
Abstract
In several ocular diseases, the vascular endothelial growth factor (VEGF) level has been found to be upregulated. Bevacizumab, an anti-VEGF drug, is the most commonly used off level drug for these conditions. Delivery of drug to the posterior site is desired for the effective management of these diseases. The present study was to develop and optimize the chitosan (CS)-coated poly(lactide-co-glycolic acid) (PLGA) nanoparticles (NPs) of bevacizumab for sustained and effective delivery to posterior ocular tissues. NPs were prepared by double emulsion solvent evaporation method and optimized for various variables (i.e., CS concentration, PLGA content, polyvinyl alcohol (PVA) concentration, and sonication time) by employing a 4-factor 3-level Box–Behnken statistical design. NPs were characterized for particle size, polydispersity index (PDI), entrapment efficiency (EE), and in vitro release. Transscleral flux was determined through goat sclera, and ocular tolerance assay was done by Hen’s Egg Test chorioallantoic membrane method. The particle size and PDI of the optimized NPs were 222.28 ± 7.45 nm and 0.19 ± 0.08, respectively. The developed NPs showed an EE of 69.26 ± 1.31% with an extended release profile. The flux was significantly higher that is, 0.3204 ± 0.026 μg/cm2/h for the NPs compared to drug solution. Thus, CS-coated PLGA NPs can be potentially useful as ocular drug carriers to target retina.
Introduction
The major ocular diseases that significantly impacted vision of many people worldwide include age-related macular degeneration (AMD), diabetic retinopathy, cataract, uveitis, keratitis, and glaucoma (Pascolini and Mariotti Citation2012). Diabetic retinopathy (DR) is the most common complication of diabetes which can lead to sight threatening ocular problems. It was suggested that one-third of the diabetic patients have signs of retinopathy (Cheung et al. Citation2010, Lamoureux and Wong Citation2011).
Anti-vascular endothelial growth factor (VEGF) therapies bind to the VEGF signaling peptide with high affinity to neutralize VEGFs downstream effect of promoting the growth of leaky immature vessels (Ferrara et al. Citation2003). Anti-VEGF treatments are currently Food and drug Administration (FDA) approved for neovascular AMD but widely used off label for DR (Simó et al. Citation2014) and other neovascularization ocular diseases (Oh et al. Citation2009, Kim et al. Citation2010). The most common delivery approach to treat posterior ocular diseases has been the direct injections into the vitreal cavity (del Amo and Urtti Citation2008).
VEGF-A is predominant in human eye and plays a key pathogenic role in the inflammation, angiogenesis, and permeability changes in diabetic eye. There are four active isomers of VEGF-A consisting of 121, 165, 189, and 206 amino acids (Ferrara et al. Citation2003). Bevacizumab (Avastin, Genentech/Roche) is a full-length recombinant humanized monoclonal antibody that binds to all VEGF isomers (van Wijngaarden et al. Citation2005). A dose of 1.25 mg of intravitreal bevacizumab has a vitreous half-life of 4.32 days in rabbit pharmacokinetic model without any toxic effect (Bakri et al. Citation2007). Therefore, the treatment of diabetic retinopathy and other neovascular diseases required several intravitreal injections for maintaining the therapeutic concentration at target tissues (Fung et al. Citation2006). However, repeated intravitreal injection elevates complications such as exposure to infection, intravitreal hemorrhage, cataract, retinal detachment and opening of the sclerotomy site (Rich et al. Citation2006, Gambrelle et al. Citation2006).
Recently, many scientists are concentrating on subconjunctival (SC) injections and SC delivery systems as a potential and effective method to target posterior ocular tissues. Drug injected into SC space has direct access into intraocular tissue through transscleral route and fast clearance via conjunctival blood and lymphatic flow (Ranta and Urtti Citation2006, Amrite et al. Citation2008). Larger accessible surface area of sclera and a high degree of hydration make it permeable to hydrophilic substances (Srikantha et al. Citation2012). Scleral matrix is having proteoglycans which contributes to negative charge on the sclera and may bind to positively charged molecules (Ranta et al. Citation2010). By exploiting the permeability of the sclera, the SC route may offer potential alternative to enhance bioavailability and tissue targeting (Hosoya et al. Citation2005).
Many nanotechnology-based drug delivery carriers have been investigated for SC delivery of drug to target posterior tissue in sight-threatening diseases. Various ocular drug delivery strategies via transscleral route include liposomes (Moon et al. Citation2006), nanoparticles (NPs) (Shmueli et al. Citation2013), implants (Molokhia et al. Citation2010, Kawashima et al. Citation2011), hydrogel (Misra et al. Citation2009, Yu et al. Citation2014, Xu et al. Citation2013), light-activated systems (Tyagi et al. Citation2013), ultrasound-triggered systems (Huang et al. Citation2014), etc., have been investigated. Intravitreal injection of bevacizumab chitosan (CS) NPs was evaluated and shown effective inhibition of VEGF expression on retina of diabetic rats and longer duration of action compared to bevacizumab alone without any toxic effect (Lu et al. Citation2014).
Recently, mucoadhesive ocular drug delivery has been investigated for topical as well as by periocular route (Lu et al. Citation2014, Kesavan et al. Citation2012, de la Fuente et al. Citation2010). CS, being hydrophilic polysaccharide with cationic properties, is widely studied for ophthalmic use (Basaran and Yazan Citation2012). The strong binding with negatively charged cellular surface together with bioadhesion to the mucin layer of conjunctiva and corneal surface makes the CS most applicable for improving bioavailability of drug to surface as well as posterior site (Kaur and Smitha Citation2002). Antibacterial activity of CS also helped to inhibit or lessen the chances of post-infection after ocular administration (Felt et al. Citation2000).
In the present study, CS-coated poly(lactide-co-glycolic acid) (PLGA) NPs were developed by double emulsion solvent evaporation technique and further optimization was done using Design Expert software. NPs were characterized for particle size, shape, zeta potential, and in vitro drug release. Permeation through sclera and in vitro toxicity assay were performed to investigate the suitability for SC delivery of the developed formulation.
Materials and methods
Materials
PLGA (PURASORB® PDLG; DL-lactide/Glycolide copolymer, ratio M/M%: 50/50; 33 kDa) was generous gift from Purac Biochem BV (Gorinchem, the Netherlands). CS (MW: 120,000, with a deacetylation degree >80%) was procured from India Sea Foods (Kochi, Kerala, India). Polyvinyl alcohol (PVA; MW 95,000) was purchased from Sigma-Aldrich (St. Louis, MO). Bevacizumab (Avastin®) was obtained on special order from a medical shop (Help Line Pharmacy, Delhi, India). Enzyme-linked immunosorbent assay (ELISA) kit for bevacizumab assay was purchased from Qayee Bio-Technology Co., Ltd, Shanghai, China. Milli-Q water collected from water purification system (Millipore, Billerica, MA) in the laboratory was used in this experiment. All other chemicals and reagents used were procured from Merck Ltd. (Mumbai, India).
Preparation of CS-coated PLGA NPs
Placebo CS-coated poly(d,l-lactide-co-glycolide) NPs were prepared by one-step oil in water emulsion, solvent evaporation method with some modification (Guo and Gemeinhart Citation2008). In brief, PLGA polymer was dissolved in 2 mL of dichloromethane (DCM). This organic solution containing polymer was dropped into aqueous solution of CS and PVA prepared in 1% acetic acid (pH 5) kept under stirring at 600 rpm using magnetic stirrer (REMI, Mumbai, India). The formed emulsion was stirred continuously for 6 h at room temperature to evaporate DCM. NPs were collected by centrifugation at 18,000 rpm (REMI Cooling Centrifuge, Mumbai, India), washed 3 times with Milli-Q water, and finally resuspended in 2 mL of Milli-Q water and dried in a lyophilizer (Heto DRYWINNER, Germany) using mannitol (1% w/v) as cryoprotectant.
Bevacizumab (Avastin) loaded CS-coated PLGA NPs was prepared by double emulsion solvent evaporation method described by Varshochian et al. (Citation2013), with some modification (Varshochian et al. Citation2013). Briefly, 100 μL of 1% w/v bevacizumab solution (equivalent to 1 mg) in phosphate buffer saline (PBS, pH 7.4) was emulsified in 2 mL of organic solution containing appropriate amount of PLGA by sonication using probe sonicator (Hielscher Ultrasonics, Berlin, Germany) at 60 W in an ice bath. The formed primary emulsion was dropped in aqueous solution bearing CS and PVA and sonicated for 5–15 min to form w/o/w emulsion. CS and PVA concentrations vary from 0.25% w/v to 0.75% w/v and 0.50% w/v to 1.0% w/v, respectively. Temperature was maintained throughout the procedure using an ice bath. The developed w/o/w emulsion was stirred for 6 h at 1000 rpm at room temperature to allow evaporation of organic solvent (DCM) and hardening of the NPs. Finally, NPs were collected and lyophilized as placebo.
Experimental design studies
Design of experiment (DoE) approach is very useful for conducting the experiments in a way that the maximum information can be gathered from the data of minimum number of experimental runs as well as considering the interactions between the independent variables or factors (Myers and Montgomery Citation2002). Box–Behnken statistical design was selected in this study due to its significant less number of designed experiments in comparison to other techniques. First, placebo formulation was optimized to save the drug.
The most critical factors affecting drug penetration through the sclera are particle size and polydispersity index (PDI) of the nanocarrier. Therefore, four quantitative variables including CS concentration (A) and PVA concentration in the aqueous phase (B), PLGA concentration in organic phase (C), and sonication time (D) were defined (). Particle size (Y1) and PDI (Y2) were selected as dependent variables (responses). The variable ranges were chosen according to the screening results. Response surface quadratic model was obtained using Design Expert 8.0.7.1 software (Stat-Ease Inc., Minneapolis, MN). From the software, 27 experiment runs were designed and formulations were prepared in triplicate (). A second-order polynomial function was employed to develop mathematical relationship of the responses Y1 and Y2 and the independent variables (A, B, C, and D) as given:
(1)
where Y is associated with the predicted response(s); β0 is the intercept; and β1,β2, β3, and β4 are linear coefficients. β11, β22, β33, and β44 represent squared coefficients and β12, β13, β14, β23, β24, and β34 are the interaction coefficients of the equation. The best fit model obtained by the Design-Expert was evaluated on the basis of analysis of variance (ANOVA). The statistical significance of the data was performed in terms of regression coefficients.
Table 1. Levels of independent and dependent variables used in experiments.
Table 2. Observed values of responses for Box–Behnken design.
The three-dimensional (3D) response surface plots were used to show graphically the relationship and interaction between the coded variables and the response. The optimum concentrations of independent variables based on the responses constrained in their minimum levels were selected. Furthermore, five formulations were selected by point prediction and the experimental responses were compared with the predicted values to validate the chosen experimental domain and polynomial equation (). All the selected formulations were prepared and evaluated for the entrapment efficiency (EE), drug loading, and in-vitro drug release kinetics.
Table 3. Formulation selected by point prediction for optimization.
Characterization of NPs
Size, shape, and morphology
The particle size, PDI, and zeta potential of NPs were determined using photon correlation spectroscopy (Zeta sizer, HAS 3000; Malvern Instruments, Malvern, UK). The analysis was performed at 25 °C with an angle of detection 90°. The surface morphology of bevacizumab-loaded CS-coated PLGA NP was determined using transmission electron microscopy (TEM) (Philips CM 10, Holland). Few microliters of samples of the NPs suspension were dropped on a carbon-coated film 300 mesh copper grid and allowed to set for 10 min until air-dried. After complete drying, the sample was stained using 2% w/v phosphotungstic acid solution and excess was removed with filter paper. Digital Micrograph and Soft Imaging Viewer software were used to perform the image capture and analysis.
Entrapment and loading efficiency
To determine the amount of bevacizumab entrapped in NPs, an indirect method was used as reported in earlier work (Gan and Wang Citation2007). Before lyophilization, the NP suspension was centrifuged at 18,000 rpm at 4 °C for 15 min using cooling centrifuge (C24, REMI Cooling Centrifuge, Mumbai, India). The clear supernatant was collected and analyzed for bevacizumab by ELISA.
The EE was calculated from the following equation:
(2)
Drug loading efficiency was determined by weighing the lyophilized NPs and was calculated using the equation:
(3)
In vitro release and permeation study
The in vitro release study was performed by dispersing a determined amount of CS-PLGA NP (equivalent to 1.25 mg of bevacizumab) in 10 mL PBS (containing 0.1% sodium azide and 0.02% thiomersal) and were kept in water bath shaker (Nirmal International, New Delhi, India) at 37° ± 1 °C under shaking at 150 rpm (Xu et al. Citation2013). At specific time intervals, 1 mL sample was taken from the glass vial, centrifuged at 8000 rpm for 5 min at 4 °C using cooling centrifuge (C24, REMI Cooling Centrifuge, Mumbai, India). The supernatant was estimated for bevacizumab concentration by ELISA test (Miyake et al. Citation2010). Each withdrawn samples were replaced with an equal amount of fresh PBS to maintain sink condition throughout the experiment. Cumulative % release of drug was plotted against time to study the release profile.
In vitro transscleral permeation studies of CS-coated NPs suspension and drug solution were carried out on goat sclera through modified Franz diffusion cell (SFDC 6, LOGAN Instruments, NJ) (Ranta et al. Citation2010, Wen et al. Citation2013). Whole eyeballs of goat were procured from slaughter’s house and kept in normal saline, maintained at 4 ± 2 °C. The sclera was carefully prepared by removing the adhering tissues and washed with cold PBS and kept in cold freshly prepared PBS of pH 7.4 before use. The excised sclera with an effective diffusion area of 0.80 cm2 was fixed between clamped donor and receptor compartments of the Franz diffusion cell in such a way that its episcleral surface faced the donor compartment. The drug solution/CS-coated NPs suspension (0.1 mL) equivalent to about 1 mg of bevacizumab was placed in donor compartment. The receiver compartment was filled with freshly prepared PBS (pH 7.4, containing 0.1% sodium azide and 0.02% thiomersal). The whole system was maintained at 37 ± 0.5 °C. At predetermined time intervals, 1 mL sample was withdrawn from the receptor compartment and an equal quantity of fresh PBS was added to the receptor compartment to maintain sink conditions throughout the experiment. The amount of bevacizumab in samples was estimated by ELISA. All experiments were conducted under light-shielding condition and the samples were stored at 4 °C before quantification.
Ex vivo mucoadhesion study
The mucoadhesive strength of CS-coated NPs was determined by incubating it with pig mucin suspension (Ameeduzzafar et al. Citation2014). A suspension of 1% pig mucin (Himedia, Mumbai, India) was prepared in PBS(0.05 M, pH 7.4) and both placebo and bevacizumab-loaded CS-coated PLGA NPs were mixed and incubated with 1 mL of mucin suspension at 37 °C for 24 h. The samples were centrifuged at 20,000 rpm for 30 min (REMI Cooling Centrifuge, Mumbai, India) and supernatant was evaluated for free pig mucin by UV spectrophotometer (Shimadzu UV-1700, Kyoto, Japan) at 251 nm. The mucoadhesion strength was calculated by the following equation:
where Ci = initial concentration of pig mucin; Cf = concentration of free pig mucin after incubation.
Toxicity to ocular tissue
Hen’s Egg Test Chorioallantoic Membrane (HET-CAM) test is an alternative method to check the ocular irritancy of any product. It is used as an alternative to the Draize Rabbit Eye Test (ICCVAM Citation2010). Briefly fertilized fresh hen’s eggs weighing between 50 and 60 g were selected and divided into three groups each containing three eggs. All the eggs were incubated in a humidified incubator at a temperature of 37 ± 0.5 °C and 55 ± 7% RH for 9 days. Eggs were rotated manually in gentle manner after every 12 h. On the 10th day, the egg shell was opened at the side of the air chamber and the inner membrane was removed without any damage to the vascular CAM. Careful formulations (0.5 mL) were instilled directly onto the CAM surface and left in contact for 5 min. NaOH (0.1 M) and normal saline (0.9%) were used as positive and negative controls, respectively. The membrane was assessed for any sign of vascular damage as hyperemia, hemorrhage, and coagulation and scored (0 to 21) according to the scoring scheme. The overall irritation score was calculated as the mean sum of individual scores of all the end points (0, 0.5, 2, and 5 min) from three replicate assays (Tavaszi and Budai Citation2006). Final image was taken to compare the results of the finding.
Statistical analysis
All the results were presented as mean ± SD, and the data were analyzed statistically using one-way ANOVA using Tukey–Kramer multiple comparison test. Differences with P < .05 were considered to be significant.
Results
Experimental design studies
The CS-coated NPs were optimized by response surface methodology using Design-Expert software. Totally, 29 runs with five center points were prepared and their responses are shown in . Best model for each response was chosen by the ANOVA by calculating F values. A full-quadratic second-order polynomial equation was found as best fitted model for both particle size and PDI. The regression analysis for responses R1, R2, and ANOVA of each model were shown in and , respectively. 3D plots showed the interaction effects of the independent variables on the responses as well as their usefulness in studying the effects of two factors on one response at a time ().
Figure 1. (A)–(D) Three-dimensional (3D) response surface plots showing relative effects of different process parameters on the particle size and PDI.
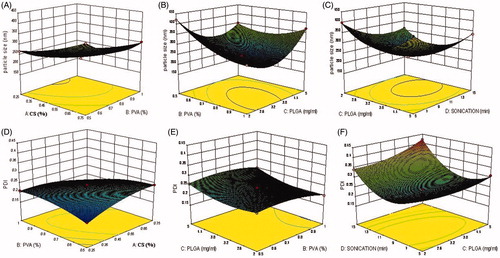
Table 4. Summary of results of regression analysis for responses R1 and R2
Table 5. Analysis of variance of calculated model for responses.
The quadratic polynomial equation for particle size (R1) is given as follows:
Particle size (R1) = +233.67 + 46.50* A − 31.33* B + 4.42* C − 32.42* D − 2.75* AB −11.75*AC +4.50*AD +42.25*BC −15.00*BD + 7.75*CD + 13.67*A2 + 39.92*B2 + 80.54*C2 + 45.04*D2
The “Model F value” of 245.87 implies the model is significant. Values of “Prob > F” less than 0.0500 indicate that model terms are significant. In this case, A, B, C, D, AC, BC, BD, CD, A2, B2, C2, and D2 are significant model terms. The “Lack of Fit F value” of 0.55 implies the Lack of Fit is not significant relative to the pure error. The “Pred. R2” of 0.9833 is in reasonable agreement with the “Adj. R2” of 0.9925, that is, the difference is less than 0.2. “Adeq Precision” measures the signal-to-noise ratio. A ratio greater than 4 is desirable. A ratio of 53.598 from the model indicates an adequate signal. This model can be used to navigate the design space. The polynomial equation of response R1 shows the combined effect of CS concentration, PVA concentration, PLGA concentration, and sonication time on particle size.
The quadratic polynomial equation for PDI (R2) is given as follows:
PDI (R2) = +0.23 + 6.667E-003*A − 9.167E-003*B + 0.019*C + 0.038*D − 0.030*AB − 0.012*AC + 0.022*AD + 5.00E-003*BC − 0.012*BD + 0.020*CD − 0.020*A2 − 0.019*B2 + 0.023*C2 + 0.087*D2
The “Model F value” of 269.00 implies the model is significant. Values of “Prob > F” less than 0.0500 indicate that model terms are significant. In this case, A, B, C, D, AB, AC, AD, BD, CD, A2, B2, C2, and D2 are significant model terms. The “Lack of Fit F value” of 0.72 implies the Lack of Fit is not significant. The “Pred. R2” of 0.9841 is in reasonable agreement with the “Adj. R2” of 0.9931, that is, the difference is less than 0.2. “Adeq Precision” measures the signal-to-noise ratio. A ratio greater than 4 is desirable. Our ratio of 68.816 indicates an adequate signal. This model can be used to navigate the design space.
The effect of variables on colloidal characteristic of the NP, that is, particle size and PDI is crucial in determining the penetrating efficiency of NP across the barriers. Hence, the aim of our study was to optimize the influence of formulation variables to obtain the NPs of desired size (within 250 nm) for their ocular application. Furthermore, they optimized for the pay load and release characteristic of the drug from the coated NPs to target the system.
Effect of CS
The CS concentration was varying from 0.25% to 0.75% w/v. The mean particle size varies from 210 ± 23.41 nm to 420 ± 38.67 nm as shown in . As the concentration of CS increased from 0.25 to 0.75% w/v, a gradual increase in the particle size was observed. A significant increase in particle size was also seen when CS concentration was increased from 0.25% to 0.50% w/v (P < .05) (). PDI increases with the increase in the CS concentration as shown in .
Effect of PVA on particle size and PDI
PVA acts as stabilizer in the formulation and in general as its concentration increases or decreases in size was observed which is statistically significant (P ≤ .05). As the concentration of PVA was increased from 0.5 to 0.75% w/v, there was less reduction in mean particle size as compared to that observed at increment of PVA from 0.75 to 1.0% w/v (). This result is in agreement with earlier work reported by Nafee et al. (Citation2007) and Mainardes and Evangelista (Citation2005). PVA acts as stabilizer and as the PVA concentration increases higher, the PDI goes on decreasing ().
Effect of PLGA content
With the rise in polymer concentration in organic phase (PLGA) from 2 mg/mL to 5 mg/mL, the NPs become larger in size as reported in earlier work (Lamprecht et al. Citation2000). While very less effect was found on PDI which is non-significant (P > .05) ()).
Effect of sonication time
To get the desired size and PDI of formulation, all were sonicated for the time duration of 5–15 min after formation of the double emulsion. Furthermore, the formulation was kept on magnetic stirrer for 3 h to reduce the agglomeration. Sonication was performed at 80 W and it was observed as the duration of sonication increases from 5 to 10 min a significant decrease in the particle size was obtained while further sonication (after 10 min) results in slight increase in the mean size and PDI. The sonication up to 10 min gives favorable results in every composition of the formulations. From the Box–Behnken design, the optimum composition was obtained which was further optimized by point prediction and five formulations were developed and evaluated for EE, drug loading, and release kinetics ().
The EE and drug loading were found in between 50–70% and 4–7, respectively. There is a significant difference in all the formulations (P < .05) ANOVA suggested the data is significant P values is .0257. More than 60% EE bevacizumab suggest good entrapment in NP showed optimized process parameters.
Characterization
Size and morphology
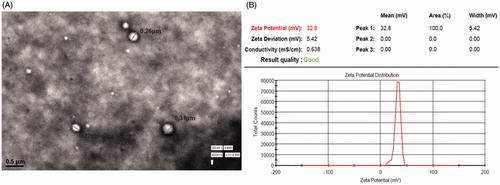
The TEM image of coated CS-PLGA NPs is shown in . It shows spherical-shaped particles with well-defined core shell structure. The CS-coated NPs have detected with positive zeta potential as expected due to the positive charge of the CS polymer. Zeta potential of the optimized formulation is +32.8 which shows coating of the PLGA NPs ().
Figure 2. (A) TEM image of chitosan-coated PLGA nanoparticles. (B) Zeta potential of the optimized formulation.
In vitro release and permeation study
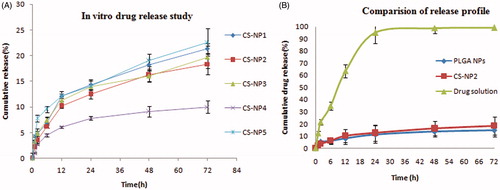
In vitro release of bevacizumab from various CS-coated PLGA NPs is illustrated in . The drug release from CS-PLGA NPs reduces the burst release of drug as can be seen in uncoated PLGA particles in (Pandit et al. Citation2014). A very slow and consistent drug release was obtained in case coated NPs. They could not achieve complete release till 72 h (maximum 25%). While in case of drug solution, more than 90% of the drug was released within 24 h which could not able to give longer effect of drug ().
Figure 3. (A) In vitro release study of different NPs. (B) Comparison of drug release profile of CS-coated NPs, PLGA NPs, and drug solution.
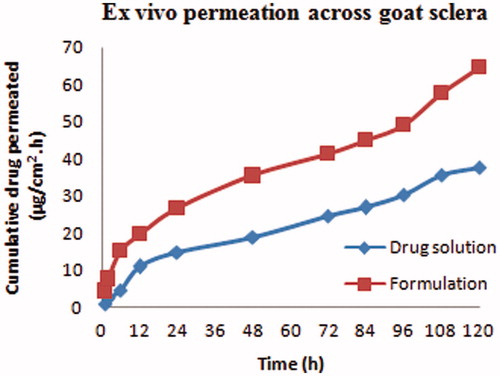
The scleral permeation was assessed for the optimized bevacizumab-loaded CS-PLGA NPs and drug solution using diffusion cell. The flux of bevacizumab from the optimized NPs was significantly (P < .01) increased (0.3204 ± 0.026 μg/cm2/h) compared to drug solution (0.2510 ± 0.015 μg/cm2/h) as calculated from the steady-state portion of the graph plotted between cumulative amount permeated per unit area verses time ().
Figure 4. Ex vivo transscleral permeation of NPs and drug solution.
Mucoadhesion study
The mucoadhesive strength of placebo CS-coated PLGA NPs was found 92.67 ± 3.12% and 89.21 ± 1.22% for bevacizumab-loaded NPs. It is due to hydrogen bond formation between positive charge amino group of CS and oligosaccharide chains of mucin.
HET-CAM
A mean score for CS-PLGA NPs, normal saline (negative control), and 0.1 M NaOH (positive control) were found to be 0.53, 0, and 12, respectively, up to 5 min (). The respective figure after incubation was shown in . clearly shows very less vascular damage compared to the positive control and the aggregated score is less than 1 for the formulation so it was non-irritant as well as well tolerated for ophthalmic use.
Figure 5. HET-CAM test image of formulation treated (A), negative control (B), and positive control (C).
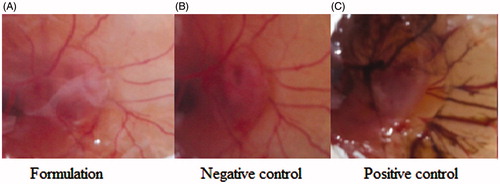
Table 6. Scores obtained in HET-CAM test.
Discussion
PLGA has been widely reported for ocular delivery of macromolecules such as DNA, siRNA, and proteins (Wang et al. Citation2013, de la Fuente et al. Citation2008). Mostly, single and double emulsion methods are used for encapsulation which gives low entrapment of the drug and a large amount of polymer (PLGA) can be exposed to organic solvents and shear stress which can lead to denaturation of the biopolymers (Mainardes and Evangelista Citation2005). Hence, surface coating of PLGA NPs can be a solution to avoid the direct exposure of biopolymer to/in the harsh conditions during the preparation stage. CS as mucoadhesive polymer has well utilized for topical use as in situ gelling agent (Katiyar et al. Citation2014), as particulate delivery system (Badawi et al. Citation2008). Jain et al. (Citation2011) have reported improved transcorneal permeation of CS-coated PLGA using rhodamine as model drug (Jain et al. Citation2011). Here, CS was selected as coating material (polymer) for PLGA NPs for SC delivery. The transscleral permeation of the drug mainly depends on the permeation across the physical barriers: sclera, choroid, and retinal pigment epithelium (RPE) (Ranta et al. Citation2010). Being a less invasive route compared to intravitreal administration, SC injection provides higher drug bioavailability (about 0.1%) in retina and vitreous compared to the topical administration (about <0.001%, less than 5% of the administered dose) (Kim et al. Citation2004).
The posterior ocular tissues drug availability by SC administration takes place primarily via drug permeation through the sclera. Sclera is proved to be permeable to large hydrophilic compounds such as protein (Watson and Young Citation2004). Sclera is a negatively charged membrane in neutral pH so being a mucoadhesive polymer (CS) coating on PLGA NPs might be expected to interact with negatively charged barrier (sclera) and hence inducing transscleral uptake and prolong the residence time on the scleral surface. This improves the drug bioavailability to retinal and vitreal tissues (Ranta et al. Citation2010).
The mean particle size diameter of CS-coated PLGA NPs was found to increase gradually with CS concentration. This increment in the particle size could be attributed to the increased viscosity of the aqueous phase (CS-PVA solution) which lowers the shear stress on organic phase (PLGA) during ultrasonication and results into larger emulsion droplets (Gan et al. Citation2005).
Another suggested reason might be the process of PLGA particle coating which is reported as solely an interfacial phenomenon so the increased particle size was due to the increased amount of adsorbed CS on the surface of PLGA NPs (Gan et al. Citation2005). Another important variable is PVA concentration. The PLGA NPs were stabilized by the layers of PVA which results from a stable network of PVA on the polymer surface and results in a slight negative zeta potential on NPs. The mechanism of PVA binding to PLGA has been suggested to be interpenetration of PVA and PLGA molecules during NPs formation. This binding takes place when the organic solvent was removed from the interface.
In case of CS-PLGA NPs which is characterized by a strong positive zeta potential and are mainly stabilized by electrostatic repulsion. CS is a weak base with β (1,4) linked monomers of D-glucosamine and N-acetyl-D-glucosamine. In acidic medium (pH 4–5), the amine group will be positively charged and results in a high surface charge to the NP (Gan et al. Citation2005).
The amount of surfactant (PVA) plays a major role in stabilization of emulsified droplets during the NPs formation. Increasing the amount of PVA in the aqueous phase caused a gradual increase in the viscosity resulted in the formation of smaller droplets which leads to small-sized NPs. However, the decrease in the particle size with PVA concentration is not linear.
PLGA concentration also plays an important role in deciding the size of NPs. There was a significant increase in the particle size of NPs with PLGA content in the organic solution. This attributed to the increased viscosity of the organic solution with PLGA concentration resulting in a poor dispersibility of the PLGA solution into the aqueous phase. Furthermore, the molecules are forming larger particle by coalesce in a concentrated solution. Thus, viscosity of the aqueous phase and organic phase has great influence on the particle size of the NPs.
Polydispersity was found to less due to the ultrasonication process. The sonication time has an effect on size and PDI of the particles. Increasing the sonication time significantly decreases the size up to 10 min while both increasing and decreasing the sonication time caused aggregation and thus increased in resultant particles size. This was in agreement with the results reported in previous investigations (Varshochian et al. Citation2013, Son et al. Citation2009). In case of PDI, as the sonication time increases from 5 to 10 min at 80 W, the value of PDI minimized and further sonication cause increase in the PDI or broad particle size distribution. The increased PDI after 10 min is due to high shear stress on the emulsion droplet which leads to agglomerate and results into broad size distribution.
In general, drug releases from any biodegradable polymeric particles follows several mechanisms such as desorption from the particles surface, degradation and erosion of the polymer, diffusion, and readsorption through the pores of the polymer network (Jain et al. Citation2010).
The coated NPs showed initial release (3–4% in 1 h) followed by slow and consistent release over a period of 72 h (). The initial burst release of drug which was found in case of PLGA NPs was slow down in CS-coated NPs. This can be attributed to the additional layer of CS present on the NPs surface which is acting as an additional barrier to drug diffusion (Durán-Lobato et al. Citation2014). This release is in agreement with the earlier published results (Yoncheva et al. Citation2009).
The permeation of drug was performed across the sclera through CS-coated NPs and drug solution. The CS-coated particles showed more permeation of bevacizumab as compared to the bevacizumab solution (). This could be illustrated due to permeation enhancing activity of CS as well as the nano size of coated NPs. The interaction of a positively charged amino group of CS with negatively charged scleral surface could be another reason for the enhanced permeation.
The binding with the mucin is the ability of CS presence on the surface of NP. This binding results from the electrostatic interaction of the negatively charged mucin to the NPs. The addition of mucin to the NPs caused a reduction in mucin concentration. This reduction was more pronounced when MilliQ water was used as incubation medium than in PBS (pH 7.4). This reflects the ionic strength of the medium which affect the surface charge and hence the binding efficiency of the mucin to the NPs. The pH of the incubation medium plays an important role in the mucin binding to NPs. The electrostatic forces involved in binding process are relatively weak at pH 7.4 due to reduced zeta potential at the surface of NPs in PBS as compared to water. Thus, lesser mucin molecule can be attracted to the surface of NP at this pH.
Irritancy potential of all ophthalmic formulation is very important before it is subjected to human use. The developed CS-coated NPs were tested on hen’s egg chorioallantoic membrane and result showed that it was non-irritant and tolerated to chorioallantoic membrane after incubation period.
The effect was found similar to the control group ().
Conclusion
CS-coated PLGA NPs were prepared by double emulsification solvent evaporation method and were optimized using 4-factor, 3-level Box–Behnken statistical design. CS concentration, PLGA content, PVA concentration, and sonication time were considered as independent variables. The optimized formulation was selected based on point prediction tool of design software which was having particle size 222.28 ± 7.45 nm with PDI of 0.19 ± 0.08. The optimized formulation showed drug loading capacity of 6.19 ± 1.07% and EE of 67.28 ± 5.18%. The in vitro drug release profile of optimized CS-coated NPs was found to be sustained and controlled over 72 h. CS-coated NPs showed better permeability of the bevacizumab across the sclera as compared to the drug solution. The coated particles were retained on the surface of sclera and hence enhance drug residence time and permeation. The drug was therefore available for longer duration and hence reduces the frequency of administration. These results provide evidence that CS-PLGA NPs are well suited as carrier for anti-VEGF (Bevacizumab) to target retina via SC route. Further studies concerning the cytotoxicity and concentration dependent reduction of the VEGF concentration in diabetic retinopathy model are underway.
Acknowledgements
The author is grateful to Council of Scientific & Industrial Research (CSIR), New Delhi, India (CSIR-SRF, sanction no. 09/591(0137)/2013-EMR-I)) for financial assistance. She is thankful to Purac Biochem BV (Gorinchem, the Netherlands) for sending PLGA (polymer) as gift samples. Authors also thankful to AIIMS, New Delhi for providing TEM facility.
Disclosure statement
The authors report no declarations of interest.
References
- Ameeduzzafar Ali J, Bhatnagar A, Kumar N, Ali A. 2014. Chitosan nanoparticles amplify the ocular hypotensive effect of cateolol in rabbits. Int J Biol Macromol. 65:479–491.
- Amrite AC, Edelhauser HF, Kompella UB. 2008. Modeling of corneal and retinal pharmacokinetics after periocular drug administration. Invest Ophthalmol Vis Sci. 49:320–322.
- Badawi AA, El-Laithy HM, El-Qidra RK, El-Mofty H. 2008. Chitosan based nanocarriers for indomethacin ocular delivery. Arch Pharm Res. 31:1040–1049.
- Bakri SJ, Snyder MR, Reid JM, Pulido JS, Singh RJ. 2007. Pharmacokinetics of intravitreal bevacizumab (Avastin). Ophthalmology. 114:855–859.
- Basaran E, Yazan Y. 2012. Ocular application of chitosan. Expert Opin Drug Deliv. 9:701–712.
- Cheung N, Mitchell P, Wong TY. 2010. Diabetic retinopathy. Lancet. 376:124–136.
- de la Fuente M, Raviña M, Paolicelli P, Sanchez A, Seijo B, Alonso MJ. 2010. Chitosan-based nanostructures: a delivery platform for ocular therapeutics. Adv Drug Deliv Rev. 62:100–117.
- de la Fuente M, Seijo B, Alonso MJ. 2008. Bioadhesive hyaluronan–chitosan nanoparticles can transport genes across the ocular mucosa and transfect ocular tissue. Gene Ther. 15:668–676.
- del Amo EM, Urtti A. 2008. Current and future ophthalmic drug delivery systems. A shift to the posterior segment. Drug Discov Today. 13:135–143.
- Durán-Lobato M, Muñoz-Rubio I, Holgado M, Álvarez-Fuentes J, Fernández-Arévalo M, Martín-Banderas L. 2014. Enhanced cellular uptake and biodistribution of a synthetic cannabinoid loaded in surface-modified poly(lactic-co-glycolic acid) nanoparticles. J Biomed Nanotech. 10:1068–1079.
- Felt O, Carrel A, Baehni P, Buri P, Gurny R. 2000. Chitosan as tear substitute: a wetting agent endowed with antimicrobial efficacy. J Ocul Pharmacol Ther. 16:261–270.
- Ferrara N, Gerber HP, LeCouter J. 2003. The biology of VEGF and its receptors. Nat Med. 9:669–676.
- Fung AE, Rosenfeld PJ, Reichel E. 2006. The International Intravitreal Bevacizumab Safety Survey: using the internet to assess drug safety worldwide. Br J Ophthalmol. 90:1344–1349.
- Gambrelle J, Kodjikian L, Abi-Ayad N, Wolff B, Grange JD. 2006. Grange, persistent unsealed sclerotomy after intravitreal injection of triamcinolone acetonide with a 30-gauge needle. J Fr Ophtalmol. 29:e22.
- Gan Q, Wang T, Cochrane C, McCarron P. 2005. Modulation of surface charge, particle size and morphological properties of chitosan-TPP nanoparticles intended for gene delivery. Colloids Surf B Biointerfaces. 44:65–73.
- Gan Q, Wang T. 2007. Chitosan nanoparticle as protein delivery carrier—systematic examination of fabrication conditions for efficient loading and release. Colloids Surf B Biointerfaces. 59:24–34.
- Guo C, Gemeinhart RA. 2008. Understanding the adsorption mechanism of chitosan onto poly(lactide-co-glycolide) particles. Eur J Pharm Biopharm. 70:597–604.
- Hosoya KI, Lee VH, Kim KJ. 2005. Roles of the conjunctiva in ocular drug delivery: a review of conjunctival transport mechanisms and their regulation. Eur J Pharm Biopharm. 60:227–240.
- Huang D, Wang L, Dong Y, Pan X, Li G, Wu C. 2014. A novel technology using transscleral ultrasound to deliver protein loaded nanoparticles. Eur J Pharm Biopharm. 88:104–115.
- ICCVAM. Appendix B3 of “ICCVAM test method evaluation report: current validation status of in vitro test methods proposed for identifying eye injury hazard potential of chemicals and products” NIH Publication, 2010. Available from: http://iccvam.niehs.nih.gov/methods/ocutox/MildMod-TMER.htm
- Jain GK, Pathan SA, Akhter S, Ahmad N, Jain N, Talegaonkar S, Khar RK, Ahmad FJ. 2010. Mechanistic study of hydrolytic erosion and drug release behaviour of PLGA nanoparticles: influence of chitosan. Polym Degrad Stab. 95:2360–2366.
- Jain GK, Pathan SA, Akhter S, Jayabalan N, Talegaonkar S, Khar RK, Ahmad FJ. 2011. Microscopic and spectroscopic evaluation of novel PLGA-chitosan nanoplexes as an ocular delivery system. Colloids Surf B Biointerfaces. 82:397–403.
- Katiyar S, Pandit J, Mondal RS, Mishra AK, Chuttani K, Aqil M, Ali A, Sultana Y. 2014. In situ gelling dorzolamide loaded chitosan nanoparticles for the treatment of glaucoma. Carbohydr Polym. 102:117–124.
- Kaur IP, Smitha R. 2002. Penetration enhancers and ocular bioadhesives: two new avenues for ophthalmic drug delivery. Drug Dev Ind Pharm. 28:353–369.
- Kawashima T, Nagai N, Kaji H, Kumasaka N, Onami H, Ishikawa Y, et al. 2011. A scalable controlled-release device for transscleral drug delivery to the retina. Biomaterials. 32:1950–1956.
- Kesavan K, Kant S, Singh PN, Pandit JK. 2012. Mucoadhesive chitosan-coated cationic microemulsion of dexamethasone for ocular delivery: in vitro and in vivo evaluation. Current Eye Res. 38:342–352.
- Kim H, Robinson MR, Lizak MJ, Tansey G, Lutz RJ, Yuan P, Wang NS, Csaky KG. 2004. Controlled drug release from an ocular implant: an evaluation using dynamic three-dimensional magnetic resonance imaging. Invest Ophthalmol Vis Sci. 45:2722–2731.
- Kim WJ, Jeong HO, Chung SK. 2010. The effect of bevacizumab on corneal neovascularization in rabbits. Korean J Ophthalmol. 24:230–236.
- Lamoureux EL, Wong TY. 2011. Diabetic retinopathy in 2011: further insights from new epidemiological studies and clinical trials. Diabetes Care. 34:1066–1067.
- Lamprecht A, Ubrich N, Pérez MH, Lehr CM, Hoffman M, Maincent P. 2000. Influences of process parameters on nanoparticle preparation performed by a double emulsion pressure homogenization technique. Int J Pharm. 196:177–182.
- Lu Y, Zhou N, Huang X, Cheng JW, Li FQ, Wei RL, Cai JP. 2014. Effect of intravitreal injection of bevacizumab-chitosan nanoparticles on retina of diabetic rats. Int J Ophthalmol. 7:1–7.
- Mainardes RM, Evangelista RC. 2005. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm. 290:137–144.
- Misra GP, Singh RS, Aleman TS, Jacobson SG, Gardner TW, Lowe TL. 2009. Subconjunctivally implantable hydrogels with degradable and thermoresponsive properties for sustained release of insulin to the retina. Biomaterials. 30:6541–6547.
- Miyake T, Sawada O, Kakinoki M, Sawada T, Kawamura H, Ogasawara K, Ohji M. 2010. Pharmacokinetics of bevacizumab and its effect on vascular endothelial growth factor after intravitreal injection of bevacizumab in macaque eyes. Invest Ophthalmol Vis Sci. 51:1606–1608.
- Molokhia SA, Sant H, Simonis J, Bishop CJ, Burr RM, Gale BK, Ambati BK. 2010. The capsule drug device: novel approach for drug delivery to the eye. Vision Res. 50:680–685.
- Moon JW, Song YK, Jee JP, Kim CK, Choung HK, Hwang JM. 2006. Effect of subconjunctivally injected, liposome-bound, low-molecular-weight heparin on the absorption rate of subconjunctival hemorrhage in rabbits. Invest Ophthalmol Vis Sci. 47:3968–3974.
- Myers RH, Montgomery DC. Response Surface Methodology Process and Product Optimization Using Designed Experiments. 2nd ed. New York: John Wiley & Sons, Inc.; 2002.
- Nafee N, Taetz S, Schneider M, Schaefer UF, Lehr CM. 2007. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine. 3:173–183.
- Oh JY, Kim MK, Wee WR. 2009. Subconjunctival and intracorneal bevacizumab injection for corneal neovascularization in lipid keratopathy. Cornea. 28:1070–1073.
- Pandit J, Sultana Y, Aqil M. 2014. Poly(lactide-co-glycolide) nanoparticles for an extended delivery of bevacizumab to retina: formulation and in vitro characterization. Adv Sci Lett. 20:1588–1593.
- Pascolini D, Mariotti SP. 2012. Global estimates of visual impairment: 2010. Br J Ophthalmol. 96:614–618.
- Ranta VP, Mannermaa E, Lummepuro K, Subrizi A, Laukkanen A, Antopolsky M, et al. 2010. Barrier analysis of periocular drug delivery to the posterior segment. J Control Release. 148:42–48.
- Ranta VP, Urtti A. 2006. Transscleral drug delivery to the posterior eye: prospects of pharmacokinetic modeling. Adv Drug Deliv Rev. 58:1164–1181.
- Rich RM, Rosenfeld PJ, Puliafito CA, Dubovy SR, Davis JL, Flynn HW, Jr, et al. 2006. Short-term safety and efficacy of intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Retina. 26:495–511.
- Shmueli RB, Ohnaka M, Miki A, Pandey NB, de Silva RL, Koskimaki JE, et al. 2013. Long-term suppression of ocular neovascularization by intraocular injection of biodegradable polymeric particles containing a serpin-derived peptide. Biomaterials. 34:7544–7551.
- Simó R, Sundstrom JM, Antonetti DA. 2014. Ocular anti-VEGF therapy for diabetic retinopathy: the role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes Care. 37:893–899.
- Son S, Lee WR, Joung YK, Kwon MH, Kim YS, Park KD. 2009. Optimized stability retention of a monoclonal antibody in the PLGA nanoparticles. Int J Pharm. 368:178–185.
- Srikantha N, Mourad F, Suhling K, Elsaid N, Levitt J, Chung PH, Jackson TL. 2012. Influence of molecular shape, conformability, net surface charge, and tissue interaction on transscleral macromolecular diffusion. Exp. Eye Res. 102:85–92.
- Tavaszi J, Budai P. 2006. The use of HET-CAM test in detecting the ocular irritation. Commun Agric Appl Biol Sci. 72:137–141.
- Tyagi P, Barros M, Stansbury JW, Kompella UB. 2013. Light-activated, in situ forming gel for sustained suprachoroidal delivery of bevacizumab. Mol Pharm. 10:2858–2867.
- van Wijngaarden P, Coster DJ, Williams KA. 2005. Inhibitors of ocular neovascularization: promises and potential problems. JAMA. 293:1509–1513.
- Varshochian R, Jeddi-Tehrani M, Mahmoudi AR, Khoshayand MR, Atyabi F, Sabzevari A, Esfahani MR, Dinarvand R. 2013. The protective effect of albumin on bevacizumab activity and stability in PLGA nanoparticles intended for retinal and choroidal neovascularization treatments. Eur J Pharm Sci. 50:341–352.
- Wang Y, Li P, Kong L. 2013. Chitosan-modified PLGA nanoparticles with versatile surface for improved drug delivery. AAPS PharmSciTech. 14:585–592.
- Watson PG, Young RD. 2004. Scleral structure, organisation and disease. A review. Exp Eye Res. 78:609–623.
- Wen H, Hao J, Li SK. 2013. Characterization of human sclera barrier properties for transscleral delivery of bevacizumab and ranibizumab. J Pharm Sci. 102:892–903.
- Xu X, Weng Y, Xu L, Chen H. 2013. Sustained release of Avastin® from polysaccharides cross-linked hydrogels for ocular drug delivery. Int J Biol Macromol. 60:272–276.
- Yoncheva K, Vandervoort J, Ludwig A. 2009. Influence of chitosan layer on the properties of surface modified poly(lactide-co-glycolide) nanoparticles. J Disper Sci Technol. 30:213–216.
- Yu J, Xu X, Yao F, Luo Z, Jin L, Xie B, et al. 2014. In situ covalently cross-linked PEG hydrogel for ocular drug delivery applications. Int J Pharm. 470:151–157.