
Abstract
This study focuses on the development of a universal mathematical model for drug release kinetics from liposomes to allow in silico prediction of optimal conditions for fine-tuned controlled drug release. As a prelude for combined siRNA-drug delivery, nanoliposome formulations were optimized using various mole percentages of a cationic lipid (1,2-dioleoyl-3-trimethylammonium-propane, DOTAP) in the presence or absence of 3 mol% distearoyl phosphoethanolamine, polyethylene glycol (PEG–2000mDSPE). Outcome parameters were particle size, zeta potential, entrapment efficiency, in vitro drug release, and tumor cell kill efficiency. The optimized formula (containing 20% DOTAP with 3% DSPE-mPEG(2000) was found to be stable for six months, with round-shaped particles without aggregate formation, an average diameter of 71 nm, a suitable positive charge, and 89% drug encapsulation efficiency (EE). The 41% drug release during 6 h confirmed controlled release. Furthermore, the release profiles as functions of pH and temperature were investigated and the kinetics of the drug release could adequately be fitted to Korsmeyer–Peppas’ model by multiple regression analysis. The statistical parameters confirmed good conformity of final models. Functionality of the novel cationic liposome formulations (± DOX) was tested on osteosarcoma (OS) cell lines. Increased OS cell toxicity (1.3-fold) was observed by the DOX-loaded vs. the free DOX. A feasibility pilot showed that siRNA could be loaded efficiently as well. In conclusion, we have established a predictive mathematical model for the fine-tuning of controlled drug release from liposomal formulations, while creating functional drug-delivery liposomes with potential for siRNA co-delivery to increase specificity and efficacy.
Graphical Abstract
Introduction
Improvement of current cancer therapies is often an urgent and unmet need, in particular in cases where toxicity of chemotherapeutics is high when applied systemically and/or chemoresistance occurs. An example is osteosarcoma, the most common primary cancerous tumour in bone, being most prevalent in children and young adults. The frequently occurring multidrug resistance (MDR) to cytotoxic therapy in OS, and occurrence of many adverse side effects when using increased chemotherapeutic dosages are accountable for the limited efficacy of current therapies [Citation1,Citation2]. To tackle these limitations, two major new strategies are the topic of research nowadays: (i) Nano drug delivery systems, and (ii) interference with the chemotherapeutic resistance mechanisms, e.g. via small interfering RNAs (siRNAs), within OS. Both are novel, promising options to reduce the harmful side effects of systemic drug administration, while reaching high(er) treatment efficiencies [Citation3]. The challenge is to develop co-delivery systems to reach synergism in treatment efficacy.
Liposomes are common vesicles for nano drug delivery systems for cancer treatment compounds because of their low toxicity, low immunogenicity, high potential for gene incorporation, and ease of preparation [Citation4]. Simulation and modelling is a useful tool for the prediction and optimization process for controlled drug release before carrying out costly and time-consuming production of liposome formulations. Development of a kinetic model as a function of important parameters helps to predict the release behaviour of drug, better process control, high quality production and decreased energy dissipation, and also plays an important role to understand the mechanisms involved in the process. Several reports on models for release kinetics have been published. Xiong et al. examined 9-(methylaminomethyl) anthracene and procaine hydrochloride release behaviour from poly(lactic acid) –F127– poly(lactic acid) aggregates in PBS (pH 7.4, 0.01 M) and developed a kinetic model [Citation5]. Mourtas et al. studied models of calcein and griseofulvin release from liposomal gels using agarose assisted immobilization as a technique to separate gels [Citation6]. Sanson et al. presented in vitro pH and temperature-dependent doxorubicin release profiles into block copolymer vesicles of poly (trimethylene carbonate) -b-poly (L-glutamic acid) [Citation7].
A doxorubicin-containing liposomal product, Doxil, is available and FDA-approved for OS treatment [Citation8]. However, adaptations to the liposomal formulation are necessary to improve the incorporation of siRNA for co-delivery systems. In this regard, cationic lipids are useful components to add to the liposomal formulation to improve gene incorporation [Citation9]. An example of this approach is the study of Chen et al., who developed a sigma-receptor targeted liposomal co-delivery system using cationic lipids (N,N-distearyl-N-methyl-N- 2-(N-arginyl) aminoethyl ammonium chloride (DSAA) or 1,2-di-(9Z-octadecenoyl)-3-trimethylammonium-propane (DOTAP)), and PEGylation using 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000 (DSPE-PEG 2000) for DOX and MDR1 siRNA co-delivery. They demonstrated the potential of this co-delivery system to overcome MDR [Citation10]. However, no studies on the optimization of the cationic lipids and PEGylation compounds have been performed so far.
The major aims of this study are two-fold: (i) optimization of DOTAP and DSPE-PEG 2000 content in liposomal formulations of doxorubicin as an effective carrier of the simultaneous gene–drug therapy in future studies; and (ii) the development of an in vitro kinetic model for drug release in relation to temperature and pH medium, to accurately predict drug release profiles for future liposomal formulations for other drugs and active compounds deliveries.
Last but not least, we performed functionality tests on the osteosarcoma cell line MG-63 to determine the toxicity effects and treatment efficacy of the resulting optimized formulation ± DOX, as well as a proof-of-principle pilot study to test siRNA loading capability.
Experimental and methods
Materials
The doxorubicin HCL (DOX) was obtained from Ebewe Pharma (Vienna, Austria). Cholesterol and 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) were obtained from Sigma-Aldrich Co (St. Louis, MO) and Avanti Polar Lipids (Alabaster, AL), respectively. The distearoyl phosphoethanolamine, polyethylene glycol (Lipoid PE 18:0/18:0 – PEG2000, DSPE-mPEG 2000) and 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) were purchased from Lipoid GmbH (Ludwigshafen, Germany). siRNA (sense strand: 5’-GUA-CGA-GGC-CUA-CAA-CAU-G99–3’; antisense strand: 5’-CAU-GUU-GUA-GGC-CUC-GUA-C99–3’) was synthesized by Eurogenetic co. (Biotechnology Company, Seraing, Belgium) [Citation11]. All other chemicals and solvents used in this investigation were of analytical grade.
Cell cultures and biochemical analysis
Human primary (short-term culture) osteoblasts Hum63 were provided by healthy patients undergoing total knee replacement after informed consent and human osteosarcoma cell line MG-63 was obtained from Pasteur Institute of Iran (Tehran, Iran). All cells were cultured in D-MEM (GIBCO, Grand Island, NY) supplemented with 10% foetal bovine serum (FBS) and 1 mg/mL Penicillin-Streptomycin (Gibco, Invitrogen, Grand Island, NY) at 37 °C and 5% CO2 in a humidified incubator.
Preparation and characterization
In this study, we extended our previous work [Citation12] to develop new formulations for gene–drug co-delivery. Liposomal DOX formulation was optimized in terms of desired particle size, controlled drug release, high encapsulation efficiency (EE) and positive ζ- Potential. Briefly, the various experiments were categorized as:
Optimization experiments of DOTAP contents (0, 10, and 20%) in different DSPE-mPEG (2000) % (0 and 3%).
Drug kinetic release assay at different values of pH and temperature (pH: 4.5, 5.4, and 7.4; Temperature: 25, 37, and 42 °C).
Development of a mathematical model based on the experimental data, followed by statistical validations.
Functionality tests: determination of the toxicity effects and treatment efficacy of the resulting optimized formulation ± DOX.
Pilot study for the assessment of the siRNA loading efficiency.
Liposomal doxorubicin was prepared with pH gradient method to improve the solubility of the drug [Citation13]. Briefly, DPPC and cholesterol (1:0.43 molar ratios) with various DOTAP contents (0, 10, and 20%) were dissolved in chloroform at 51 °C. All formulations had 0 or 3% DSPE-mPEG2000. Hydration was performed with 1300 μL ammonium sulphate for 45–60 min at 55 °C using rotary instrument (Heidolph, Schwabach, Germany). Prepared vesicles were then sonicated to reduce the vesicles mean size. Doxorubicin (0.5 mg/mL) was loaded into the blank liposomes at 55 °C. The zeta potential, size distribution, and polydispersity index of liposomal DOX were evaluated using dynamic laser scattering technique (DLS) by Brookhaven Corp Instruments (Holtsville, NY). All measurements were carried out in triplicate and at room temperature and their mean values were reported. Characterization of surface morphology and internal structure were determined by Field Emission Scanning Electron Microscopy (FESEM, KYKY-EM3200–30KV, China), Cryo-transmission electron microscopy (cryo-TEM, FEI Tecnai 20, type Sphera, Oregon). Chemical integrity of the nano liposome was also evaluated by Fourier Transform Infrared (FTIR) spectroscopy [Citation12].
Doxorubicin liposome EE was determined as described previously [Citation12]. Doxorubicin-loaded formulation was purified from free doxorubicin by dialysis tubing. The amounts of drug entrapped in the liposomes were calculated with a UV spectrophotometer at 480 nm.
Also the release kinetic of drug was monitored against PBS for 6 h at 37 and 42 °C temperature and pH 7, 5 and 4. The physical stability of nano formulation during 180 d storage was evaluated in terms of vesicle size, PDI, zeta potential, and EE.
Mathematical modelling of drug release kinetic
The normalized drug release from nanocarriers was calculated by the following EquationEquation (1)(1) :
(1)
where Mt and Mf are the cumulative amounts of drug released at any time (t) and final amounts of drug released, respectively.
The normalized drug release mainly accrued for early stage (0–8 h) that can be described by mathematical and semi-empirical model, Peppa’s model [Citation14]:
(2)
where k depends on the structural characteristics of the liposome and the drug and n is the release exponent is relative to the drug release mechanism (Fickian diffusion or non-Fickian diffusion) and the geometry of the carrier. Dimension of k is (time−n) and n is dimensionless.
Also nonlinear regression analysis was performed using MATLAB software version 7.8 (Mathworks Inc., Natick, MA) in order to fit the experimental drug release with models.
Root mean square error (RMSE) [Citation15] and R-square (R2) [Citation16] were used to determine the quality of the fit. Higher values of R2 and lower values of RMSE indicate a better fit. These can be calculated with expression (Citation3) and (Citation4):
(3)
(4)
where release (%)exp,i is the experimental release found in any measurement, Release (%)pre,i is predicted release for this measurement, N is the number of observations, Z is the number of model constants, Release (%)exp is the total average data, and i is ith data. It is assumed that there is no resistance in buffer solution to transfer drug.
In order to generalize the drug release kinetic model, coefficients of drug release model were developed with various parameters such as buffer's pH and temperature by multiple regression technique.
Functionality test: evaluation of cytotoxic activity
The Alamar blue assay was applied for cell viability analysis [Citation12]. Briefly, MG-63 cells were seeded at a density of 104 cells per well into 96-well plates with medium and 10% FBS. Medium was subsequently replaced with media containing dilution series of blank liposome, DOX and DOX loaded-liposome varied from 0.1 to 10 mg/mL, and cells were further incubated for 72 h. Thereafter, each well was rinsed with PBS, followed by incubation for 3 h with 250 μL of Alamar Blue stock solution. For quantification, the Alamar blue solution was transferred to a fresh 96-well plate, and absorption was recorded by an automated microplate spectrophotometer (EPOCH Microplate Spectrophotometer Synergy HTX, Bio Tek, Winooski, VT), with an excitation and emission wavelength of 530 and 590 nm, respectively. The mean standard deviation of three results was calculated.
Also Alamar blue assays were employed to represent the nontoxic effect of prepared liposome formulation on human primary (short-term culture) osteoblasts. So the various concentrations of blank liposome (100–0.1 mg/mL) were exposed to cells during 72 h according to Alamar blue cytotoxicity protocol and cell viability was measured.
Evaluation the formation of siRNA liposomal DOX complex
The siRNA targeting the JIP1-gene [Citation17] was used as a model to evaluate the ability of formation of siRNA-liposomal DOX complex to develop as co-delivery system for future research. The optimized formulation of liposomal DOX was incubated with siRNA for 30 min. Untrapped siRNA was separated using ultracentrifugation at 35,000 rpm (Beckman Coulter Optima LK90, Indianapolis, IN). The zeta potential of prepared complex of gene-liposomal DOX was measured using DLS and the difference between the zeta potential of gene-liposomal DOX complex and liposomal DOX formulation represents the capacity of gene incubation of optimized formulation.
Result and discussion
Optimization liposomal DOX formulation
Various liposomal doxorubicin formulations were evaluated in order to determine the optimal formulation in terms of high EE, high positive charge, low PDI, controlled drug release (at 37 °C and pH =7) and small vesicle size ().
Table 1. Effect of PEGylation and DOTAP addition on liposomal DOX characteristics.
As can be seen, PEG addition into liposomes (F1→F4, F2→F5, and F3→F6) improved the physical characteristics of doxorubicin-loaded liposomes, such as vesicle size (small diameter), polydispersity index and ζ- Potential (positive charged), drug release (controlled release), and EE (high %EE). Similarly, Kim et al. and Li et al. found that the vesicle size was slightly decreased by the addition of PEG due to the reduction of aggregation of liposome together. They also found that PEG addition improved positive ζ- Potential and release kinetics of drug, followed by slower release rate [Citation18,Citation19]. In fact, for anionic formulation (F1 and F4) PEG addition reduced absolute value of ζ- Potential that was frequently explained by i) an enhancement of drag force against the motion of vesicle due to PEG chain on the surface of liposome ii) a shift further distances away of shear plane the from the liposome surface. However, for cationic formula (F2, F3, F5 and F6), data is not support that results of anionic formula and related mechanism is indistinct, but the change in ζ- Potential for by PEG addition is subtle compared to the DOTAP addition hence PEG effect on ζ- Potential for cationic formula is not considered. The same observations have not been reported previously. Our result about increasing EE by PEG addition was in contrast with Li et al. (2001) [Citation19].
Increase in DOTAP concentration increases the EE and ζ- Potential for PEGylated liposome and non-PEGylated once (i.e. F1→F2 or F5→F6). With the DOTAP addition increase EE, decrease vesicle mobility and also vesicles size attributed to decreasing in cholesterol content [Citation12].
Also the drug release decreases due to decrease in cholesterol content. The number of positive charge agent increased with 10% increase in molar amount of DOTAP which in addition to improving the positive charge. This effect also decreases polydispersity index relate it to further mutual repel force between the particles with the same-sign charge in the suspension system.
It can be concluded that the optimal formula contains 20% DOTAP with 3% DSPE-mPEG(2000), which led to optimal controlled release and highest EE.
Moreover, sufficient value for ζ- Potential was achieved at 20% DOTAP addition which makes it an appropriate carrier for drug-gene delivery. The PDI (0.159) of proposed formula (F6) was less than 0.2, which shows that nanoparticles had homogeneous size.
According to results presented in , encapsulation of drug in liposomal formula (F6), increased the vesicle size, ζ- Potential (positive charge) and PDI. These results are in agreement with previous works [Citation20]. They also observed that DOX encapsulation into liposomes makes liposomes larger and more positively charged.
Table 2. Effect of drug encapsulation.
Chemical interaction
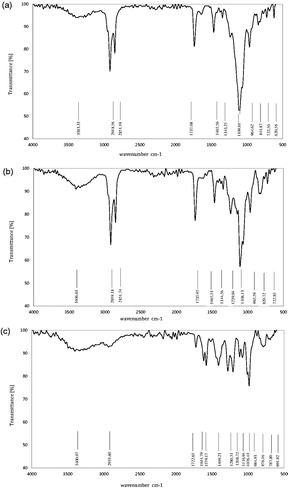
) shows the FTIR spectrum of optimum liposomal formula (F6), after and before drug loading and free drug (DOX). According to , there were characteristic peaks of phospholipid, DSPE-mPEG and cholesterol at 3383.33 cm−1 (O–H stretching in phenols and N–H stretching in 1°, 2° amines, amides), 2918.56 cm−1 (–CH3 asymmetric and symmetric stretching), 2851.19 cm−1 (–CH2 asymmetric and symmetric stretching), 1343.25 cm−1 (–CH3 bending), 1465.56 cm−1 (–CH2 bending), 1737.08 cm−1 (ester stretching), 1109.61 cm−1 (ether stretching in ester group) and 841.87 cm−1 (P–O). Other peaks were related to C–H “oop” aromatics (722.50 cm−1) and –C–H bend alkenes (650–1000 cm−1). All peaks are repeated in , which indicated that there is no chemical interaction between doxorubicin and DPPC, cholesterol, and DSPE-mPEG in liposome formulation.
Figure 1. FTIR spectra of optimal formula (F6) (a): after drug loading, (b): before drug loading, and (c): DOX.
Morphology of optimum formula
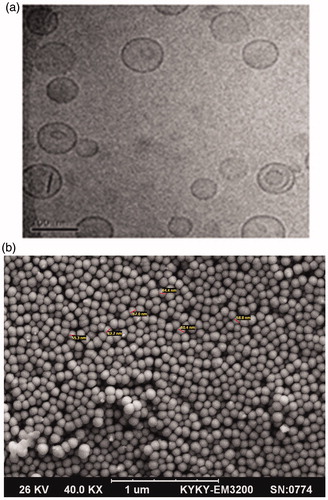
TEM and FESEM micrograph of the liposomal DOX formula (F6) are indicated in ). As can be seen, the optimal formula was round, smooth surfaced, spherical shaped and without aggregates. Also the rigid boundaries of liposomes and their bilayer structure were illustrated.
Figure 2. (a) TEM (bar = 100 nm) and FESEM (b) micrographs of optimum formula (F6 + DOX).
Physical stability of optimal formula
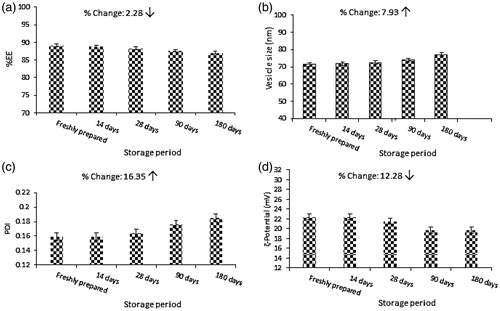
The confirmation of physical stability of liposomal DOX formula (F6) is shown in ) in terms of EE, vesicle size, PDI, ζ- Potential. From the analysis of results it is evident that no significant changes in physical characteristics of F6 were accrued. Results indicated that the change of EE, vesicle size, PDI and ζ- Potential were 2.28, 7.93, 16.35 and 12.28%, respectively after 6 months of storage.
Figure 3. Physical stability study of optimal formula (F6 + DOX) during 6 months. (a) Encapsulation efficiency, (b) Vesicle size, (c) PDI and (d) ζ- Potential.
Cytotoxicity assay
Cytotoxicity of various concentrations of liposomal DOX on primary bone cells and the MG-63 osteosarcoma cell line was evaluated with Alamar Blue assay. The results are shown in .
Figure 4. Cell viability on human primary osteoblasts and MG-63 cell line of liposomal formulations with various concentrations, determined after 72 h using Alamar Blue colorometric assay.
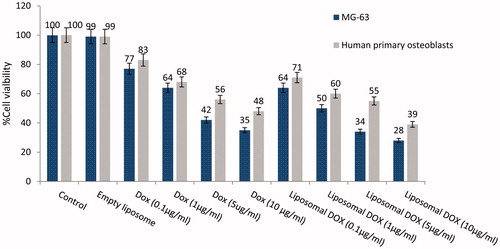
Empty liposomes do not show any toxicity on human primary osteoblasts cells. However in comparison to our previous works, DOTAP addition has caused a slightly increased toxicity [Citation12].
Cytotoxicity on MG-63 cells of free DOX, empty liposome and liposomal DOX in various concentrations was also assessed with Alamar Blue colorimetric assay, after 72 h. Results are depicted in , too.
Cells grown were inhibited by doxorubicin in both entrapped and free forms but the liposomal form of doxorubicin appeared to decrease MG-63 cell viability more effectively, i.e. by 1.24-fold for Alamar Blue. This can be attributed to slow release properties of liposomal formulation that enhanced the toxicity for MG-63 osteosarcoma cells.
High concentration of small molecule of free doxorubicin could transfected into cell by diffusion through the cell membrane but the sudden uptake of high concentration of doxorubicin into part of cells maybe leads to prevent expose to doxorubicin for other parts of the cells. Although controlled release strategy of doxorubicin by liposome is limited by slow release of entrapped doxorubicin and gradually diffuses through the MG-63 cells membrane but uniform expose for all cells was occurred in long term.
Also in comparison of on human primary osteoblasts and MG-63 cell line, there was clear and identified difference between cell viability of primary bone cell and MG-63 cells (16%↑ for free DOX and 23%↑ for liposomal DOX) against DOX due to pH and thermo-sensitivity properties of prepared formulation.
Evaluation the formation of siRNA-liposomal DOX complex
The siRNA was incubated as a gene model into liposomal DOX (F6) and the zeta potential of gene-drug formulation was reduced to 7–8 mv (). These results confirmed that the siRNA was successfully incubated with liposomal DOX. The slight positive charge of prepared co-delivery system could be improving the interaction between cytoplasm and carrier, and increasing cellular transfection (data were not reported). Prepared gene-drug model formulation were tested on osteosarcoma but could be used for other cancerous cells delivery due to its physical and biological characteristics.
Figure 5. The capacity of gene incubation evaluation by zeta potential measurement during siRNA incubation. (a) Before siRNA incubation. (b) After siRNA incubation.
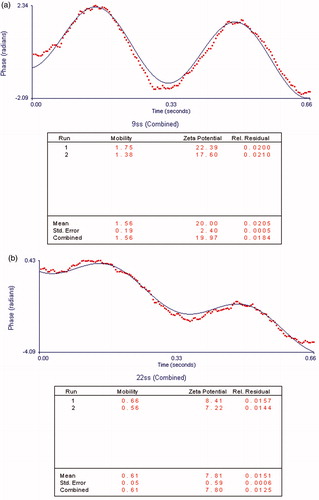
The toxicity of peppered carrier with combination of siRNA-DOX would be enhanced, leads to increase killing efficiency of endosomal cells and also increase in vivo side effects against normal cells. Though the pH and thermo sensitivity properties of purposed formula contribute in local targeting of osteosarcoma cells but the systemic targeting of osteosarcoma cells using peptide would be more effective that are being developed in our lab for DOX-siRNA delivery [Citation21].
Release kinetics
The in vitro drug release studies of F6 formula at various pH values (a) (4, 5.4, and 7.4) and temperatures (b) (25, 37, and 42) are shown in . Free DOX is a very small molecule that could be easily passes through the membrane. Thus the slow release present profile was not dependent on limitation of DOX transfer. The release profiles of F6 were clearly delayed approximately posts 96 h due to liposomal membrane induced barrier effect of DOX. The long-term sustained release behaviour of DOX governed by three mechanisms i) the slow rate of solubilisation of aggregated DOX inside the liposome, ii) the diffusion of dissolved DOX from liposome and transfer to outside the dialysis tube and iii) dispersion DOX molecule by convection mechanism due to gentle shaking of PBS solution [Citation22].
Figure 6. In vitro kinetic release of drug for F6 formula in various pH (a) and temperatures (b).
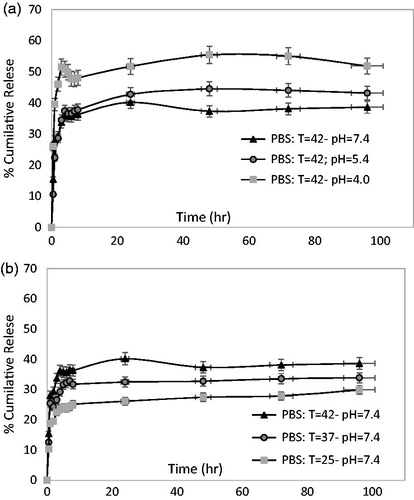
Interestingly, as can be deduced from , the slow drug release rate accrued at higher value of pH and lower value of temperature. So that, the 43% accumulated release reached at 96 h for 42 °C and pH = 5.4 while just 33% of DOX was released for 37 °C and pH = 7.4.
This phenomenon demonstrated that at low pH (5.4) to increase hydrophilicity of DOX and disolubilization inside the liposome, thus drug release accelerated. Also another observation about the fast drug release at high temperature (42 °C) is likely to be attributing to increase the permeability inside the liposome membrane. With rule of thumb and by taking the results of cell viability assay and release kinetic it could be concluded that just 44% would be release during 72 h and leads to kill approximately 56% of MG-63 cells at pH = 5.4 and 42 °C. This despite the fact that, total release of free DOX leads to 45.5% cells kill.
The results confirm that F6 formula has good controlled release properties, effective doxorubicin-delivery abilities under endosomal pH values, and stability at room temperature conditions.
Mathematical modelling of release kinetics
Average of various statistical parameters was calculated for all of the tests according to EquationEquations (3(3) and Equation4)
(4) of Peppa’s model. The average R2 and RMSE are 0.9587 and 0.0653, respectively. Coefficients of Peppa’s model and its statistical parameters have been calculated for all tests (). According to , high value of average R2 and low value of RMSE indicate that predicted data are very close to experimental data.
Table 3. Proposed model coefficients (expression 2).
Such mathematical analysis was conducted for under study DOX-loaded liposome system. Determined Exponent values for all under study condition were less than 0.5 and Fickian diffusion was observed for DOX release period. The drug release in higher temperature and lower pH was characterized by lower release exponent values (n).
Nonlinear regression analysis was performed to develop a comprehensive model as a function of temperature and pH. Coefficients of Peppa’s model as the best fitting model involve the drug release variable such as temperature (T) and pH of medium that were determined as follows:
(5)
The model constants of expression 5 are reported in .
Table 4. Model constants and validation of model coefficient.
Validation of these two functions was evaluated with %E (Mean of relative error percent, expression 6), RMSE, and chi-square (χ2-expression 7) [Citation23]. Low values of %E, RMSE, and χ2 show good fit of purposed coefficient model with presented model (expression 5). These values are reported in .
(6)
(7)
By comparing computed release with measured ones, the validation of finally established model for release kinetics of doxorubicin with coefficients as the function of process parameters was made in all tests. R2 and slopes had been provided for all tests for measured release versus predicted release in . A good agreement between model and the experimental data is shown in .
Table 5. Slope and R2 for predicted release vs. experimental release.
Conclusions
In conclusion, this study suggests a novel, optimized formulation of cationic PEGylated liposomal doxorubicin for drug-gene delivery. We used small molecule JNK-interacting protein 1 (JIP1) as a model of siRAN for evaluation the ability of gene-drug encapsulation as a co-delivery system.
Cytotoxicity assay performed on MG-63 and primer bone cells with Alamar blue colorimetric method for this optimal formula, showed that the IC50s of doxorubicin loaded liposome for MG-63 cells were 1.29-fold lower than the free form of doxorubicin. The drug release profile confirms that prepared formulation acts as local targeting dependent on temperature and pH difference between the normal and cancer cells. In fact drug release was more rapid at 42 °C and acidic pH that are proportional to pH levels in the endosomes of cells. The results demonstrated that experimental data were adequately fitted by the Peppa’s kinetic mathematical model as the function of temperature and pH.
Acknowledgements
The authors thank Dr. Mohammad-Amin Moradi, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB, Eindhoven, The Netherlands, for helping us to use the Cryogenic transmission electron microscopy.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- M.N.H. Fateme Haghiralsadat, Ghasem Amoabediny, Behrouz Zandieh-doulabi, Posthuma de Boer Jantine, Treatment of osteosarcoma metastases: Codelivery of siRNA and small molecule anticancer drugs based Nano-biotechnology: a review, Biotechnol. Adv. (2016).
- Gumulec J, Fojtu M, Raudenska M, et al. Modulation of induced cytotoxicity of doxorubicin by using apoferritin and liposomal cages. Int J Mol Sci. 2014;15:22960–22977.
- MajidiZeinali Sehrig S, Samiei F, Milani M, etet al. Magnetic nanoparticles: applications in gene delivery and gene therapy, Artif. Cells Nanomedicine Biotechnol. 2016;44:1186–1193.
- Singh B, Garg T, Goyal AK, et al. Recent advancements in the cardiovascular drug carriers. Artif Cells Nanomed Biotechnol. 2016;44:216–225.
- Xiong XY, Tam KC, Gan LH. Release kinetics of hydrophobic and hydrophilic model drugs from pluronic F127/poly (lactic acid) nanoparticles. J Control Release. 2005;103:73–82.
- Mourtas S, Fotopoulou S, Duraj S, et al. Liposomal drugs dispersed in hydrogels: effect of liposome, drug and gel properties on drug release kinetics. Colloids Surf Biointerfaces. 2007;55:212–221.
- Sanson C, Schatz C, Le Meins J-F, et al. A simple method to achieve high doxorubicin loading in biodegradable polymersomes. J Control Release. 2010;147:428–435.
- Fateme Haghiralsadat B, Amoabediny ZG, Sheikhha MH, et al. A novel approach on drug delivery: investigation of new nano-formulation of liposomal doxorubicin and biological evaluation of entrapped doxorubicin on various osteosarcomas cell lines. Cell J. 2017 (in press). http://celljournal.org/journal/article/abstract/4502
- Kim B-K, Seu Y-B, Bae Y-U, et al. Efficient delivery of plasmid DNA using cholesterol-based cationic lipids containing polyamines and ether linkages. IJMS. 2014;15:7293–7312.
- Chen Y, Bathula SR, Li J, et al. Multifunctional nanoparticles delivering small interfering RNA and doxorubicin overcome drug resistance in cancer. J Biol Chem. 2010;285:22639–22650. doi:10.1074/jbc.M110.125906.
- Boer JPD. Towards targeted treatment for osteosarcoma. Amsterdam, Netherlands: VU University Medical Center; 2014.
- HaghiralsadatAmoabediny FG, Sheikhha M, Zandieh-doulabi H, et al. New liposomal doxorubicin nanoformulation for osteosarcoma: drug release kinetic study based on thermo and pH sensitivity. Chem Biol Drug Des. 2017. [Epub ahead of print].doi: 10.1111/cbdd.12953
- Madden TD, Harrigan PR, Tai LCL, et al. The accumulation of drugs within large unilamellar vesicles exhibiting a proton gradient: a survey. Chem Phys Lipids. 1990;53:37–46.
- Korsmeyer RW, Gurny R, Doelker E, et al. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35.
- MJ. Procopio An experimental analysis of classifier ensembles for learning drifting concepts over time in autonomous outdoor robot navigation, ProQuest, Boulder, 2007.
- B. Vidakovic Statistics for bioengineering sciences: with MATLAB and WinBUGS Support, 2nd ed. Springer Science & Business Media, 2011.
- De Boer JP, van Egmond PW, Helder MN, et al. Targeting JNK-interacting protein 1 (JIP1) sensitises osteosarcoma to doxorubicin. Oncotarget. 2012;3:1169–1181.
- Kim J-Y, Kim J-K, Park J-S, et al. The use of PEGylated liposomes to prolong circulation lifetimes of tissue plasminogen activator. Biomaterials. 2009;30:5751–5756.
- Li Y-P, Pei Y-Y, Zhang X-Y, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J Control Release. 2001;71:203–211.
- Xiao W, Chen X, Yang L, et al. Co-delivery of doxorubicin and plasmid by a novel FGFR-mediated cationic liposome. Int J Pharm. 2010;393:120–127.
- Posthumadeboer J, Piersma SR, Pham TV, et al. Surface proteomic analysis of osteosarcoma identifies EPHA2 as receptor for targeted drug delivery. Br J Cancer. 2013;109:2142–2154.
- Nie Y, Ji L, Ding H, et al. Cholesterol derivatives based charged liposomes for doxorubicin delivery: preparation, in vitro and in vivo characterization. Theranostics. 2012;2:1092–1103.
- R, Peck J. Devore Statistics: The Exploration & Analysis of Data, 7th ed. Cengage Learning: Boston, 2011.