p-nitrophenylsulfonamido) benzoxazole (compound 1d) inhibited the human GST P1–1 enzyme with similar potency like the reference drug ethacrynic acid (Ertan-Bolelli et al. Citation2014). As is well known, hGST P1–1 enzyme is over-expressed in many cancers and contributes to multi-drug resistance by directly conjugating with anticancer agents etoposide (Mathew et al. Citation2006, Schisselbauer et al. Citation1990, Tsuchida and Sato Citation1992, Waxman Citation1990) However, compounds 1a, 1b, and 1c did not show more the inhibitory effect for the hGST P1–1 enzyme than ethacrynic acid (Ertan-Bolelli et al. Citation2014). As is well known, the inhibition of DNA Topo II is one of the important targets for cancer therapy (Wu et al. Citation2011). Therefore, we used molecular docking method in order to predict the interactions with DNA Topo II enzyme. In this study, all tested compounds (1a–d) were docked into active site of Topo II (PDB ID: 3QX3) by using CDocker method (Wu et al. Citation2003, Discovery Studio 3.5, 2012). Firstly, etoposide, which is the well-known specific inhibitor of Topo II enzyme, was docked. After then molecular docking studies were applied for the compounds (1a–d). The molecular docking studies of the tested compounds showed that their binding to Topo II active site with the position and orientation was found to be very close to the result of the crystal structure of etoposide in the enzyme. All the docking scores were given in . According to the docking studies, etoposide revealed H bonds with Asp479, Gly478, DC8, DT9, DG10, and DG13, which are in accordance with the X-ray structure binding features (3QX3). As seen in and , one of the most active sulfonamido benzoxazole 1d showed two H bond interactions with an active site residue Lys456 and DT9. While Lys456 bound with nitro group at the para position of benzene sulfonamido moiety, DT9 bound NH of sulfonamido. There is an electrostatic interaction between Asp479 and nitro group of compound 1d, as well. Besides, DA12, DT9, DG13, and DC8 interrelated with benzoxazole ring of compound 1d using pi–pi interactions. Nitro group of para position phenyl and nitrogen atom of oxazole of another the most effect compound 1b interacted with Gly478 and DA12 using H bond, respectively. Additionally, the benzoxazole ring of compound 1b indicated pi–pi interactions with DT9, DA12, and DG13. Compound 1c showed H bond interaction between Pro819 and the nitro group. Derivative 1c had also pi–pi interactions with DC8, DT9, and DG13 of DNA. Moderate active compound 1a has three H bonds with active site residues Gly478, DT9, DA12, and three pi–pi interactions with DC8, DT9, and DA12. While DC8 interacted to phenyl ring, DT9 and DA12 interrelated to benzoxazole of compound 1a. Binding energies of these compounds 1a, 1b, 1c, 1d, and etoposide are −36.73, −37.33, −38.68, −43.20, and −83.39, respectively. Even if derivative 1a docked into Topo II enzyme (3QX3) well, the binding energy of it is higher than the other, as seen in . This can be pointed why compound 1a did not show more effect than etoposide.
Figure 4. (A) Docking pose of etoposide in Topo II enzyme (PDB code:3QX3) nucleotides are seen as parent colors: Deoxy adenine (DA) is red, Deoxy guanine (DG) is green, Deoxy cytosine (DC) is purple, Deoxy thymine (DT) is blue. (B) Docking position of compound 1b. (C) Docking position of compound 1d (hydrogen bond is shown as green dashed line).
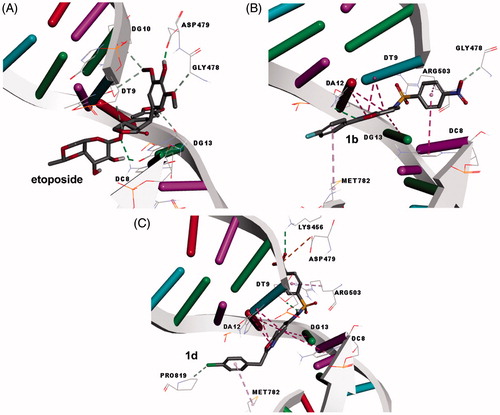
Table 3. Docking results.
In summary, the main objective for treatment of cancer is to synthesize the compounds that show promising activity as chemotherapeutic agents that are more effective and have lower toxicity. The results of this study point out that the activity of the reported compounds (1b, 1c, and 1d) supports its promise as a component of therapeutic strategies for cancer.
Conclusions
Recently we have reported, that among the tested sulfonamido benzoxazole derivatives, only compound 1d (2-(p-chlorobenzyl)-5-(p-nitrophenylsulfonamido)benzoxazole was found to be a significantly potent inhibitor like reference drug ethacrynic acid against human hGST P1–1.
In this study, we have screened some of the previously synthesized 2-(p-subtitutedbenzyl/phenyl)-5-(p-nitrophenylsulfonamido)benzoxazole derivatives for their antitumor activities against the HL-60 human leukemia cell line. Then, computational studies using the DFT methods have been done in order to understand the difference in antitumor activity against HL-60 cells of the tested sulfonamido benzoxazole derivatives. Moreover, all tested compounds (1a–d) were docked into active site of DNA Topo II (PDB ID: 3QX3) by using CDocker method for predicting the interactions into Topo II enzyme.
According to the result of the anti-proliferative activity against HL-60 cancer cell line of the sulfonamide benzoxazoles, bearing a substituent of the para position of benzyl/phenyl ring of the second carbon of benzoxazole bicyclic structure showed very important activity. Even these compounds (1b, 1d, and 1c) were found to be more potent than well-known drug etoposide. In here, specially, it is established that a halogen such as chlorine, fluorine on the R position play a very significant role in order to enhance the anti-proliferative activity against HL-60 cells.
The quantum chemical calculations showed a good correlation with biological activity for compounds 1b and 1d. Moreover, it has been obtained that smaller dipole moment caused higher antitumor effect against HL-60 cell line.
According to molecular docking studies, it can be considered that tested sulfonamide benzoxazole compounds would be shown their antitumor activity on the HL-60 cell line via the inhibition of Topo II enzyme. Furthermore, we need to conclude that the mechanism of the antitumor activity of compound 1d could be via either the inhibition of GST P1–1 enzyme as reported by us and DNA Topo II enzyme.
In conclusion, it can be considered that these compounds (1b, 1c, and 1d) could be potential drug candidates as new antitumor agents and provide facilities us for developing more effective anticancer drugs.
Disclosure statement
The authors have declared no conflicts of interest.
References
- Abdelgawad MA, Abdellatif KRA, Ahmed OM. 2014. Design, synthesis and anticancer screening of novel pyrazole derivatives linking to benzimidazole, benzoxazole and benzothiazole. Med Chem. S1:001. doi:10.4172/2161-0444.S1-001.
- Akbal O, Erdal E, Vural T, Kavaz D, Denkbaş EB. 2016. Comparison of protein- and polysaccharidebased nanoparticles for cancer therapy: synthesis, characterization, drug release, and interaction with a breast cancer cell line. Artif Cells Nanomed Biotechnol.17:1–11. doi:10.3109/21691401.2016.1170694.
- Akbay A, Oren I, Temiz-Arpaci O, Aki-Sener E, Yalcin I. 2003. Synthesis and HIV-1 reverse transcriptase inhibitor activity of some 2,5,6-substituted benzoxazole, benzimidazole, benzothiazole and oxazolo(4,5-b)pyridine derivatives. Arzneim Forsch. 53:266–271.
- Akurathi V, Dubois L, Lieuwes NG, Chitneni SK, Cleynhens BJ, Vullo D, et al. 2010. Synthesis and biological evaluation of a 99mTc-labelled sulfonamide conjugate for in vivo visualization of carbonic anhydrase IX expression in tumor hypoxia. Nucl Med Biol. 37:557–564.
- Baldwin EL, Osheroff N. 2005. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 5:363–372.
- Becke AD. 1993. Density‐functional thermochemistry. III. The role of exact exchange. J Chem Phys. 98:5648–5652.
- Caleta I, Kralj M, Marjanovic M, Bertosa B, Tomic S, Pavlovic G, et al. 2009. Novel cyano- and amidinobenzothiazole derivatives: synthesis, antitumor evaluation, and X-ray and quantitative structure-activity relationship (QSAR) analysis. J Med Chem. 52:1744–1756.
- Chai CD, Head-Gordon M. 2008. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys. 10:6615–6620.
- Chandak N, Bhardwaj JK, Sharma R, Sharma PK. 2013. Inhibitors of apoptosis in testicular germ cells: synthesis and biological evaluation of some novel IBTs bearing sulfonamide moiety. Eur J Med Chem. 59:203–208.
- Crespo R, de Bravo MG, Colinas PA, Bravo RD. 2010. In vitro antitumor activity of N-glycosyl sulfonamides. Bioorg Med Chem Lett. 20:6469–6471.
- Discovery Studio 3.5, Accelrys Inc. 2012.
- El-Hady HA, Abubshait SA. 2015. Synthesis of imidazolinone and benzoxazole derivatives, and evaluation of their anticancer activity. Res Chem Intermediat. 41:1833–1841.
- Ertan T, Yildiz I, Tekiner-Gulbas B, Bolelli K, Temiz-Arpaci O, Ozkan S, et al. 2009. Synthesis, biological evaluation and 2d-QSAR analysis of benzoxazoles as antimicrobial agents. Eur J Med Chem. 44:501–510.
- Ertan-Bolelli T, Musdal Y, Bolelli K, Yilmaz S, Aksoy Y, Yildiz I, Aki-Yalcin E, Yalcin I. 2014. Synthesis and biological evaluation of 2-substituted-5-(4-nitrophenylsulfonamido)benzoxazoles as human GST P1-1 inhibitors, and description of the binding site features. ChemMedChem. 9:984–992.
- Ertan-Bolelli T, Yildiz I, Ozgen-Ozgacar S. 2016. Synthesis, molecular docking and antimicrobial evaluation of novel benzoxazole derivatives. Med Chem Res. 25:553–567.
- Fleming I. 1976. Frontier Orbitals and Organic Chemical Reactions. Trowbridge, Wiltshire: Wiley Redwood Books, pp. 1.
- Gautam MK. Sonal, Sharma NK, Priyanka, Jha KK. 2012. Pharmacological profile and pharmaceutical importance of substituted benzoxazoles: a comprehensive review. Int J ChemTech Res. 4:640–650.
- Gaussian 09, Revision D.01, Frisch, Trucks GV, Schlegel HB, Scuseria GE, et al. 2009. Wallingford, CT: Gaussian Inc.
- Jagani H, Kasinathan N, Meka SR, Josyula VR. 2016. Antiapoptotic Bcl-2 protein as a potential target for cancer therapy: a mini review. Artif Cells Nanomed Biotechnol. 44:1212–1221.
- Kamal A, Reddy KS, Khan MN, Shetti RV, Ramaiah MJ, Pushpavalli SN, et al. 2010. Synthesis, DNA-binding ability and anticancer activity of benzothiazole/benzoxazole-pyrrolo[2,1-c][1,4]benzodiazepine conjugates. Bioorg Med Chem. 18:4747–4761.
- Kim JSQ, Gatto B, Yu C, Liu A, Liu LF, La Voie EJ. 1996. Structure-activity relationships of benzimidazoles and related heterocycles as topoisomerase I poisons. Bioorg Med Chem. 6:163–172.
- Klimesová V, Kocí J, Waisser K, Kaustová J, Möllmann U. 2009. Preparation and in vitro evaluation of benzylsulfanyl benzoxazole derivatives as potential antituberculosis agents. Eur J Med Chem. 44:2286–2293.
- Krishna R, Mayer LD. 2000. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 11:265–283.
- Lage H, Aki-Sener E, Yalcin I. 2006. High antineoplastic activity of new heterocyclic compounds in cancer cells with resistance against classical DNA topoisomerase II-targeting drugs. Int J Cancer. 119:213–220.
- Lee C, Yang W, Parr RG. 1988. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter. 37:785–789.
- Mathew N, Kalyanasundaram M, Balaraman K. 2006. Glutathione-S-transferase inhibitors. Expert Opin Ther Pat. 16:431–444.
- Mossmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 65:55–63.
- Nitiss JL. 2009. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 9:338–350.
- Oksuzoglu E, Tekiner-Gulbas B, Alper S, Temiz-Arpaci O, Ertan T, Yildiz I, et al. 2008. Some benzoxazoles and benzimidazoles as DNA topoisomerase I and II inhibitors. J Enzyme Inhib Med Chem. 23:37–42.
- Omar MA, Shaker YM, Galal SA, Ali MM, Kerwin SM, Li J, et al. 2012. Synthesis and docking studies of novel antitumor benzimidazoles. Bioorg Med Chem. 20:6989–7001.
- Oren I, Temiz O, Yalcin I, Sener E, Akin A, Ucarturk N. 1997. Synthesis and microbiological activity of 5 (or 6)-methyl-2- substituted benzoxazole and benzimidazole derivatives. Arzneim Forsch/Drug Res. 47:1393–1397.
- Oren I, Temiz O, Yalcin I, Sener E, Altanlar N. 1998. Synthesis and antimicrobial activity of some novel 2,5- and/or 6-substituted benzoxazole and benzimidazole derivatives. Eur J Pharm Sci. 7:153–160.
- Owa T, Yoshino H, Okauchi T, Yoshimatsu K, Ozawa Y, Sugi NH, et al. 1999. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J Med Chem. 42:3789–3799.
- Padalkar VS, Borse BN, Gupta VD, Phatangare KR, Patil VS, Umape PG, et al. 2012. Synthesis and antimicrobial activity of novel 2-substituted benzimidazole, benzoxazole and benzothiazole derivatives. Arab J Chem. doi:10.1016/j.arabjc.2011.12.006.
- Pinar A, Yurdakul P, Yildiz-Oren I, Temiz-Arpaci O, Acan NL, AkiSener E, Yalcin I. 2004. Some fused heterocyclic compounds as eukaryotic topoisomerase II inhibitors. Biochem Biophys Res Commun. 317:670–674.
- Plemper RK, Erlandson KJ, Lakdawala AS, Prussia A, Boonsombat J, Aki-Sener E, et al. 2004. A target site for template-based design of measles virus entry inhibitors. Proc Natl Acad Sci USA. 101:5628–5633.
- Rida SM, Ashour FA, El-Hawash S, El-Semary M, Badr MH, Shalaby MA. 2005. Synthesis of some novel benzoxazole derivatives as anticancer, anti-HIV-1 and antimicrobial agents. Eur J Med Chem. 40:949–959.
- Sato S, Kajiura T, Noguchi M, Takehana K, Kobayashi T, Tsuji T. 2001. AJI9561, a new cytotoxic benzoxazole derivative produced by Streptomyces sp. J Antibiot. 54:102–104.
- Satyendra RV, Vishnumurthy KA, Vagdevi HM, Rajesh KP, Manjunatha H, Shruthi A. 2011. Synthesis, in vitro antioxidant, anthelmintic and molecular docking studies of novel dichloro substituted benzoxazole-triazolo-thione derivatives. Eur J Med Chem. 46:3078–3084.
- Schisselbauer JC, Silber R, Papadopoulos E, Abrains K, LaCreta FP, Tew KD. 1990. Characterization of glutathione S-transferase expression in lymphocytes from chronic lymphocytic leukemia patients. Cancer Res. 50:3562–3568.
- Seenaiah D, Ramachandra Reddy P, Mallikarjuna Reddy G, Padmaja A, Padmavathi V, Siva Krishna N. 2014. Synthesis, antimicrobial and cytotoxic activities of pyrimidinyl benzoxazole, benzothiazole and benzimidazole. Eur J Med Chem. 77:1–7.
- Shi DF, Bradshaw TD, Wrigley S, McCall CJ, Lelieveld P, Fichtner I, et al. 1996. Antitumor benzothiazoles. 3. Synthesis of 2-(4-aminophenyl)benzothiazoles and evaluation of their activities against breast cancer cell lines in vitro and in vivo. J Med Chem. 39:3375–3384.
- Tsuchida S, Sato K. 1992. Glutathione transferases and cancer. Crit Rev Biochem Mol Biol. 27:337–384.
- Varga A, Aki-Sener E, Yalcin I, Temiz-Arpaci O, Tekiner-Gulbas B, Cherepnev G, Molnar J. 2005. Induction of apoptosis and necrosis by resistance modifiers benzazoles and benzoxazines on tumour cell line mouse lymphoma L5718 Mdr + cells. In Vivo. 19:1087–1092.
- Waxman DJ. 1990. Glutathione S-transferases: role in alkylating agent resistance and possible target for modulation chemotherapy—a review. Cancer Res. 50:6449–6454.
- Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YS, et al. 2011. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 333:459–462.
- Wu G, Robertson DH, Brooks CL III, Vieth M. 2003. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J Comput Chem. 24:1549–1562.
- Xiang P, Zhou T, Wang L, Sun C, Hu J, Zhao Y, Yang L. 2012. Novel benzothiazole, benzimidazole and benzoxazole derivatives as potential antitumor agents: synthesis and preliminary in vitro biological evaluation. Molecules. 17:873–883.
- Yildiz-Oren I, Tekiner-Gulbas B, Yalcin I, Temiz-Arpaci O, Aki-Sener E, Altanlar N. 2004b. Synthesis and antimicrobial activity of new 2-[p-substituted-benzyl]-5-[substituted-carbonylamino]benzoxazoles. Arch Pharm Med Chem. 337:402–441.
- Yildiz-Oren I, Yalcin I, Aki-Sener E, Ucarturk N. 2004a. Synthesis and structure–activity relationships of new antimicrobial active multisubstituted benzazole derivatives. Eur J Med Chem. 39:291–298.