
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
To strengthen the anti-tumour efficacy and weaken the side effects, a nano targeted drug delivery system was constructed. The nanostructured lipid carriers (NLCs) were prepared by the melt-emulsification method. Modified with the octaarginine, thiolytic cleavable polyethylene glycol (PEG) and targeting peptide simultaneously on the surface, this multifunctional NLC could not only actively target to tumour tissues, but also control the cell penetration effect of the octaarginine easily by a safe reducing agent l-cysteine (l-Cys). In the present study, the pharmaceutical characteristics, the cytotoxicity and cellular uptake on NCI-H1299 cells in vitro, the biodistribution and targeting effect and anti-tumour ability in vivo were employed to evaluate the formulations. As the results revealed, various NLCs had a mean particle size of about 40 nm and a positive zeta potential of about 10 mV. The optimum density of cleavable PEG was confirmed as 10% and the best concentration of l-cysteine was determined as 20 mM via the qualitative and quantitative cellular uptake study. Based on these outcomes, the multiply decorated NLC manifested a great cell growth inhibition with the increased concentration of paclitaxel (PTX). Moreover, it preferred to accumulate at tumours, but not normal organs in vivo. Compared with Taxol®, this preparation demonstrated stronger anti-tumour efficacy and better security. Therefore, the multifunctional NLC can be considered as a promising drug delivery system targeting to tumours.
Introduction
Cancer is still one of the main causes of human death worldwide, ranking second to cardiovascular diseases [Citation1,Citation2]. Traditional treatments frequently cause serious side effects due to the toxicity of solvent system (ethanol, Cremophor EL and Tween-80) [Citation3]. To enhance the anti-tumour efficacy and simultaneously reduce the adverse effects, researchers have employed multifarious drug carriers, especially several nanocarriers such as nanostructured lipid carriers (NLCs), liposomes, drug-polymer conjugates, polymeric nanoparticles and polymeric micelles [Citation4–11]. The size of these nano preparations is usually between 20 nm and 200 nm, and this characteristic could lead to the accumulation of preparations in the tumour area, benefiting from the enhanced permeability and retention (EPR) effect [Citation12,Citation13]. However, the above drug delivery systems are lack of specificity, and are easy to be cleared via reticuloendothelial system (RES), which partly limits their application. In order to overcome these limitations, many researchers have adopted various functional moieties to modify the nanocarriers on the surface in recent years.
Pegylation is one kind of common decorated methods. This strategy can assist the nanocarriers in reducing the interactions with plasma proteins to obtain a “stealth” effect, and can extend in vivo circulation time by decreasing the clearance of RES [Citation14]. Nevertheless, the polyethylene glycol (PEG) also prevents the interaction between the nanocarriers and cell surface, which can influence the cellular uptake [Citation15]. To surmount this predicament, we hope that PEG remains on the surface during circulation but is separated from the surface when concentrating at tumour area. Therefore, the PEG which can be cleaved under particular conditions is in demand. A variety of ways have been reported about the cleavage of PEG, including pH sensitive [Citation16,Citation17], MMP sensitive [Citation18,Citation19], esterase sensitive [Citation20] or reductive potential sensitive chemical bonds [Citation14,Citation21]. And among these methods, constructing the reductive potential sensitive PEG is a good choice, because some redox sensitive chemical bonds such as disulfide bonds are easy to establish and can be cracked precisely by a secure reducing agent l-cysteine (l-Cys). It is reported that the concentration of reductive chemicals in extracellular tumour tissue is too low to cleave the disulfide linkages [Citation15], so the exogenous l-Cys is usually necessary.
Moreover, an ideal tumour targeted drug delivery system should not only accumulate drugs in tumour tissue but also deliver the drugs into tumour cells effectively [Citation15]. The cell penetrating peptides (CPPs) [Citation22] have captured our attention due to their excellent enhancement on cellular uptake [Citation23], and the octamer of arginine (R8), a member of the CPP family, is familiar in nowadays studies. Nevertheless, drug delivery systems modified with R8 or other CPPs could not distinguish malignant cells from the healthy ones, leading to the hidden danger in systemic administration. Their non-specificities are regarded as the fatal shortcoming. To overcome this drawback, one feasible strategy is to shield R8 by the thiolytic cleavable PEG. During circulation, positively charged R8 could not interact with the negatively charged cells of blood vessels because of the steric hindrance of PEG chains, and when nanocarriers appear in tumour area, R8 on the surface can be exposed to take effect via relieving from the masking of PEG in the presence of enough l-Cys, thus nonspecific binding is able to be avoided [Citation24]. In the process, we hold the view that the cleavable PEG is more like a switch and could response to different environments, which is defined as the “on-off” effect. Consequently, this tactic indirectly offsets the non-specificity of R8.
In addition, for further strengthening specific tumour cellular recognition and internalization, all kinds of active targeting moieties are used to modify the nanocarriers, such as peptides [Citation25,Citation26], folic acid [Citation27–29], hyaluronic acid [Citation30], etc. Among them, we pick out one type of peptide which could bind efficiently to epidermal growth factor receptor (EGFR) over-expressed tumour cells in vitro, since EGFR is proved as an effective target for therapeutic intervention in patients with cancer on basis of previous work [Citation31,Citation32]. This small peptide is named AEYLR (Ala–Glu–Tyr–Leu–Arg) which is derived from the C terminus of the EGFR [Citation33]. Therefore, the preparations with AEYLR could demonstrate more accumulation and uptake at tumour tissue.
In this study, the NLCs [Citation34] were chosen as the drug carriers because of their high physical stability and low toxicity [Citation35]. Furthermore, we explored surface density of the thiolytic cleavable PEG to achieve optimal screening effect for R8. Subsequently, the suitable concentration of l-Cys for breakage was also considered. On the basis of these consequences, cell penetrating peptide R8 and targeted peptide AEYLR were co-modified on the surface to construct a multifunctional NLC. The drug delivery system we developed was not merely decorated with a single moiety, but the cleavable PEG, R8 and AEYLR were put into use at the same time. Therefore, the multiply modified NLC could not only selectively target to tumours and penetrate tumour cells efficiently, but also manifest low toxic side effect and prolonged retention in vivo after systemic administration. The pharmaceutical properties like mean particle size and zeta potential were examined. Besides, the in vitro cytotoxicity and cellular uptake, including qualitative and quantitative ways on NCI-H1299 cells which were EGFR over-expressed were also assessed. And we mainly focused on selecting the appropriate density of cleavable PEG and deciding the concentration of l-Cys on cellular uptake. As for the in vivo researches, the active targeting effect of AEYLR was explored on tumour-bearing mice and DiR, one kind of fluorescent dye, was used to measure the in vivo distribution of preparations by the imaging technology. In addition, paclitaxel (PTX) was chose to be the model drug, and then in vivo, we employed S180 tumour-bearing mice to evaluate anti-tumour activity of the PTX-loaded multifunctional NLC.
Materials and methods
Materials
Stearyl-polyarginine (R8) and stearyl-PEG2000-AEYLR (AEYLR) with greater than 98% purity were synthesized by TeraBio Technology Co., Ltd. (Guangzhou, China). DOPE-PEG5000 and DOPE-S-S-PEG5000 were synthesized by Xi’an Ruixi Biological Technology Co., Ltd. (Xi’an, China). Glycerin monostearate (GMS), Kolliphor HS15 and Kolliphor ELP were generously presented by BAFS Co., Ltd. (Ludwigshafen, Germany). Medium chain triglyceride (MCT) was purchased from Yuhao Chemical Co., Ltd. (Hangzhou, China). Compritol 888 ATO was kindly donated by Gattefosse Co., Ltd., Shanghai, China. Coumarin 6 (Cou6) was purchased from J&K Scientific Ltd. (China). PTX was purchased from Jiangsu Hengrui Pharmaceutical Co., Ltd. (Jiangsu, China). DiR iodide (DiR) was purchased from Beyotime Co., Ltd. (Shanghai, China).
NCI-H1299 cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI-1640 medium (Hyclone Co., Ltd., Logan city, Utah; Thermo Fisher Scientific, Shanghai, China), supplemented with 10% foetal bovine serum (Sijiqing Co., Ltd., Hangzhou, China). The cells were maintained at 37 °C in a humidified incubator with 5% CO2. Besides, all the animal experiments were carried out in compliance with guidelines for the Care and Use of Laboratory Animals of Shenyang pharmaceutical university.
All other chemicals and reagents were of analytical or cell culture grade.
Methods
Preparation of NLCs
NLCs were prepared by the melt-emulsification method as described previously [Citation36]. Briefly, GMS, Compritol 888 ATO, MCT and Kolliphor ELP were mixed and heated under moderate stirring at 85 °C to get a transparent and uniform oil phase. Meanwhile, Kolliphor HS15 was dispersed in purified water at the same temperature to obtain the aqueous phase which was dropped into the oil phase to get the emulsion. After that, the emulsion was cooled immediately at 4 °C to get the blank NLC. To get the NLCs modified with different materials, such as R8, AEYLR, DOPE-PEG5000, DOPE-S-S-PEG5000 or several of these, the formulated materials were added into the oil phase, and the process was as above.
The NLC decorated with R8 was named R-NLC, and the one modified with AEYLR was named A-NLC. And the NLC decorated with DOPE-PEG5000 (non-cleavable PEG) was named P-NLC, and the one modified with DOPE-S-S-PEG5000 (cleavable PEG) was named PS-NLC. AEYLR and R8 dual-modified NLC was named AR-NLC. Similarly, the NLC co-decorated with DOPE-PEG5000, AEYLR and R8 was named PAR-NLC, and the one co-modified with DOPE-S-S-PEG5000, AEYLR and R8 was named PSAR-NLC. When the NLC was decorated with different PEG density, it was named n% P-NLC. As an example, the 10% PSAR-NLC represented that it was modified with 10% DOPE-S-S-PEG5000, AEYLR and R8 simultaneously.
Model drug PTX or fluorescent dye Cou6 was mixed with the oil phase when preparing PTX-loaded NLCs or Cou6-loaded NLCs.
Size and zeta potential
The mean particle size and zeta potential of various NLCs were determined using a Malvern Zetasizer Nano (Malvern Instruments Ltd., Malvern, UK). Prior to measurement, all the samples were diluted 10 times, and each measurement was made in triplicate.
Quantitative analysis of cellular uptake of various Cou6 loaded NLCs
Coumarin 6 was selected to be the fluorescent material which was loaded in various NLCs to investigate the cellular uptake. NCI-H1299 cells were employed in the in vitro cellular studies, and were cultured in RPMI-1640 medium supplemented with 10% FBS. The cells were trypsinised and passaged every other day at the ratio of 1:3. Besides, the safe reducing agent L-Cys was dissolved in Hepes buffer (pH 7.4) for further testing the breakage of disulfide bonds in cleavable PEG.
The cells were seeded in 24-well plates at a density of 1 × 105 cells/well. After cultivation for 24 h, the culture medium was then replaced by FBS free fresh RPMI-1640 medium containing various Cou6-loaded NLCs, and the concentration of Cou6 maintained at 2 μg/mL in each well. The Cou6-loaded PSAR-NLCs, including 2.5% PSAR-NLC, 5% PSAR-NLC, 10% PSAR-NLC and 20% PSAR-NLC, which were different at the density of cleavable PEG were pretreated with l-Cys in Hepes buffer (pH 7.4), and the final concentration of l-Cys was set at 0, 5, 10, 20 and 40 mM in each well respectively when these processed formulations were added. After incubation at 37 °C under 5% CO2 for 2 h, the culture medium was discarded and cells were washed three times with cold PBS. 100 μL RIPA Lysis Buffer (Beyotime, China) was added into each well to lyse the cells, and 25 μL lysate was used to assay the protein content using BCA Protein Assay Kit (Beyotime, China). The residue was measured by a Microplate Reader (Thermo Scientific, Shanghai, China) to determine the fluorescence intensity in each well. The intracellular Cou6 amount was calculated and divided by the protein content to standardize the results and obviate the error due to different cell amounts. Finally, the results were expressed as Cou6/pro.
Confocal laser scanning microscopy
NCI-H1299 cells were cultured on sterile cover slips at the density of 1 × 105 cells/well in 24-well plates till 80% confluence, then, the culture medium was abandoned, followed by addition of fresh FBS free RPMI-1640 culture medium containing different Cou6-loaded NLCs (2.5% PSAR-NLC, 5% PSAR-NLC, 10% PSAR-NLC or 10% PAR-NLC), and the concentration of Cou6 maintained at 2 μg/mL. The formulations were preprocessed with l-Cys in Hepes buffer (pH 7.4), and the final concentration of l-Cys was 0, 5, 10 and 20 mM in each well respectively when the treated formulations were added. Cells were washed three times with cold PBS after incubation for 2 h at 37 °C, then fixed by 4% paraformaldehyde for 20 min at room temperature, and the nuclei was stained using DAPI. The cells were observed by a confocal laser scanning microscopy (CLSM).
Cytotoxicity assay
Cytotoxicity of PTX-loaded NLCs was determined utilizing MTT assay. NCI-H1299 cells were cultured in the 96-well plate at a density of 1 × 104 cells/well and incubated overnight to allow the cells attachment. At the second day, the medium was replaced with 200 μL of fresh medium containing different PTX concentrations (0.07, 0.15, 0.30, 0.63, 1.25 and 2.50 μg/mL) of PTX-loaded NLC, AR-NLC or 10% PSAR-NLC (pretreated with l-Cys) for further incubation in a 5% CO2 atmosphere at 37 °C for 24 h. Besides, the cytotoxicity of various blank NLCs was also tested, so the replaced fresh medium included different concentrations (170, 680 and 2700 μg/mL) of blank x-NLC (NLC, R-NLC, A-NLC, AR-NLC, 10% P-NLC, 10% PS-NLC, 10% PAR-NLC or 10% PSAR-NLC). After incubation, the MTT (0.5 mg/mL) solution was added to each well and the plate was incubated for 4 h. After that, the culture medium was replaced by 150 μL DMSO to dissolve Formazan crystals and the absorbance was measured at 490 nm. Cells treated without any preparations were set to be the controlled group. The relative cell viability was calculated by the following formula:
In vivo distribution and targeting study
The in vivo distribution and tumour-targeting of different NLCs were assessed by tumour-bearing mice using the FX Pro in vivo imaging system (Carestream Health, Shanghai, China). The near infrared fluorescence dye DiR was used to label preparations. Tumour-bearing mice were established by injecting 0.2 mL S180 ascites cell suspension in the left flank of the mice, containing 1 × 106 S180 cells. Mice were subjected to treatment when the subcutaneous tumours approximately reached 200–300 mm3. Various DiR-loaded NLCs were injected intravenously through the tail vein into the tumour-bearing mice to monitor the tumour accumulation and distribution in vivo. Mice were photographed by the imaging system at 1, 4, 8, 12, 24 and 48 h post-injection (n ≥ 3), and were sacrificed by cervical dislocation. The organs and tumours were removed and photographed after 48 h post-injection.
In vivo anti-tumour efficacy
The anti-tumour efficacy in vivo was evaluated in S180 tumour-bearing mice. Briefly, the mice were subcutaneously injected in the left flank with 0.2 mL of cell suspension including 1 × 106 S180 cells, and then divided into three groups (n = 6) randomly. When the tumours reached about 200–300 mm3, each group of mice were treated with physiological saline, a 10 mg/kg dose of Taxol® and PASR-PTX-NLC via intravenous injection respectively every three days and after 4 h, Hepes buffer containing l-Cys (pH 7.4) was administrated by intratumour injection in each group. The tumour volume was measured by calliper every three days and calculated based on the equation (a × b2)/2, where a and b equalled the length and width of the tumour, respectively. And the mice were also weighed every three days during the experimental period. After 15 days, the mice were killed, and the tumours were removed, photographed and weighed.
Statistical analysis
Student’s t-test was used to perform the differences, and differences were considered statistically significant at p < .05. All values were reported as mean ± standard deviation (SD).
Results
Preparation and characterization of NLCs
The AR-NLC, PASR-NLCs, PAR-NLC and Cou6-loaded preparations were prepared by the melt-emulsification method. The mean particle size, polydispersity index (PDI) and zeta potential of various NLCs were all tested in thrice. The results were listed in . The mean particle size of the NLCs was all less than 50 nm, and a slight decrease was found because of the increased ratio of PEG, but the size increased a bit when Cou6 was loaded. Besides, the zeta potential of the NLCs mainly floated up and down at 10 mV. In particular, the positive value of zeta potential might due to the R8 peptide decorated on the surface, which was rich in positively charged arginine.
Table 1. Size, PDI and zeta potential of various NLCs.
Quantitative analysis of cellular uptake
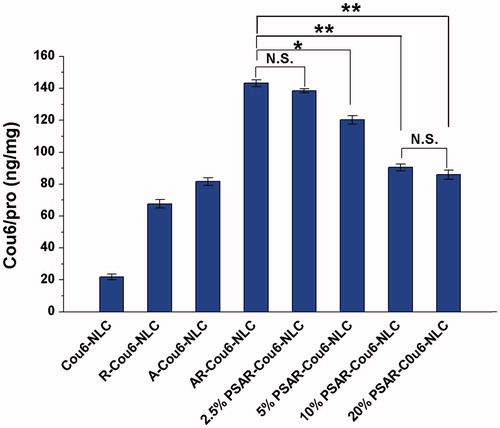
represents the cellular uptake of various NLCs on the EGFR positive NCI-H1299 cells. As it showed, the cellular uptake of R-Cou6-NLC, A-Cou6-NLC and AR-Cou6-NLC was three times, 3.7 times and 6.6 times higher than the uptake of Cou6-NLC, respectively. Obviously, a reducing tendency emerged when the density of cleavable PEG increased from 2.5% to 20%. When the density reached 10%, compared to AR-Cou6-NLC, the cellular uptake amount of PSAR-Cou6-NLC significantly decreased, which manifested this density had shielding effect (p < .01). Therefore, we took the attitude that the optimal density of cleavable PEG was 10% in the investigated range and this ratio could be used in the latter experiments.
Figure 1. The cellular uptake of various Cou6-loaded NLCs. The data are presented as the mean ± SD (n = 3). *p < .05; **p < .01; NS: not significant.
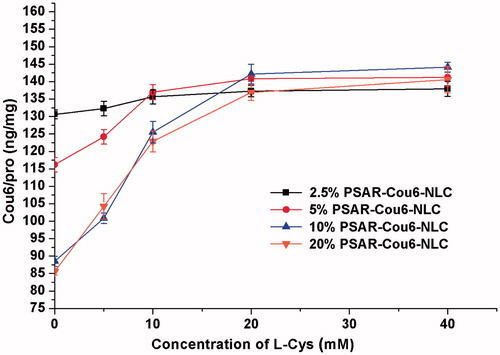
After confirming the density of cleavable PEG, a suitable concentration of l-Cys used to break the disulfide bonds was also considered. At the same PEG density, the cellular uptake quantity rose clearly with the increased concentration of l-Cys from 0 mM to 40 mM (). The more l-Cys, the less steric hindrance effect of PEG performed, and thus the “functional molecule” R8 was exposed, allowing the NLCs to enter tumour cells with high efficiency. When the concentration of l-Cys was 20 mM, there was a good exposure of R8 at different PEG density. So a 20 mM l-Cys was applied in the following studies.
Figure 2. The cellular uptake of PSAR-Cou6-NLCs modified with different PEG density in the presence of different l-Cys concentration. The data are presented as the mean ± SD (n = 3).
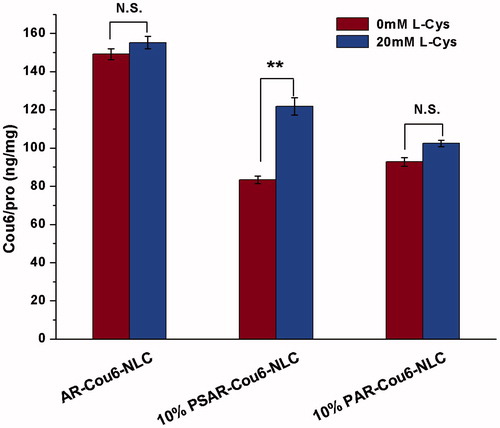
To further make sure whether l-Cys could influence the effects of AEYLR and R8 on cellular uptake, and whether the “on–off” effect was caused by the disulfide bond of cleavable PEG, we prepared AR-Cou6-NLC, 10% PSAR-Cou6-NLC and 10% PAR-Cou6-NLC to survey their uptake in the absence or in the presence of l-Cys (). As a result, there was no significant change after the addition of 20 mM l-Cys for AR-Cou6-NLC, and this proved that l-Cys was unable to disturb the behaviours of two small peptides. Moreover, 10% cleavable PEG modified AR-Cou6-NLC exhibited 1.5 times higher (p < .01) uptake efficiency after adding l-Cys, as compared to the uptake without l-Cys. However, 10% non-cleavable PEG modified formulation did not cause this result, which clearly demonstrated that the disulfide linkage was the key to producing the “on–off” effect.
Figure 3. The cellular uptake of Cou6-loaded AR-NLC, 10% PSAR-NLC and 10% PAR-NLC before and after the treatment of l-Cys. The data are presented as the mean ± SD (n = 3). **p < .01; NS: not significant.
Confocal laser scanning microscopy
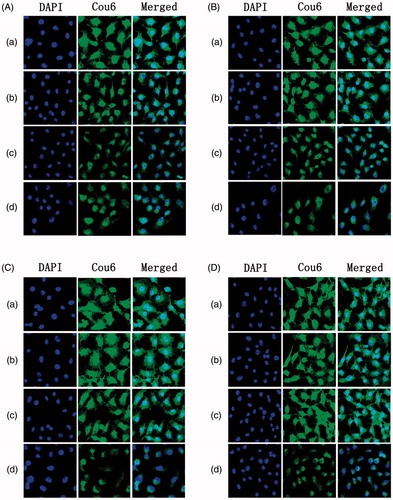
Besides the quantitative analysis, we also employed the CLSM to visualize the cellular uptake of different PEG density modified NLCs under the environment of various concentrations of l-Cys in order to demonstrate the shielding effect of PEG and the contribution to unmasking function of l-Cys. As shown from the CLSM images (), the green fluorescence (the fluorescence of Cou6) became gradually weak following the increase of PEG without l-Cys, which indicated that the shielding effect was caused by PEG due to its steric hindrance effect. When the PEG density was 10%, the fluorescence intensity was the weakest, so this density was suitable. For another, with the concentration of l-Cys varying from 0 mM to 20 mM (), different degrees of enhancement on the green fluorescence appeared in the preparations except the 10% PAR-NLC, and this phenomenon was in accordance with the consequence from above quantitative analysis. Therefore, the 20 mM was an applicative concentration for cleaving the PEG.
Figure 4. The CLSM images treated with Cou6-loaded 2.5% PSAR-NLC (a), 5% PSAR-NLC (b), 10% PSAR-NLC (c) and 10% PAR-NLC (d) in the presence of different l-Cys concentrations: (A) 0 mM, (B) 5 mM, (C) 10 mM and (D) 20 mM.
Cytotoxicity
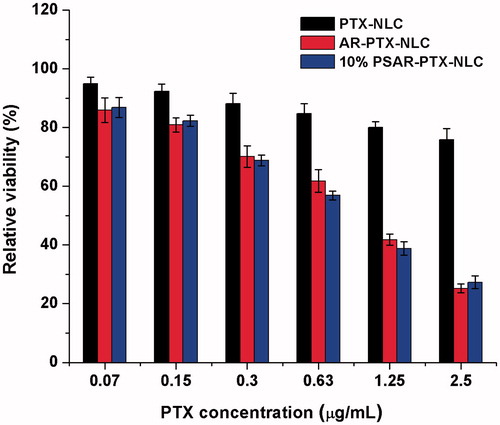
Cytotoxicity is an important index for the in vitro anti-tumour evaluation about drugs and preparations. In the present study, we employed PTX as the model drug to prepare the PTX-loaded NLC, AR-NLC and 10% PSAR-NLC (pretreated with l-Cys), and the cytotoxicities of them were evaluated after incubation with the NCI-H1299 cells for 24 h (). As a result, there was a reducing trend on relative cell viability in the three groups with the increase of concentration of PTX. Especially, AR-PTX-NLC and 10% PSAR-PTX-NLC (pretreated with l-Cys) exhibited stronger growth inhibition effects than PTX-NLC at the same PTX concentrations. When the PTX concentration was 2.5 μg/mL, the cellular viability of PTX-NLC was still as high as above 75%, while those in the other two groups were both under 30%. And the results were consistent with previous cellular uptake studies.
Figure 5. In vitro cytotoxicity of PTX-loaded NLC, AR-NLC and 10% PSAR-NLC. The data are presented as the mean ± SD (n = 6).
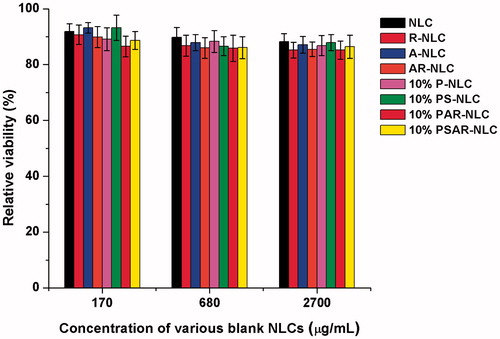
To further survey whether the various blank carriers we used could produce extra cytotoxicity, the blank NLCs (NLC, R-NLC, A-NLC,AR-NLC, 10% P-NLC, 10% PS-NLC, 10% PAR-NLC, 10% PSAR-NLC) were prepared and set the concentrations at 170, 680 and 2700 μg/mL, which were equivalent to PTX concentration at 0.15, 0.63 and 2.50 μg/mL, respectively. As shown in , the relative cell viability of blank NLCs was all above 85%, which indicated the blank nanocarriers and materials used for decorating caused little cytotoxicity. In conclusion, the depressing effect of preparations was achieved by the loaded PTX, but not by the cargo.
Figure 6. In vitro cytotoxicity of various blank NLCs. The data are presented as the mean ± SD (n = 6).
In vivo distribution and targeting study
To investigate whether the small peptide AEYLR could play an important role on improving the accumulation in tumour, the in vivo targeting abilities of the NLC, AR-NLC, 10% PAR-NLC and 10% PSAR-NLC were tested in S180 tumour bearing model. As shown in , for DiR-NLC treated group, the fluorescence in the tumour area appeared at 8 h post-administration. Meanwhile, the fluorescence intensity increased as time goes on, and reached the strongest at 24 h post-administration. Compared to DiR-NLC, DiR loaded AR-NLC not only performed its existence earlier at 4 h post-treatment, but also revealed much brighter fluorescence all the time even at 48 h post-treatment. The results indicated that AEYLR could efficiently enhance the targeting effect to tumour. In addition, in contrast with AR-DiR-NLC, two groups which were respectively treated with pegylated preparations (PAR-NLC and PSAR-NLC) did not show evident difference in the targeting behaviour, proving that this PEG density was suitable.
Figure 7. In vivo biodistribution of NLCs at 1, 4, 8, 12, 24 and 48 h after intravenous tail injection in tumour-bearing mice. (a) DiR-NLC, (b) AR-DiR-NLC, (c) 10% PAR-DiR-NLC and (d) 10% PSAR-DiR-NLC.

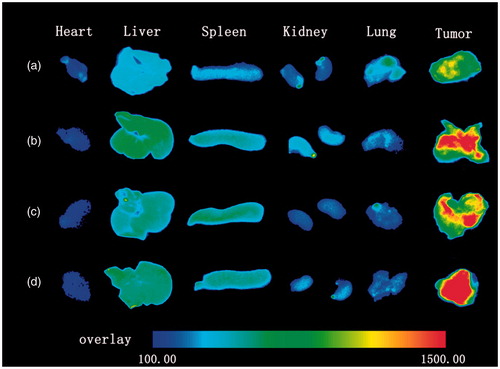
Moreover, according to , the DiR-NLC distributed little in organs and this confirmed the safety of nanocarriers. By contrast, although more fluorescence was observed in tumour by treating with the AR-DiR-NLC, but the accumulations increased in liver and spleen were undesired. In addition, after further modifying with PEG, what we noticed was the decreased fluorescence intensity in liver and most preparations still accumulated in tumour as compared to the AR-DiR-NLC. The results were probably attributed to the long circulation effect and shielding function of PEG.
Figure 8. Fluorescence imaging of major organs and tumour taken from tumour-bearing mice after 48 h post-injection of DiR-NLC (a), AR-DiR-NLC (b), 10% PAR-DiR-NLC (c) and 10% PSAR-DiR-NLC (d), respectively.
In vivo anti-tumour efficacy
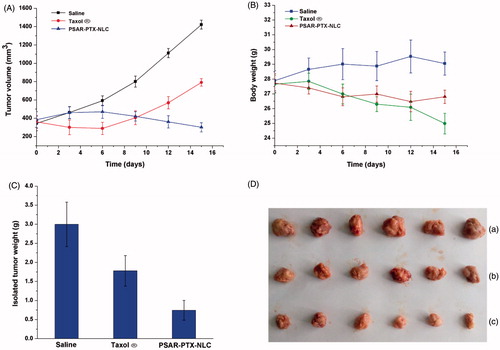
The in vivo anti-tumour activity of the PTX-loaded PSAR-NLC was investigated in S180 tumour bearing mice. From , the tumour volume grew rapidly and reached about 1400 mm3 on the 15th day in the control group (treated with saline), but compared to saline-treated group, the growth of tumours in mice treated with Taxol® and PSAR-PTX-NLC was evidently inhibited and the tumour volumes were respectively about 800 and 300 mm3 at last. Especially, the multiply modified formulation PSAR-PTX-NLC revealed a larger potential for inhibiting the increase of tumours at later period. Furthermore, the isolated tumour weight of mice () treated with PSAR-PTX-NLC was about 20 and 40% of those treated with saline and Taxol®, respectively, which was in agreement well with the results of tumour volume. In addition, the photograph in more intuitively indicated the in vivo anti-tumour efficacy in three groups. In respect to safety evaluation, the body weight of mice was monitored during the research (). The weight loss of mice was found both in Taxol® group and PSAR-PTX-NLC group. More than 11% of weight loss in the Taxol®-treated group was possibly due to the non-targeted characteristic and the toxicity of solvent system of formulations (ethanol and nonionic surfactant Tween-80). However, PTX-loaded PSAR-NLC showed less weight loss than Taxol®, suggesting that this preparation could decrease the systematic toxicity of PTX to ensure the security for therapy.
Figure 9. The in vivo anti-tumour study of PTX-loaded 10% PSAR-NLC in tumour-bearing mice. (A) Changes of tumour volume, (B) Body weight changes for the tumour-bearing mice, (C) The weights of the removed tumours were measured after the completion of the in vivo assays. The data are presented as the mean ± SD (n = 6). (D) Photograph of the solid tumours removed from different treatment groups at the study termination. (a) Saline, (b) Taxol® and (c) 10% PSAR-PTX-NLC.
Discussion
This study aimed at developing a nano drug delivery system that could not only target and penetrate tumour cells efficiently to deliver drugs, but also act on normal tissue as little as possible to avoid the potential toxicity. Therefore, the two small peptides AEYLR and R8 were introduced to modify the surface of NLCs. To decrease the non-selective damage on healthy cells, we chose the PEG to shield the R8 because of its steric hindrance effect. However, the PEG is a double-edged sword, which is beneficial to accumulate the NLCs at tumour tissue but unbeneficial for the uptake of the preparations by tumour cells [Citation15]. For solving this predicament, a thiolytic cleavable PEG was employed to decorate the NLCs, and it was just dissociated from the surface by exogenous l-Cys at tumour area. So this delivery system was easily controlled by the l-Cys to realize the “on–off” effect.
In order to obtain the optimal shielding effect on R8 and the maximum breakage of the cleavable PEG, the density of PEG and the concentration of l-Cys were determined by the cellular uptake qualitatively and quantitatively. As expected, the cellular uptake of the PSAR-NLC lowered with the increase of PEG, and when the PEG density was 10%, it seemed to have a satisfying result on shielding effect. Facing such results, we assumed that the PEG modified on the surface was a soft long-chain, and it could lead to a “stealth” behaviour on R8 [Citation37]. Thus, the 10% density of PEG was screened out to use subsequently. In addition, we adopted different concentrations of the l-Cys to cleave the DOPE-S-S-PEG5000 to examine the exposure of R8. A concentration of 20 mM l-Cys was an advisable selection, since this concentration could cleave the PEG with high efficiency.
To make certain whether the controlled manner only referred to the cleavable PEG and the l-Cys, we selected the NLC modified with the DOPE-PEG5000 without disulfide bond as a contrast. As a result, the cellular uptake of the PAR-NLC did not show noticeable enhancement in the presence of l-Cys, which indicated the disulfide bonds in the DOPE-S-S-PEG5000 was the pivotal matter. Besides, after the addition of l-Cys, there was no substantial change on the uptake of AR-NLC, implying no influence on the effects of AEYLR and R8.
After confirming the ratio of PEG and the concentration of l-Cys, we continued to evaluate the preparations via the cytotoxicity and other in vivo studies. Not surprisingly, the 10% PSAR-PTX-NLC pretreated with l-Cys revealed wonderful growth inhibition of tumour cells in vitro. Meanwhile, the safety of blank carriers was also proved by testing the cytotoxicity of various blank formulations. Accumulating as many drugs as possible in tumour tissue, but not in normal organs, is vital to an ideal tumour targeted drug delivery system. Consequently, we monitored the targeting effect and biodistribution of different NLCs in vivo. Depending on the EPR effect, the NLC remained in tumour tissue passively, and the weak fluorescence intensity appeared at 8 h post-treatment in the DiR-NLC group. Nevertheless, the preparations modified with the AEYLR showed stronger fluorescence brightness and appeared earlier at tumour site without exception, including AR-NLC, 10% PAR-NLC and 10% PSAR-NLC. And these were attributed to the active targeting of AEYLR. The receptor-mediated way made the “travel” in the body purposeful. Besides, we observed that the participation of the PEG didn’t impact the function of AEYLR, which further suggested this PEG density was suitable.
To our knowledge, although the environment of tumour area was more potentially reductive, the concentration of reducing agents was too low to cleave the disulfide bonds [Citation21]. Thus, the administration of exogenous and nontoxic l-Cys was indispensable. According to the results of in vivo distribution and targeting study, the accumulation of 10% PSAR-NLC was clearly observed at 4 h post-administration, so we chose 4 h as a desired time point to inject the l-Cys intratumorally. As for in vivo anti-tumour study, compared to the Taxol®, the 10% PSAR-PTX-NLC could more effectively control the development of tumours in vivo, benefiting from the synergistic potentiation of AEYLR and R8. And beyond that, the multifunctional NLC also manifested the higher security, perhaps due to the great selectivity towards tumours of this drug delivery system and the avoidance of toxic solvent system.
Conclusions
In the present study, we developed a multifunctional NLC simultaneously modified by AEYLR, R8 and DOPE-S-S-PEG5000 (PSAR-NLC), which could not only active target to tumour cells and enhance cellular uptake but also achieve “on–off” effect under the control of l-Cys. The cooperation of the cleavable PEG and cell-penetrating peptide R8 could overcome the disadvantage of pegylation and the non-specificity of R8, indirectly. Besides in vitro studies on cellular uptake and cytotoxicity which were widely used to predict the anti-tumour ability, researches on the distribution, targeting and anti-tumour efficacy in vivo were carried out. These findings suggested that the PSAR-NLC was able to be accumulated as many as possible in tumour tissue but not in normal organs, and the PTX-loaded preparation performed higher efficacy and safety than Taxol®. Therefore, the PSAR-NLC can be considered as a promising drug delivery system targeting to tumours.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Perez-Herrero E, Fernandez-Medarde A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur J Pharmaceut Biopharmaceut. 2015;93:52–79.
- Aberoumandi SM, Mohammadhosseini M, Abasi E, et al. An update on applications of nanostructured drug delivery systems in cancer therapy: a review. Artif Cells Nanomed Biotechnol. 2016. [Epub ahead of print]. DOI:10.1080/21691401.2016.1228658.
- Immordino ML, Brusa P, Arpicco S, et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release. 2003;91:417–429.
- Gao W, Xiang B, Meng TT, et al. Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials. 2013;34:4137–4149.
- Sawant RR, Torchilin VP. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012;14:303–315.
- Torchilin VP. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J. 2007;9:128–147.
- Lee E, Kim H, Lee IH, et al. In vivo antitumor effects of chitosan-conjugated docetaxel after oral administration. J Control Release. 2009;140:79–85.
- Cho HJ, Yoon HY, Koo H, et al. Self-assembled nanoparticles based on hyaluronic acid-ceramide (HA-CE) and pluronic(R) for tumor-targeted delivery of docetaxel. Biomaterials. 2011;32:7181–7190.
- Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Coll Surf B Biointerf. 2010;75:1–18.
- Lee SW, Yun MH, Jeong SW, et al. Development of docetaxel-loaded intravenous formulation, nanoxel-PM using polymer-based delivery system. J Control Release. 2011;155:262–271.
- Ostacolo L, Marra M, Ungaro F, et al. In vitro anticancer activity of docetaxel-loaded micelles based on poly(ethylene oxide)-poly(epsilon-caprolactone) block copolymers: do nanocarrier properties have a role?. J Control Release. 2010;148:255–263.
- Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151.
- Asadi N, Davaran S, Panahi Y, et al. Application of nanostructured drug delivery systems in immunotherapy of cancer: a review. Artif Cells Nanomed Biotechnol. 2017;45:18–23.
- Maeda T, Fujimoto K. A reduction-triggered delivery by a liposomal carrier possessing membrane-permeable ligands and a detachable coating. Coll Surf B Biointerf. 2006;49:15–21.
- Kuai R, Yuan W, Qin Y, et al. Efficient delivery of payload into tumor cells in a controlled manner by TAT and thiolytic cleavable PEG co-modified liposomes. Mol Pharmaceutics. 2010;7:1816–1826.
- Koren E, Apte A, Jani A, et al. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264–273.
- Sonawane SJ, Kalhapure RS, Govender T. Hydrazone linkages in pH responsive drug delivery systems. Eur J Pharmaceut Sci. 2016;99:45–65.
- Mok H, Bae KH, Ahn CH, et al. PEGylated and MMP-2 specifically dePEGylated quantum dots: comparative evaluation of cellular uptake. Langmuir. 2009;25:1645–1650.
- Hatakeyama H, Akita H, Kogure K, et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14:68–77.
- Xu H, Deng Y, Chen D, et al. Esterase-catalyzed dePEGylation of pH-sensitive vesicles modified with cleavable PEG-lipid derivatives. J Control Release. 2008;130:238–245.
- McNeeley KM, Karathanasis E, Annapragada AV, et al. Masking and triggered unmasking of targeting ligands on nanocarriers to improve drug delivery to brain tumors. Biomaterials. 2009;30:3986–3995.
- Raucher D, Ryu JS. Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol Med. 2015;21:560–570.
- Mussa Farkhani S, Asoudeh Fard A, Zakeri-Milani P, et al. Enhancing antitumor activity of silver nanoparticles by modification with cell-penetrating peptides. Artif Cells Nanomed, Biotechnol. 2016. [Epub ahead of print]. DOI:10.1080/21691401.2016.1200059.
- Vives E, Schmidt J, Pelegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim Biophys Acta. 2008;1786:126–138.
- Yuan H, Luo K, Lai Y, et al. Novel poly(l-glutamic acid) dendrimer based drug delivery system with both pH-sensitive and targeting functions. Mol Pharmaceutics. 2010;7:953–962.
- Gao Y, Wang ZY, Zhang J, et al. RVG-peptide-linked trimethylated chitosan for delivery of siRNA to the brain. Biomacromolecules. 2014;15:1010–11018.
- Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2012;64:342–352.
- Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13:256–262.
- Wang F, Wang Y, Ma Q, et al. Development and characterization of folic acid-conjugated chitosan nanoparticles for targeted and controlled delivery of gemcitabinein lung cancer therapeutics. Artif Cells Nanomed Biotechnol. 2016. [Epub ahead of print]. DOI:10.1080/21691401.2016.1260578.
- Park JH, Cho HJ, Yoon HY, et al. Hyaluronic acid derivative-coated nanohybrid liposomes for cancer imaging and drug delivery. J Control Release. 2014;174:98–108.
- Han C, Li Y, Sun M, et al. Small peptide-modified nanostructured lipid carriers distribution and targeting to EGFR-overexpressing tumor in vivo. Artif Cells Nanomed Biotechnol. 2014;42:161–166.
- Grünwald V, Hidalgo M. Developing inhibitors of the epidermal growth factor receptor for cancer treatment. J Natl Cancer Inst. 2003;95:851–867.
- Han CY, Yue LL, Tai LY, et al. A novel small peptide as an epidermal growth factor receptor targeting ligand for nanodelivery in vitro. Int J Nanomed. 2013;8:1541–1549.
- Jaiswal P, Gidwani B, Vyas A. Nanostructured lipid carriers and their current application in targeted drug delivery. Artif Cells Nanomed Biotechnol. 2016;44:27–40.
- Fernandes RS, Silva JO, Monteiro LO, et al. Doxorubicin-loaded nanocarriers: a comparative study of liposome and nanostructured lipid carrier as alternatives for cancer therapy. Biomed Pharmacother. 2016;84:252–257.
- Zhang W, Liu J, Zhang Q, et al. Enhanced cellular uptake and anti-proliferating effect of chitosan hydrochlorides modified genistein loaded NLC on human lens epithelial cells. Int J Pharm. 2014;471:118–126.
- Jokerst JV, Lobovkina T, Zare RN, et al. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond). 2011;6:715–728.