
Abstract
The objective of this work was to develop a simple, selective, and sensitive LC–MS/MS method for the quantitation of the mepivacaine in Chinese biological matrix. The calibration curve of mepivacaine ranged from 0.5 to 2000 ng/mL with the lower limit of quantitation being 0.5 ng/mL. This sensitivity was high enough to describe the profile of blood mepivacaine level versus time. Thereby it was very desirable for the pharmacokinetic study because of its high sensitivity and accuracy. The study used a single-dose two-period crossover design principle. For the pharmacokinetic analysis of plasma, the mean (SD) values obtained were as follows: t1/2, 1.63 (0.43) h; Cmax, 435.3 (67.4) ng/ml; AUC0-t, 1546.9 (339.7) ng/ml·h; AUC0-∞, 1982.3 (421.4) ng/ml·h; Tmax, 0.62 (0.31) h. The validated method has been successfully applied to assess the pharmacokinetic study of mepivacaine after a single administration to Chinese volunteers.
Introduction
Local anesthetics are drugs that can temporarily, completely and reversibly block nerve conduction. Local anesthesia is to use local anesthetic in certain areas of the body to result in sensory loss and prevent muscle activity by reversibly blocking nerve conduction. However, when local anesthetic is absorbed into the bloodstream or directly injected into the blood circulation, it can affect the central nervous system, cardiovascular system and other organ function. The extent and nature of the impact depend on the dose of local anesthetic into the blood circulation per unit of time (Eng et al. Citation2014). Excessive use of local anesthetic is likely to cause poisoning or, in severe cases, even death (Bailard et al. Citation2014). The local anesthetic amide drug, mepivacaine (pKa = 7.6), was a member of the pipecoloxylidide group (). Its mechanism is to suppress nerve membrane ion flow to stabilize nerve cell membranes and then prevent impulse generation and conduction (Su et al. Citation2014). This drug was extensively metabolized by the liver and excreted in the urine of animals and human (Baselt Citation2002).
Figure 1. The structure of mepivacaine (A) and lidocaine (B).
GC methods including tandem mass spectrometry detection and liquid chromatographic methods are mainly used for quantification of mepivacaine in biological fluids, either in humans or in animals (Baniceru et al. Citation2004; Hattori et al. Citation1991; Nieddu et al. Citation2007; Ohshima and Takayasu Citation1999; Watanabe et al. Citation1998). However, these methods do not meet modern drug development needs with respect to an efficient extraction procedure, shorter run time and high sensitivity. LC-MS/MS has become a method of the choice for the determination of small molecules in biological matrices because of its superior LLOQ (Lower Limit of quantitation), sensitivity, and improved selectivity. In current pharmacokinetic studies, LC–MS/MS is playing an important role for quantification of different kinds of drugs. Hence, it is necessary to develop a robust, sensitive, and simple LC–MS/MS method for the pharmacokinetic study of mepivacaine. By searching the PubMed database, it is found that pharmacokinetic studies on LC–MS/MS for the determination of mepivacaine in biological samples have not yet been reported.
Materials and methods
Chemicals and reagents
Mepivacaine (purity 93.7%) was supplied by Chenghui Shuangda Pharmaceutical Co. Ltd., (Jinan, Shandong, China). The mepivacaine standard (purity 99.1%) and internal standard (IS), lidocaine (pKa =7.9, purity 98.6%), were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Acetonitrile, ammonium acetate, formic acid, methyl tert butyl ether, sodium hydroxide were supplied by Shanghai chemical reagents company. Ultra-pure water was obtained using a Milli-Q UF-Plus system (Millipore, Germany) with a resistivity of at least 18.2 MΩ cm− 1 at 25°C and TOC value below 5 ppb.
Instrumentation
A Varian LC–MS/MS system (Palo Alto, CA) consists of a ProStar 410 auto-sampler (ProStar, DE, USA), two ProStar 210 pumps, and a 1200 L triple quadrupole mass spectrometer equipped with an electrospray ionization source. Varian MS workstation version 6.3 software (Palo Alto, CA) was used for data acquisition and processing.
Chromatographic conditions
The chromatographic separation was performed on a 5-μm ODS-3 column (150 mm × 4.6 mm). The mobile phase consisted of 10 mmol/L ammonium acetate (contained 0.5% formic acid) and acetonitrile (60:40, v/v) at a flow-rate of 0.5 ml/min and the column temperature was maintained at 20 °C. Before use, the mobile phase was filtered through a 0.45 μm nylon membrane filter. The injection volume was 10 μL and the analysis time was 4 min per sample.
Mass spectrometer conditions
The positive ion mode was performed in our analysis method. LC-MS/MS was carried out using nitrogen to assist nebulization. The MRM (multiple reaction monitoring) transitions were m/z 247.3 → 98.1 for mepivacaine and m/z 235.2 → 86.1 for internal standard (IS). The mass spectrometer was tuned for unit mass resolution in the mass range used in the experiments. The optimal ESI-MS/MS parameters were as follows: the ion spray voltage and source temperature were 4000 V and 300°C respectively. The gas rates of nitrogen for nebulizing gas, turbo gas, curtain gas and collision gas were set to 40, 50, 16 and 5 psi, respectively. Drying gas flow was set at12 L/min and the quadrupole temperature, 150 °C.
Standard solutions and calibration curves
Two stock solutions of mepivacaine for standard solution and quality control (QC) were prepared from independent preparations by dissolving mepivacaine in methanol to yield a concentration of 0.1 mg/mL. The working solutions of mepivacaine were prepared by the serial dilution of stock solution with methanol to obtain the following mepivacaine concentrations: 0.5, 5, 50, 100, 500, 1000 and 2000 ng/mL. Working solutions for QC samples with concentrations of 5, 100 and 1000 ng/mL were prepared in the same manner. IS working solution was also prepared by diluting the IS stock solution (0.1 mg/mL) to a final concentration of 500 ng/mL with methanol. All the stock solutions were kept at −20°C, and the working solutions, at 4°C before use.
Calibration standards of mepivacaine were prepared at a concentration ranging from 0.5 to 2000 ng/mL by spiking working solutions into proper blank human plasma. QC samples were prepared in the same manner at three concentration levels of 5, 100 and 1000 ng/mL. All of the spiked blood samples were then treated according to the sample preparation procedure. Both the calibration standard samples and the QC samples were applied in the method validation and the pharmacokinetic study.
Sample preparation
Plasma samples were thawed in a water-bath at 37 °C. 200 μl volume of the plasma sample was transferred to a 10 ml plastic test tube together with 10 μl of IS solution (500 ng/ml) and 100 μl NaOH aq (1 mol/L). After vortex shaking for 1 min (Eppendorf, 5432 vortex mixer, Germany), 3 ml of methyl tert butyl ether was added and the mixture was vortexed for 5 min. After centrifugation at 12000 rpm for 10 min (Thermo IEC, Micromax, MA, USA), the upper organic layer was quantitatively transferred to a 10-ml glass tube and evaporated to dryness using an evaporator at 40 °C. The residue was reconstituted in 100 μl of the mobile phase and then vortex-mixed. After centrifugation at 12000 rpm for 5 min, 10 μl aliquot of the solution was injected into the LC-MS/MS system for analysis.
Assay validation
Validation of assay performance was carried out according to FDA guidelines for bioanalytical method validation (FDA Guidelines, 2013). The selectivity was assessed using six different batches of blank human plasma prepared and analyzed as described in addition to incurred samples collected from the volunteers at zero time, before the administration of mepivacaine. The linearity was evaluated using a zero sample processed without analyte but with IS and nine non-zero samples covering a range of 0.5–2000 ng/mL and analyzed in six replicates. Peak area ratios of mepivacaine to the IS were plotted versus the corresponding concentrations, the linearity was evaluated and the LLOQ was determined. The accuracy and precision of the assay were determined via the analysis of spiked samples at four concentration levels (LLOQ, LQC, MQC and HQC) using six replicates. Within-run precision was evaluated via the analysis of the samples in six replicates in the same day while the between-run precision was evaluated via repeated analyses over three days. Calibration curves prepared and constructed in the same batch were employed in all determinations. The recovery was determined by comparing the peak areas of the analytes spiked into blank plasma at the three QC levels to those of the analytes in neat solvent. The matrix effect was evaluated via the analysis of the QC samples prepared in the post-extraction blank plasma obtained from six different batches. The recovery percentage and variability were determined by comparing the mean peak areas of mepivacaine prepared in extracted plasma samples to those prepared inne at solvent. Stability of mepivacaine in human plasma was evaluated at the storage conditions below and results were compared to the initial concentration at cycle zero. Stock solution stability was assessed at 4–8 °C over 15 h. Freeze–thaw stability was assessed in triplicate via analysis of the QC samples following three cycles of freezing (at −80 °C for 24 h) and thawing (unassisted at room temperature). Benchtop stability was assessed at room temperature for a period of 24 h while long-term stability was evaluated at −80°C for a period of 30 days. QC samples were processed and stored in the auto-sampler at 2–8 °C for 15 h to determine the possible effect of occasional delay in injection or reinjection of samples on their stability.
Clinical pharmacokinetic study
Subjects
Eight healthy male volunteers were recruited from the Department of Pharmacology of our hospital. Aged from 20 to 35, they are non-smokers and non-drinkers. The mean body weight was within the normal range (BMI from 18.2 kg/m2 to 23.6 kg/m2). On the basis of medical history, clinical examination and laboratory investigation (hematology, blood biochemistry, and urine analysis), all volunteers were deemed in good health. The volunteers were instructed to abstain from taking any medication including over the counter (OTC) drugs for at least 2 weeks prior to and during the study period and avoid any alcohol or xanthine containing food and beverages 36 h prior to, or during the course of the study. Inclusion and exclusion criteria were in line with the requirements established in the Chinese guidelines for clinical pharmacokinetic study. All qualified participants signed an informed consent form and eleven blood samples from volunteers were withdrawn at different time points until 12 h after the administration of the medication. This study protocol was approved by the ethical committee in the university, and followed the principles of the Declaration of Helsinki.
Study design
Eight subjects were prohibited to eat after 8:00 pm on the night before the experiment. The test was started at 8:00 am on the following day. 1.0 mL of 2% mepivacaine injection (total dose of drug was 20 mg) was taken as anesthesia and was administrated by infiltration on one side of lip and cheek surface on the volunteering subjects. The process was conducted by professional dentists. During anesthesia, lips should be opened as much as possible to expose the lip surface. The needle was injected into lip and cheek sulcus. The needle was held parallel with the long axis of the tooth. The needle length was of 1–2 cm. The needle and mucosa were beveled. 1.0 mL of 2% mepivacaine was slowly injected between the mucosal and periosteum over about 10 s. Tissues should not be expanded. After drug injection, the needle should be withdrawn slowly (after administration of anesthesia, the needle was used to test the anesthesia effect so as to record the onset time and duration of anesthesia).
A standard meal was served 4 h after dosing. Before drug administration, a catheter was placed in a suitable forearm vein of each subject and a 5-ml blood sample was drawn into a heparinized tube. Additional blood samples (5 mL) were obtained at designated time points (pre-dose and 0.167, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12 h after administration) and were transferred into tubes. The plasma was separated by centrifugation at 12000 rpm (10 min) and stored at −20 °C until further analysis. Pharmacokinetic parameters were calculated against the plasma concentration-time data. The elimination half-life (T1/2) was determined by linear regression of the terminal portion of the plasma concentration-time data. The area under the plasma concentration-time curve from zero to the last measurable plasma concentration point (AUC0–t) was calculated by the linear trapezoidal method. Extrapolation to infinite time (AUC0–∞) was calculated as follows: AUC0–∞ = AUC0–t + Ct/ke, where Ct is the last measurable plasma concentration and ke is the terminal elimination rate constant. The results were expressed as the mean ± the standard deviation (SD). Pharmacokinetic parameters were calculated using the software Excel 2013 with 3P97 (Microsoft office, NE, USA).
Results
Optimization of sample preparation
The method reported herein, with a quantitative limit of 0.500 ng/mL and a shorter retention time (3.5 min), is simple, reproducible, good for analysis of analyte, and could satisfy the measurement requirement of in vivo drug concentrations. This method has been successfully used in measuring the concentration of mepivacaine in human plasma. Assay validation was done according to Capron et al. Citation2014, Lendoiro et al. Citation2012 and Nouman et al. Citation2015.
Selectivity
Results of the analysis of six different batches of blank plasma indicated the absence of endogenous interference at the retention times of the studied drugs and IS. This confirmed the selectivity of the assay toward the studied drugs in the presence of matrix components. The selectivity was further assessed using incurred samples, collected at zero time from the volunteer and patient groups in order to ensure that co-administered midications do not interfere with assay performance.
Linearity and lower limit of quantitation
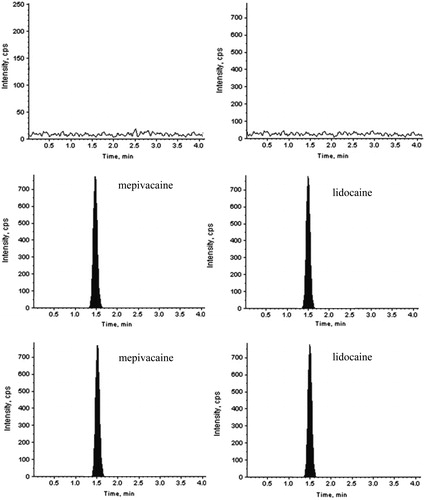
Linearity was evaluated using the average of six determinations at nine concentration levels covering a range of 0.5–2000 ng/mL for mepivacaine. shows representative chromatograms for human plasma spiked with the studied drugs and IS along with their corresponding blank plasma samples. The mean values for the regression equations were Y = 0.0062X + 0.0044, r = 9997, where Y is the peak area ratio of the analyte to the internal standard and X is the concentration of the analyte in ng/mL. Blank and zero samples were included in the analysis in order to verify the absence of interference and reproducibility of the sample preparation procedure. The LLOQ (0.50 ng/mL) was defined as the lowest concentration of analyte that could be quantitatively determined with acceptable precision and accuracy and was included while studying various assay validation parameters. For each calculated standard concentration, deviation from the nominal value should not be more than ±15%, except that for the LLOQ, a ± 20% deviation was deemed acceptable.
Figure 2. LC-MS/MS chromatograph of mepivacaine. Blank plasma (A), and blank plasma added to mepivacaine, internal standard (B) and sample (C).
Accuracy and precision
Analysis of the spiked plasma samples at four concentration levels (LLOQ, LQC, MQC and HQC) revealed that the within-run accuracy of the assay was with an RSD of 7.83–12.9% for mepivacaine (). The between-run accuracy was with an RSD of 9.25–10.62% for mepivacaine (). Obtained results showed clearly that the assay possessed adequate accuracy and precision. The evaluation of precision was based on the criteria that the RSD at each concentration level should be within ±15% except that for the LLOQ, a ± 20% is acceptable.
Table 1. Accuracy and precision for the determination of mepivacaine (n = 6).
Recovery and matrix effect
Liquid–liquid extraction using methyl tert butyl ether was found to be the optimum approach to achieve consistent recovery of the analytes and IS. The recoveries of mepivacaine were measured at three QC levels in six replicates and the results were summarized in . The recovery of each analyte needed not to be 100%, however variability should be consistent among different samples. The matrix effect was investigated as described in order to reveal possible ionization suppression or enhancement caused by matrix components. Adequate recoveries for the analytes and IS indicated that the current sample processing protocol successfully removed the matrix interference as shown in . The matrix effect was examined and the mean peak areas of mepivacaine in three QC samples (LQC, MQC and HQC) prepared in extracted plasma were compared to those obtained from analysis of neat standards of equivalent concentrations. Mean percentage recovery was within the range of 91.6–93.6% and RSD of 6.76–8.96% as summarized in . The recovery of IS was 87.6 ± 6.82% (n = 6).
Table 2. Recovery and matrix effect for the determination of mepivacaine (n = 6).
Stability
According to ICH guidelines for bioanalytical method validation: (i) conditions used in stability experiments should reflect situations likely to be encountered during actual sample handling and analysis and (ii) samples should be prepared from a freshly made stock solution of the analyte in appropriate analyte-free, interference-free biological matrix. Initially, stock solution stability during short term storage for 15 h at 2–8 °C was assessed. The response obtained using the LC–MS/MS assay was compared to that of the freshly prepared solution that demonstrated adequate stability of the stock solution. Results confirmed that three freeze–thaw cycles of the QC samples did not affect the quantification of mepivacaine. Thawing of the frozen samples and their being kept at room temperature for 24 h did not result in notable degradation of the analytes. The QC samples were stored frozen at −80°C and remained stable for at least 30 days. The extracted samples were also analyzed after being stored in the auto-sampler (2–8 °C) for at least 24 h. Results summarized in suggested that human plasma samples containing mepivacaine could be handled under normal laboratory conditions without any significant degradation of the studied drugs.
Table 3. Summary of stability of mepivacaine in human plasma (n = 3).
Clinical pharmacokinetic study
The assay has been successfully utilized to analyze samples obtained from subjects administered single dose of mepivacaine. Mean plasma concentration–time curves are shown in . The pharmacokinetic parameters calculated are summarized in . The present method was proved to be suitable for determining the plasma concentrations of mepivacaine for up to 12 h following a single dose in a small sample volume (200 μL). Therefore, a sufficient number of samples can be obtained in human without any impairment inflicted to the physiological state. As was shown in , drug plasma concentration reached a higher level (more than 400 ng/mL) in the initial 0.5 h after single administration of mepivacaine injection. Then the drug concentration decreased markedly after 1 h and stayed at a lower level of 45–200 ng/mL.
Figure 3. Mean plasma concentration–time curve in 8 adult, healthy Chinese subjects when administered single dose of mepivacaine (linear plot). The vertical lines were error bars and the small triangles represented the plasma concentration at specific time point.
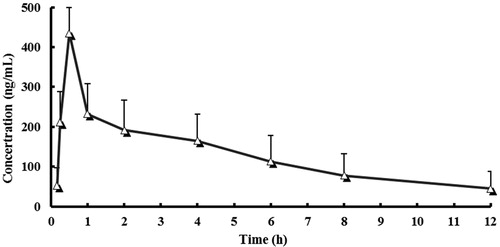
Table 4. Pharmacokinetic parameters for eight volunteers after administration of a single dose of mepivacaine.
Discussion
Liquid chromatography and mass spectrometric conditions
To effectively separate lidocaine, mepivacaine and endogenous substances in plasma, it was found in the pre-test study that aqueous phase pH changes had a large influence on the separation effect. Thus, system optimization was performed on the aqueous phase pH and it was found that the best pH result was achieved when 0.5% formic acid was added into 10 mmol/L ammonium acetate to effectively separate drug, internal standard and impurities. Moreover, the choice of adding acetonitrile system in the mobile phase aimed at increasing ionization in the component to be measured. When the aqueous phase ratio was relatively high, the ionization had lower efficiency and weaker response. Considering all the facts, acetonitrile-10 mmol/L ammonium acetate (contained 0.5% formic acid) (40:60) was chosen as the mobile phase. Under these conditions, the chromatographic behavior was moderate and had no matrix interferences.
In the optimization of the MS conditions, because local anesthetics structural formula all had amide bond, mass response appeared under both positive and negative ion detection mode. However, after investigation, it was believed that the positive ion mode detection had better response than its negative counterpart. Thus, the positive ion mode detection was used during measurement (Kakinohana and Okuda Citation1995). In the selection of ion source, these two compounds were found to have better response and linearity under the ESI source than the APCI source. Under the ESI +ionization, lidocaine and mepivacaine mainly generated excimer ion peak m/z 235.2 and 247.2 in Q1 full scan spectra. No other solvent or metal processing and ion were found. Mass spectrometry of product ion system was performed on [M+H]+. The main fragment ions of lidocaine and mepivacaine were m/z 86.0 and 98.0 respectively. When m/z 86.0 and 98.0 were chosen for quantitative analysis of product ion monitoring, there was no crosstalk or substrates interference among channels. Meanwhile, in order to improve the specificity and detection sensitivity of the method, multiple reaction monitoring (MRM) was applied, and the parameters of mass spectrometer were optimized to determine the final MS conditions.
Optimization of sample preparation
Biological samples contain a large number of endogenous substances such as proteins, peptides, and inorganic salts. Simple processing of biological samples, such as the protein precipitation method, may cause a large number of endogenous substances still remaining in the injection solution and produce obvious ion suppression (Błażewicz et al. Citation2014; Luo et al. Citation2014; Thomas et al. Citation2015) . This experiment applied the method of ‘sample was basified and then extracted by methyl tert-butyl ether’, which reduced the possibility of ion suppression. No significant matrix effects were found during the sample measurement process. Most of the local anesthetics were alkalescence amine, and usually in salt form. Therefore, samples can only be effectively extracted after dissociate target. Alkalizing agents and its pH had a notable effect on the extraction recovery of such drugs. This study compared the alkalizing agents at pH 8, 10, and 13. In the end, the study chose 1 mol/L of NaOH at pH 13 as the alkalizing agent. At the same time, a systematic investigation was performed on the alkalizing agents at this concentration level. The extraction recovery was used as an index to determine the final amount of alkalizing agent to be added. The author also tested ether, ethyl acetate and other organic solvents. Based on blank interference and substrate influence, methyl tert-butyl ether was finally decided to be used for extract.
Therefore, the advantages of this research method are mainly reflected in the following aspects: (1) a simple sample preparation procedure (one-step liquid-liquid extraction); (2) short analysis time (only 5 min); (3) Strong method specialization.
Conclusion
To the best of our knowledge, there is no validated LC–MS/MS method available to date for the quantitation of the mepivacaine in Chinese biological matrix. In the present study, we have developed and validated a simple, selective, and sensitive LC–MS/MS method. The calibration curve of mepivacaine ranged from 0.5 to 2000 ng/mL with a lower limit of quantitation being 0.5 ng/mL. This sensitivity was high enough to describe the profile of blood mepivacaine level versus time. The sample preparation procedure involved a simple one-step liquid–liquid extraction and only 200 μL of blood sample and 10 μL of injection volume were required for the analysis. It was very desirable for the pharmacokinetic study because of its high sensitivity and accuracy. The validated method has been successfully applied to assess the pharmacokinetic study of mepivacaine after a single administration to Chinese volunteers.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Bailard NS, Ortiz J, Flores RA. 2014. Additives to local anesthetics for peripheral nerve blocks: Evidence, limitations, and recommendations. Am J Health Syst Pharm. 71:373–385.
- Baniceru M, Croitoru O, Popescu SM. 2004. Determination of some local anesthetics in human serum by gas chromatography with solid-phase extraction. J Pharm Biomed Anal. 35:593–598.
- Baselt RC (Ed.). 2002. Disposition of Toxic Drugs and Chemicals in Man (6th ed.). CA USA: Biomedical Publications, Vol. 126–129, pp. 627–629, 944–945.
- Błażewicz A, Klatka M, Dolliver W, Kocjan R. 2014. Determination of total iodine in serum and urine samples by ion chromatography with pulsed amperometric detection - studies on analyte loss, optimization of sample preparation procedures, and validation of analytical method. J Chromatogr B Analyt Technol Biomed Life Sci. 962:141–146.
- Capron A, Destree J, Maiter D, Wallemacq P. 2014. Validation of a rapid liquid chromatography-tandem mass spectrometric assay for the determination of octreotide plasma concentrations. Clin Biochem. 47:139–141.
- Eng HC, Ghosh SM, Chin KJ. 2014. Practical use of local anesthetics in regional anesthesia. Curr Opin Anaesthesiol. 27:382–387.
- FDA, Guidelines: Bioanalytical Method Validation, 2013 [Internet]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf
- Hattori H, Yamamoto S, Yamada T, Suzuki O. 1991. Determination of local anaesthetics in body fluids by gas chromatography with surface ionization detection. J Chromatogr. 564:278–282.
- Kakinohana O, Okuda Y. 1995. The plasma concentration measurement of local anesthetics, thiobarbiturates and mexiletine by HPLC with automatic pre-treatment system. Masui. 44:1165–1170.
- Lendoiro E, Cordeiro C, Rodríguez-Calvo MS, Vieira DN, Suárez-Peñaranda JM, López-Rivadulla M, Muñoz-Barús JI. 2012. Applications of Tandem Mass Spectrometry (LC-MSMS) in estimating the post-mortem interval using the biochemistry of the vitreous humour. Forensic Sci Int. 223:160–164.
- Luo Q, Chen X, Wei Z, Xu X, Wang D, Wang Z. 2014. Simultaneous and high-throughput analysis of iodo-trihalomethanes, haloacetonitriles, and halonitromethanes in drinking water using solid-phase microextraction/gas chromatography-mass spectrometry: an optimization of sample preparation. J Chromatogr A. 1365:45–53.
- Nieddu M, Boatto G, Serra D, Soro A, Lorenzoni S, Lubinu F. 2007. HPLC-DAD determination of mepivacaine in cerebrospinal fluid from a fatal case. J Forensic Sci. 52:1223–1224.
- Nouman EG, Al-Ghobashy MA, Lotfy HM. 2015. Development and validation of LC-MSMS assay for the determination of the prodrug dabigatran etexilate and its active metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 989:37–45.
- Ohshima T, Takayasu T. 1999. Simultaneous determination of local anesthetics including ester-type anesthetics in human plasma and urine by gas chromatography-mass spectrometry with solid-phase extraction. J Chromatogr B Analyt Technol Biomed Life Sci. 726:185–194.
- Su N, Liu Y, Yang X, Shi Z, Huang Y. 2014. Efficacy and safety of mepivacaine compared with lidocaine in local anaesthesia in dentistry: a meta-analysis of randomised controlled trials. Int Dent J. 64:96–107.
- Thomas J, Khanam R, Vohora D. 2015. A validated HPLC-UV method and optimization of sample preparation technique for norepinephrine and serotonin in mouse brain. Pharmaceut Biol. 53:1539–1544.
- Watanabe T, Namera A, Yashiki M, Iwasaki Y, Kojima T. 1998. Simple analysis of local anaesthetics in human blood using headspace solid-phase microextraction and gas chromatography-mass spectrometry-electron impact ionization selected ion monitoring. J Chromatogr B Analyt Technol Biomed Life Sci. 709:225–232.