
Abstract
The glutathione transferases (GSTs) are a family of widely distributed Phase II detoxification enzymes. GST P1-1 is frequently overexpressed in rat and human tumours. It is suggested that overexpression of hGST P1-1 by human tumor cells may play a role in resistance to cancer chemotherapy. Hence, hGST P1-1 can be a promising target for cancer treatment. In this study, new hGST P1-1 inhibitors, 2-(4-substitutedphenyl/benzyl)-5-(4-trifluoromethylphenylsulphonamido) benzoxazole derivatives (Va–Vk) have been designed and synthesized. Surprisingly, in vitro hGST P1-1 enzyme inhibition studies demonstrated that all of the tested compounds except Vj had better activity than the reference drug EA and it is also correlated with the docking results. Additionally we compared the interactions with hGST P1-1 enzyme of newly synthesized compound Vh (bearing CF3 group) and previously synthesized compound 5f (bearing NO2 group). According to the docking results, compound Vh bound to the hGST P1-1 enzyme with a higher affinity compared to 5f. Therefore, we can consider that these data make a sense and can explain its higher activity. The compounds that obtained from this research could be used as scaffolds in design of new potent hGST P1-1 inhibitors useful in the treatment of the resistance of cancer chemotherapy.
Graphical Abstract
Introduction
The glutathione transferases (GSTs) are a family of widely distributed Phase II detoxification enzymes that catalyse the conjugation of a broad variety of reactive electrophiles to the nucleophilic sulphur atom of tripeptide glutathione (ɣ-l-glutamyl-l-cysteinylglycine, GSH) [Citation1,Citation2]. GSTs catalyze many other types of reaction such as isomerization and reduction involved in many biological processes (steroid and prostaglandin biosynthesis, tyrosine catabolism, and cell apoptosis). There are three distinct super-family members of GSTs in mammals: the membrane microsomal, mitochondrial, and cytosolic forms. Cytosolic ones were classified into seven classes, Alpha, Mu, Omega, Pi, Sigma, Theta, and Zeta, which have a promiscuous substrate specificity and are localized in different tissues with organ-specific expression patterns [Citation1,Citation3].
GSTs are soluble proteins and their masses are around 50 kDa, each composed of two polypeptide subunits. Each subunit contains an independent catalytic site composed of two components. One of them is G site, formed from a conserved group of amino acid residues in the amino terminal domain of the protein and specific for GSH or a closely related homolog. The other one is H site, binds the hydrophobic substrate which is formed largely from residues in the carboxy terminal domain and is much more structurally variable [Citation4].
Human GST P1-1 (hGST P1-1) is the most prevalent isoform of the mammalian cytosolic GSTs. GST P1-1 is frequently overexpressed in rat and human tumours, including carcinoma of the colon, lung, kidney, ovary, pancreas, oesophagus, and stomach. It is suggested that overexpression of hGST P1-1 by human tumor cells may play a role in resistance to anticancer drugs such as adriamycine, cisplatin, melphalan, and etoposide; especially if it is associated with overexpression of the cell membrane GSH conjugate transporters multidrug resistance protein MRP1 and/or MRP2 [Citation5–7]. Additionally, they can inhibit the Jun N-terminal kinase that is part of the apoptosis control system and they are also able to bind non-catalytically a wide range of endogenous and exogenous ligands. hGST P1-1 defends tumor cells by direct detoxification of anticancer drugs and by blocking apoptosis through its effects on JNK kinase [Citation8,Citation9]. Because of these functions, hGST P1-1 can be a promising target for inhibition in cancer treatment and considerable efforts have been undertaken to find specific inhibitors of hGST P1-1.
An approved drug, ethacrynic acid (EA, [2,3-dichloro-4-(2-methylenebutyryl)phenoxy]acetic acid), inhibits hGST P1-1 by binding directly to the substrate binding site (H site) of the enzyme, as well as by depleting its cofactor (GSH) via conjugation by way of Michael addition to the thiol group of GSH [Citation10,Citation11]. It is reported that sulphonamide derivatives of various compounds serve as H site substrates for GSTs [Citation12]. GSTs can display a sulphonamidase activity by catalyzing the GSH-mediated hydrolysis of sulphonamide bonds [Citation13,Citation14].
Recently, we reported some sulphonamidobenzoxazole derivatives, which exhibit hGST P1-1 enzyme inhibitory activities, and one of these compounds showed potency similar to the reference drug EA [Citation15,Citation16]. As an ongoing research, herein we report the design and synthesis of new 2-substituted-5-(4-trifluoromethylphenylsulphonamido)-benzoxazole derivatives, which may lead to the development of new potent inhibitors of the hGST P1–1 enzyme.
Materials and methods
Molecular docking
The crystal structure of the hGST P1-1, complexed with GSH, was extracted from the Protein Data Bank (pdb ID: 6GSS) [Citation17]. For preparation of protein and ligands Accelrys Discovery Studio 3.5, software was used [Citation18]. Crystal structure of hGST P1-1 was taken, hydrogens were added and optimized using the all atom CHARMm forcefield and the Adopted Basis set Newton–Raphson method until the root mean deviation (RMS) gradient was <0.05 kcal/mol/Å2. The minimized protein was defined as the receptor using the binding site module. The binding site was defined from the cavity finding method and modified which contain all of the important active site residues in the H site of the enzyme. Binding sphere was selected from the active site using the binding site tools (6.64, 3.67, 26.88, and 9.312). Novel synthesized benzoxazole derivatives, previously synthesized compound 5f and the reference drug EA were sketched; all atom CHARMm forcefield parameterization was assigned and minimized as described above. Conformational searches of the ligands were carried out using a simulated annealing molecular dynamics (MD) approach. The ligands were heated to a temperature of 700 K and then annealed to 200 K. CDocker method was performed by using Discovery Studio 3.5 [Citation19]. The hGST P1-1 enzyme was held rigid but the ligands were allowed to be flexible during refinement. Molecular docking studies were performed and all docked poses were scored by applying the Analyze Ligand Poses subprotocol. Binding energies were calculated by using in situ ligand minimization step (ABNR method) and using implicit solvent model (GBMV) in Discovery Studio 3.5. The lowest binding energies were taken as the best-docked conformation of the compound to the macromolecule. The pictures were taken by using Discovery Studio 4.1 visualizer.
Chemistry
All solvents and chemicals were purchased from commercial vendors and used without purification. The progress of the reaction was monitored on readymade silica gel plates (Merck, Darmstadt, Germany). The melting points were measured with a capillary melting point apparatus (Buchi B540, Essen, Germany) and are uncorrected. Yields were calculated after recrystallization. The 1H NMR and 13 C NMR spectra were recorded employing a VARIAN Mercury 400 MHz FT spectrometer, chemical shifts (d) are in parts per million relative to TMS, and coupling constants (J) are reported in hertz. Mass spectra were taken on a Waters Micromass ZQ using the ESI method (Waters Corp., Milford, MA). Elemental analyses were performed by Leco CHNS-932 CHNS-O analyzer. The results of the elemental analyses (C, H, N, S) were within ±0.4% of the calculated amounts.
General procedure for the preparation of 2-(4-substitutedphenyl/benzyl)-5-amino-benzoxazoles (IIIa–IIIk): The derivatives were synthesized by heating 2,4-diaminophenol dihydrochloride (I) (0.01 mol) with suitable acid (IIa–IIk) (0.01 mol) in polyphosphoric acid (PPA) (24 g) and stirring at 170–200 °C for 1.5–2.5 h. At the end of the reaction period, the residue was poured into an ice-water mixture and neutralized with an excess of 10% solution of NaOH. The residue was filtered and boiled with charcoal (200 mg) in ethanol and filtered. After the evaporation of solvent in vacuo, the crude product was obtained and recrystallized from ethanol–water (1:3) mixture (Scheme 1) [Citation20].
Scheme 1. Synthetic pathway of 5-(4-trifluoromethylphenylsulphonamido)benzoxazole derivatives. Reagents and conditions: (A) PPA, 170–200 °C, 1.5–2.5 h; (B) Pyridine and dichloromethane, RT, 16 h.
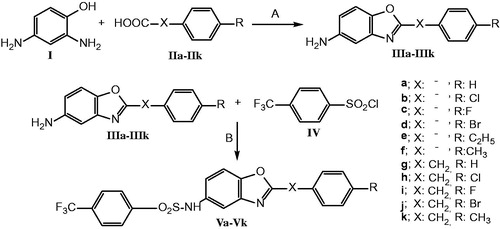
General procedure for the preparation of 5-(4-nitrophenylsulphonamido)-benzoxazole derivatives (Va–Vk): To a solution of 2-(4-substitutedphenyl/benzyl)-5-aminobenzoxazole (IIIa–IIIk) (0.048 mmol), in dichloromethan (2 ml), pyridine (0.95 mmol) and 4-trifluoromethylbenzenesulphonyl chloride (IV) (0.52 mmol) was added and the reaction mixture was stirred at room temperature for 16 h. At the end of the reaction, the residue was filtered and washed with saturated solution of CuSO4 and NaHCO3 in water [Citation21]. Then, recrystallized with a mixture of ethyl acetate/n-hexan (1:5). The crystals were dried in vacuo (Scheme 1). All of the compounds are new.
2-Phenyl-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Va): 30.3% yield; mp 217–218 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 7.13 (1H, dd, J = 8.4 Hz, J = 2.0 Hz, H-6), 7.50 (1H, d, J = 2.0 Hz, H-4), 7.56–7.62 (2H, m, H-4′, H-7), 7.68 (2H, d, J = 8.8 Hz, H-3′, H-5′), 7.95 (4H, q, H-2′, H-6′, H-3′′, H-5′′), 8.13 (2H, dd, J = 8.0 Hz, J = 1.6 Hz, H-2′′, H-6′′), 10.62 (1H, s, NH); 13C NMR (DMSO-d6, 100 MHz): δ = 111.43, 112.29, 119.64, 123.31 (JC-F = 271.2 Hz), 126.19, 126.53 (JC-F = 3.2 Hz), 127.27, 127.73, 129.28, 132.09, 132.53 (JC-F = 32 Hz), 134.14, 141.96, 143.08, 147.61, 163.35; MS (ESI) m/z 419.27 [M + H]+ (100); Anal. Calcd. for C20H13F3N2O3S: C, 57.42; H, 3.13; N, 6.70; S, 7.66. Found: C, 57.33; H, 3.41; N, 6.87; S, 7.60.
2-(4-Chlorophenyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vb): 41.2 yield; mp 233–235 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 7.14 (1H, dd, J = 8.8 Hz, J = 2.0 Hz, H-6), 7.50 (1H, d, J = 2.0 Hz, H-4), 7.64–7.67 (2H, m, H-3′, H-5′), 7.68 (1H, d, J = 9,2 Hz, H-7), 7.92–7.97 (4H, m, H-2′, H-6′, H-3′′, H-5′′), 8.11–8.15 (2H, m, H-2′′, H-6′′), 10.63 (1H, s, NH); 13 C NMR (DMSO-d6, 100 MHz): δ = 111.53, 112.28, 119.82, 123.30 (JC-F = 271.9 Hz), 124.98, 126.55 (JC-F = 3.2 Hz), 127.72, 129.02, 129.47, 132.55 (JC-F = 32.7 Hz), 134.18, 136.89, 141.87, 142.99, 147.64, 162.44; MS (ESI) m/z 453.29 [M + H]+ (100), 455.26 [M + H + 2]+ (35); Anal. Calcd. for C20H12ClF3N2O3S: C, 53.05; H, 2.67; N, 6.19; S, 7.08. Found: C, 53.17; H, 2.88; N, 6.48; S, 7.03.
2-(4-Fluorophenyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vc): 31.4% yield; mp 228–229 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 7.12 (1H, dd, J = 8.4 Hz, J = 2.0 Hz, H-6), 7.43 (2H, t, H-3′, H-5′), 7.49 (1H, d, J = 2.4 Hz, H-4), 7.68 (1H, d, J = 8.8 Hz, H-7), 7.95 (4H, t, J = 9.6 Hz, H-2′, H-6′, H-3′′, H-5′′), 8.19 ppm (2H, q, H-2′′, H-6′′), 10.62 (1H, s, NH); 13 C NMR (DMSO-d6, 100 MHz): δ = 111.44, 112.45, 116.54 (JC-F = 22 Hz), 119.60, 122.78 (JC-F = 3.2 Hz), 123.31 (JC-F = 271.3 Hz), 126.55 (JC-F = 3.2 Hz), 127.72, 129.95 (JC-F = 9.6 Hz), 132.54 (JC-F = 32.1 Hz), 134.12, 141.92, 143.01, 147.65, 162.54, 164.30 (JC-F = 248.7 Hz); MS (ESI) m/z 437.44 [M + H]+ (100); Anal. Calcd. for C20H12F4N2O3S: C, 55.05; H, 2.77; N, 6.42; S, 7.35. Found: C, 54.96; H, 3.03; N, 6.62; S, 7.22.
2-(4-Bromophenyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vd): 43.5% yield; mp 245–246 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 7.14 (1H, dd, J = 8.8 Hz, J = 2.0 Hz, H-6), 7.50 (1H, d, J = 2.0 Hz, H-4), 7.67 (1H, d, J = 8.8 Hz, H-7), 7.80 (2H, d, J = 8.8 Hz, H-3′, H-5′), 7.95 (4H, s, H-2′, H-6′, H-3′′, H-5′′), 8.05 (2H, d, J = 8.8 Hz, H-2′′, H-6′′), 10.63 (1H, s, NH); 13C NMR (DMSO-d6, 100 MHz): δ = 111.54, 112.28, 119.85, 123.31 (JC-F = 271.3 Hz), 125.32, 125.85, 126.56 (JC-F = 3.9 Hz), 127.72, 129.15, 132.40, 132.70, 134.22, 141.87, 143.01, 147.63, 162.56; MS (ESI) m/z 497.31 [M + H]+ (100), 499.24 [M + H + 2]+ (98); Anal. Calcd. for C20H12BrF3N2O3S. 0.1 H2O: C, 48.13; H, 2.46; N, 5.61; S, 6.42. Found: C, 47.78; H, 2.60; N, 5.86; S, 6.49.
2-(4-Ethylphenyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Ve): 51.2% yield; mp 193–195 °C; 1H NMR (CDCl3, 400 MHz): δ = 1.28 (3H, t, CH3), 2.73 (2H, q, CH2), 7.12 (1H, dd, J = 8.4 Hz, J = 2.0 Hz, H-6), 7.34 (2H, d, J = 8.4 Hz, H-3′, H-5′), 7.46 (2H, t, H-4, H-7), 7.69 (2H, d, J = 8.4 Hz, H-2′, H-6′), 7.90 (2H, d, J = 8.0 Hz, H-3′′, H-5′′), 8.11 (2H, d, J = 8.4 Hz, H-2′′, H-6′′); 13C NMR (CDCl3, 100 MHz): δ = 15.14 (CH3), 28.96 (CH2), 111.10, 114.56, 120.82, 120.93, 123.07 (JC-F = 271.9 Hz), 123.87, 126.26 (JC-F = 3.9 Hz), 127.84, 128.57, 132.38, 134.72 (JC-F = 33.3 Hz), 142.34, 142.69, 148.98, 149.02, 164.78; MS (ESI) m/z 447.33 [M + H]+ (100); Anal. Calcd. for C22H17F3N2O3S: C, 59.19; H, 3.84; N, 6.27; S, 7.18. Found: C, 59.10; H, 4.11; N, 6.52; S, 7.24.
2-(4-Methylphenyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vf): 54.7% yield; mp 230–232 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.38 (3H, s, CH3), 7.10 (1H, dd, J = 8.8 Hz, J = 2.4 Hz, H-6), 7.39 (2H, d, J = 8.0 Hz, H-3′, H-5′), 7.47 (1H, d, J = 2.4 Hz, H-4), 7.66 (1H, d, J = 8.4 Hz, H-7), 7.92-7.97 (4H, m, H-2′, H-6′, H-3′′, H-5′′), 8.02 (2H, d, J = 8.4 Hz, H-2′′, H-6′′), 10.60 (1H, s, NH); 13C NMR (DMSO-d6, 100 MHz): δ = 21.59 (CH3), 111.78, 112.66, 119.89, 123.79 (JC-F = 271.3 Hz), 123.84, 127.01 (JC-F = 3.8 Hz), 127.72, 128.19, 130.33, 133.00 (JC-F = 32 Hz), 134.46, 142.49, 142.84, 143.49 (JC-F = 1.5 Hz), 148.01, 164.01; MS (ESI) m/z 433.37 [M + H]+ (100); Anal. Calcd. for C21H15F3N2O3S: C, 58.33; H, 3.50; N, 6.48; S, 7.41. Found: C, 58.07; H, 3.68; N, 6.73; S, 7.41.
2-Benzyl-5-(4-trifluoromethylphenylsulphonamido)-benzoxazole (Vg): 50.2% yield; mp 132–133 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 4.28 (2H, s, CH2), 7.03 (1H, dd, J = 8.4 Hz, J = 2.4 Hz, H-6), 7.26–7.34 (5H, m, phenyl′), 7.38 (1H, d, J = 2.0 Hz, H-4), 7.55 (1H, d, J = 9.2 Hz, H-7), 7.92 (4H, s, phenyl′′), 10.53 (1H, s, NH); 13C NMR (DMSO-d6, 100 MHz): δ = 34.10 (CH2), 111.06, 112.32, 119.12, 123.31 (JC-F = 271.2 Hz), 126.52 (JC-F = 3.9 Hz), 127.06, 127.68, 128.63, 129.06, 132.34 (JC-F = 32.1 Hz), 133.58, 134.88, 141.34, 143.02, 147.79, 166.62; MS (ESI) m/z 433.31 [M + H]+ (100); Anal. Calcd. for C21H15F3N2O3S: C, 58.33; H, 3.50; N, 6.48; S, 7.41. Found: C, 58.06; H, 3.64; N, 6.66; S, 7.46.
2-(4-Chlorobenzyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vh): 58.8% yield; mp 161–163 °C; 1H NMR (CDCl3, 400 MHz): δ = 4.20 (2H, s, CH2), 7.04–7.08 (2H, m, H-3′, H-5′), 7.26–7.32 (3H, m, H-6, H-2′, H-6′), 7.36 (1H, d, J = 8.4 Hz, H-7), 7.40 (1H, d, J = 2.4 Hz, H-4), 7.68 (2H, d, J = 8.4 Hz, H-3′′, H-5′′), 7.86 (2H, d, J = 8.0 Hz, H-2′′, H-6′′); 13C NMR (CDCl3, 100 MHz): δ = 34.57 (CH2), 111.11, 114.83, 120.92, 123.05 (JC-F = 271.8 Hz), 126.27 (JC-F = 3.9 Hz), 127.79, 129.05, 130.40, 132.18, 132.62, 133.54, 134.73 (JC-F = 32.4 Hz), 141.95, 142.33 (JC-F = 1.3 Hz), 149.35, 166.31; MS (ESI) m/z 467.38 [M + H]+ (100), 469.41 [M + H + 2]+ (36); Anal. Calcd. for C21H14ClF3N2O3S: C, 54.03; H, 3.02; N, 6.00; S, 6.87. Found: C, 54.02; H, 3.15; N, 6.30; S, 6.74.
2-(4-Fluorobenzyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vi): 64.4% yield; mp 146–147 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 4.26 (2H, s, CH2), 7.02 (1H, dd, J = 8.4, J = 2.0, H-6), 7.11–7.15 (2H, m, H-3′, H-5′), 7.35–7.38 (3H, m, H-4, H-2′, H-6′), 7.52 (1H, d, J = 8.4 Hz, H-7), 7.89 (4H, s, phenyl′′); 13C NMR (DMSO-d6, 100 MHz): δ = 33.18 (CH2), 111.04, 112.27, 115.33 (JC–F = 21.8 Hz), 119.11, 123.28 (JC–F = 271.3 Hz), 126.48 (JC–F = 3.9 Hz), 127.35, 127.67, 131.04 (JC-F = 8.3 Hz), 132.50 (JC–F = 32.1 Hz), 133.59, 141.29, 143.02, 147.75, 161.29 (JC–F = 241 Hz), 166.51; MS (ESI) m/z 451.34 [M + H]+ (100); Anal. Calcd. for C21H14F4N2O3S. 0.1H2O: C, 55.78; H, 3.17; N, 6.20; S, 7.09. Found: C, 55.56; H, 3.37; N, 6.41; S, 7.20.
2-(4-Bromobenzyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vj): 54.6% yield; mp 168–170 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 4.27 (2H, s, CH2), 7.02 (1H, dd, J = 8.8 Hz, J = 2.4 Hz, H-6), 7.29 (2H, d, J = 8.4 Hz, H-3′, H-5′), 7.36 (1H, d, J = 2.0 Hz, H-4), 7.50–7.55 (3H, m, H-7, H-2′, H-6′), 7.87–7.92 (4H, m, phenyl′′), 10.52 (1H, s, NH); 13C NMR (DMSO-d6, 100 MHz): δ = 33.25 (CH2), 109.47, 111.03, 112.23, 119.10, 120.25, 123.26 (JC-F = 271.2 Hz), 126.47 (JC-F = 3.8 Hz), 127.62, 131.40 (JC-F = 6.1 Hz), 132.44 (JC-F = 32.8 Hz), 133.57, 134.25, 141.22, 142.97, 147.71, 166.17; MS (ESI) m/z 511.13 [M + H]+ (100), 513.21 [M + H + 2]+ (97); Anal. Calcd. for C21H14BrF3N2O3S: C, 49.33; H, 2.76; N, 5.48; S, 6.27. Found: C, 49.13; H, 2.89; N, 5.78; S, 6.27.
2-(4-Methylbenzyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole (Vk): 65.4% yield; mp 139–141 °C; 1H NMR (DMSO-d6, 400 MHz): δ = 2.26 (3H, s, CH3), 4.22 (2H, s, CH2), 7.03 (1H, dd, J = 8.8 Hz, J = 2.0 Hz, H-6), 7.13 (2H, d, J = 8.0 Hz, H-3′, H-5′), 7.22 (2H, d, J = 8.0 Hz, H-2′, H-6′), 7.38 (1H, d, J = 2.0 Hz, H-4), 7.54 (1H, d, J = 8.4 Hz, H-7), 7.89–7.94 (4H, m, phenyl′′), 10.52 (1H, s, NH); 13 C NMR (DMSO-d6, 100 MHz): δ = 20.59 (CH3), 33.72 (CH2), 111.03, 112.32, 119.10, 123.31 (JC-F = 271.2 Hz), 126.51 (JC-F = 3.3 Hz), 127.68, 128.91, 129.17, 131.79, 132.50 (JC–F = 32.1 Hz), 133.53, 136.20, 141.35, 143.01, 147.79, 166.80; MS (ESI) m/z 447.31 [M + H]+ (100); Anal. Calcd. for C22H17F3N2O3S. 0.3H2O: C, 58.48; H, 3.93; N, 6.20; S, 7.10. Found: C, 58.10; H, 4.00; N, 6.30; S, 7.43.
In vitro hGST P1–1 inhibitory activity
Expression and purification of human GST P1-1
Recombinant hGST P1-1 was expressed in Escherichia coli strain XL-1 Blue at 37 °C and purified by using S-hexylglutathione-Sepharose 6B [Citation22]. Escherichia coli XL-1 cells containing pKXHP1 plasmid were grown overnight in 50 ml 2YT media (16 g tryptone, 10 g yeast extract, 5 g NaCl, and 100 mg/liter ampicillin) and then transferred to 500 ml 2YT media and incubated at 37 °C in the shake incubator. Incubation was kept until the absorbance of the culture at 600 nm was 0.2–0.4. Then 0.2 mM isopropyl-β-d-thiogalactopyranoside was added to induce the expression of hGST P1–1. The cells were further grown for 16 h and then centrifuged at 1500 g for 10 min. The pellets were kept at –80 °C for 30 min. The pellets were resuspended in lysis buffer (10 mm Tris HCl, 1 mm EDTA, 0.2 mm dithiothreitol (DTT) pH 7.0 and protease inhibitor cocktail, 0.2 mg/ml lysozyme) and mixed gently on ice for 30 min and then disrupted by sonication five times for 20 s. Phenylmethanesulphonyl fluoride (170 μm) was added and the supernatant fraction was obtained by centrifugation at 15,000 g, 4 °C for 1 h.
An affinity matrix consisting of S-hexylglutathione immobilized on epoxy-activated-Sepharose 6B was used for purification of hGST P1-1 [Citation23]. The supernatant fraction was applied to the matrix equilibrated with binding buffer, and stirred gently for 40 min on ice. The matrix containing bound hGST P1-1 was washed with Buffer A (10 mm Tris HCl, pH 7.8, 1 mm EDTA, 0.2 m NaCl, 0.2 mm DTT) to eliminate non-bound proteins. The matrix was packed on top of a Sephadex G-25 column equilibrated with Buffer A in the cold room. The enzyme was then eluted with Buffer B (10 mm Tris HCl, pH 7.8, 1 mm EDTA, 0.2 m NaCl, 0.2 mm DTT, 5 mm S-hexylglutathione). The fractions containing the GST activity were concentrated on ice and then dialyzed with Buffer A without NaCl. The purity of the enzyme was validated by SDS-PAGE applying both optimal and excessive protein amounts for analysis to visualize possible impurities.
hGST P1-1 in vitro inhibition assay
hGST P1-1 in vitro inhibitory activity of the synthesized sulphonamidobenzoxazoles was measured on a Shimadzu UV-2501 PC spectrophotometer (Shimadzu Corp., Kyoto, Japan) by measuring the initial rate of absorbance change at 340 nm with CDNB. Standard enzymatic assay conditions consisted in order of 0.1 M phosphate buffer, pH 6.5 containing 1 mM EDTA, 1 mM GSH, and 1 mM CDNB (Δɛ340 = 9.6 mM−1 cm−1) at 30 °C [Citation24]. For the inhibition studies, compounds were added just before CDNB and the absorbance change was measured for 1 min in 1 ml quvarz cuvette. The reaction system contained 5% ethanol from the CDNB (20 mM stock) and 5% DMSO (2 mM stock), but the solvents had negligible inhibitory effect on the enzyme activity. The enzymatic reaction was obtained by subtracting the non-enzymatic rate from the rate measured in the presence of enzyme.
Data analysis
All activity measurements were made in triplicate of three different concentrations of synthesized compounds. The IC50 value represents the concentration of the inhibitor that gives 50% inhibition of the enzymatic activity. These IC50 values of the compounds were determined by regression analysis using Graphpad Prism 4.0 software (Graphpad Inc., San Diego, CA).
Results and discussion
Designing of the new hGST P1-1 inhibitors
The hGST P1-1 enzyme is composed of two polypeptide subunits. Each subunit has two binding sites (), a glutathione binding site (G site) located within domain-1 and a binding site for electrophilic substrates (H site) including residues from domain-1 and -2 notably Tyr8, Phe9, Pro10, Val11, Arg13, Val35, Ile104, and Tyr108. It is reported that the H site residue Tyr108 plays an important role for the activity of hGST P1-1 enzyme [Citation11,Citation25].
Figure 1. (a) Binding sites of dimeric hGST P1-1 enzyme (pdb ID 2GSS and 6GSS): the protein structure is represented in solid ribbon secondary type. (b) Docking pose of compound 5f and Vh in 6GSS. (c) Docking pose of Vh*: sulphonyl group revealed H bonds with SH and glycine NH of GSH, CF3 group of the compound showed halogen bond with Cys101, compound had interactions with Pro9 (alkyl hydrophobic), Val35 (pi-alkyl), Ile104 (pi-alkyl), and Tyr108 (pi–pi). (d) Docking pose of 5f*: sulphonyl group revealed H bond with Gln51 and Tyr108, compound had interactions with Arg13 (electrostatic), Lys44 (alkyl hydrophobic), Tyr108 (pi–pi), and GSH (pi-sulfur). (e) Docking pose of Vd*: sulphonyl group revealed H bonds with Tyr108 and glycine NH of GSH and CF3 group revealed H bond with Tyr109, compound had interactions with Phe8 (pi–pi), Val10 (pi-alkyl), Val35 (pi-alkyl), and Tyr108 (pi–pi). (f) Docking pose of Vk*: sulphonyl group revealed H bond with Glycine NH of GSH, CF3 group of the compound showed halogen bond with Ile104, compound had interactions with Phe8 (pi–pi), pro9 (pi-alkyl and alkyl hydrophobic), Ile104 (pi-alkyl and alkyl hydrophobic), Tyr108 (pi–pi), and GSH (pi-sigma). *The protein structure is represented in flat ribbon style, GSH and all the ligands are seen in stick representation.
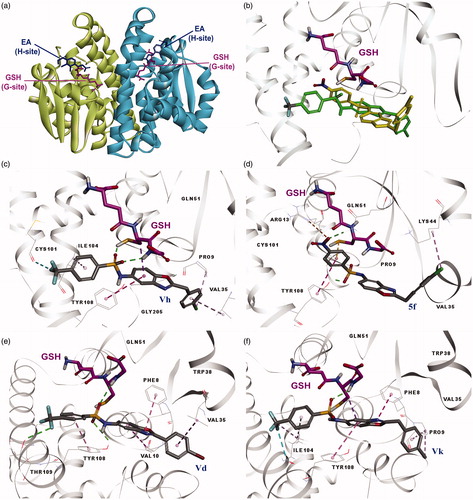
In our earlier study, we reported that one of the synthesized compounds 2-(4-chlorobenzyl)-5-(4-nitrophenylsulphonamido)benzoxazole (5f) showed potency similar to the reference drug EA [Citation16]. Herein, for seeing the substituent effect on hGST P1-1 enzyme, we replaced the NO2 group with the CF3 group, which has H-bond and halogen bond acceptor ability and a significant electronegativity. Then, we used molecular docking method in order to predict the interactions and their possible activities theoretically. The CDocker method was applied for both the known inhibitor EA, previously synthesized compound 5f and the newly designed 2-substituted-5-(4-trifluoromethylphenyl-sulphonamido)benzoxazoles (Va–Vk) under the same conditions. According to these studies, EA revealed H bonds with Arg13 and SH proton of GSH and showed hydrophobic interactions with Phe8 and Tyr108 (). The position of EA was found to be very close to the X-ray structure of EA complex with hGST P1-1 enzyme (pdb ID: 2GSS). The calculated binding energy of EA was −10.65 kcal/mol. All the designed compounds revealed H bonds or pi interactions with Tyr108. Additionally, they showed interactions with GSH and other H site residues as shown in . The results of the molecular docking studies of some compounds were as follows: Vh revealed H bonds with SH and glycine NH of GSH, CF3 group of the compound revealed halogen bond with Cys101, compound had interactions with Pro9 (alkyl hydrophobic), Val35 (pi-alkyl), Ile104 (pi-alkyl), and Tyr108 (pi–pi) (C); previously synthesized compound 5f which include a NO2 group instead of CF3 according to Vh, revealed H bonds with Gln51 and Tyr108, compound had interactions with Arg13 (electrostatic), Lys44 (alkyl hydrophobic), Tyr108 (pi–pi), and GSH (pi-sulphur) (). Superimposition of 5f and Vh is shown in . Vd revealed H bonds with Tyr108 and glycine NH of GSH and CF3 group of the compound showed H bond with Tyr109. Compound Vd had also interactions with Phe8 (pi–pi), Val10 (pi-alkyl), Val35 (pi-alkyl), and Tyr108 (pi–pi) (); Vk revealed H bond with glycine NH of GSH, CF3 group of the compound showed halogen bond with Ile104, compound showed interactions with Phe8 (pi–pi), pro9 (pi-alkyl and alkyl hydrophobic), Ile104 (pi-alkyl and alkyl hydrophobic), Tyr108 (pi–pi), and GSH (pi-sigma) (). Binding energies of all the synthesized compounds were between −6.01 and −21.08 kcal/mol (). As shown in silico studies, the designed compounds demonstrated strong interactions with the H site of hGST P1-1 enzyme. Therefore, it was deemed worthy of synthesizing of 2-(substitutedphenyl/benzyl)-5-(4-trifluoromethyl-phenylsulphonamido)benzoxazole derivatives.
Table 1. Docking results.
Surprisingly, in vitro hGST P1-1 enzyme inhibition studies demonstrated that all the tested compounds except Vj had better inhibitory activity than the reference drug EA and thus also correlated with the docking results.
Chemistry
2-(4-Substitutedphenyl/benzyl)-5-(4-trifluoromethylphenylsulphonamido)benzoxazole derivatives were synthesized as new structures via two steps synthetic pathway. First, 5-amino-2-(4-substitutedphenyl/benzyl)benzoxazole derivatives (IIIa–IIIk) were obtained by heating appropriate acid with 2,4-diaminophenol in polyphosphoric acid (PPA) [Citation20,Citation21,Citation26]. In the second step, 5-amino-2-(4-substitutedphenyl/benzyl)-benzoxazole derivatives (IIIa–IIIk) and 4-trifluoromethylbenzenesulphonyl chloride (IV) were treated in dichloromethane and pyridine to efford 5-(4-trifluoromethylphenylsulphonamido)benzoxazole derivatives (Va–Vk) as given in Scheme 1 [Citation16]. The newly synthesized compounds were characterized by spectral data. 1H NMR, 13C NMR, mass spectra, and elemental analysis results are in agreement with the proposed structures. In the 1H NMR spectra of the compounds, NH protons were observed at 10.52–10.63 ppm, and benzylic CH2 protons were appeared at 4.20–4.28 ppm. Besides, all the aromatic protons were observed at 7.02–8.19 ppm. 13C NMR spectra of these compounds were appropriate to their structures. Mass spectra showed M++H peaks in accordance with their formulas, since the electrospray ionization method was employed. The elemental analyses’ results (C, H, N, S) were within ±0.4% of the calculated amounts.
hGST P1-1 in vitro inhibitory activity
Sulphonamide derivatives of various aliphatic or aromatic compounds have been shown to be H site substrates for GSTs [Citation12]. In this study, newly synthesized 2-substituted-5-(4-trifluoromethylphenylsulphonamido)benzoxazole derivatives (Va–Vk) were examined for their inhibitory effects on hGST P1-1 enzyme. According to enzyme inhibition studies, all the tested compounds except Vj showed better inhibitory activity than the reference drug EA as shown in .
Table 2. The structures of the tested sulfonamidobenzoxazoles and their in vitro hGST P1-1 inhibitory effects.
Conclusion
Widely variable structures are apparently accommodated by the H site of GST. For example, sulphonamide derivatives of various aliphatic or aromatic compounds have been shown to be substrates for GSTs [Citation12]. In this study, we designed and synthesized new 2-substituted-5-(4-trifluoromethylphenylsulphonamido)benzoxazole derivatives. All the synthesized compounds were evaluated for their in vitro inhibitory activities on the hGST P1-1 enzyme. According to docking results, newly synthesized compounds had strong interactions with the H site of the enzyme. All of them showed H bonds or pi interactions with Tyr108, which is an important residue for the enzymatic process. Moreover, they also indicated interactions with GSH and other H site residues with lower binding energies as shown in . Surprisingly, we obtained satisfactory results from in vitro hGST P1-1 enzyme inhibition studies. All the newly synthesized sulphonamidobenzoxazoles except Vj were found to be more potent than the reference drug EA and it was found to be a good correlation between theoretical and experimental studies.
Moreover, inhibitory activity of newly synthesized compound Vh was found to be better than previously synthesized compound 5f, which include a NO2 group instead of CF3 at the para position of the benzene ring, bound to sulphonamide moiety. Herein, we compared the interactions with hGST P1-1 enzyme of these two compounds. According to the docking results, the SO2 group of the compound Vh revealed two H bonds with the GSH and the CF3 group showed a halogen bond with Cys101 (C) differently from 5f. Additionally, as seen in , the binding energies of Vh and 5f were found to be −21.92 and −15.79, respectively. These results showed us that compound Vh (bearing the CF3 group) bound to the hGST P1-1 enzyme with a higher affinity compared with 5f (bearing the NO2 group). Therefore, we can consider that these data make a sense and can explain its higher activity.
In conclusion, the compounds that obtained from this research could be used as scaffolds in design of new potent hGST P1-1 inhibitors useful in overcoming the resistance in cancer chemotherapy.
Acknowledgements
This work was supported by the Swedish Cancer Society and the Swedish Research Council.
Disclosure statement
The authors declare that they have no conflict of interest.
Additional information
Funding
Related Research Data
References
- Josephy PD, Mannervik B. Molecular toxicology. 2nd ed. New York (NY): Oxford University Press; 2006.
- Mathew N, Kalyanasundaram M, Balaraman K. Glutathione S-transferase (GST) inhibitors. Expert Opin Ther Pat. 2006;16:431–444.
- Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88.
- Kazemnejad S, Rasmi Y, Sharifi R, et al. Class-Pi of glutathione S-transferases. Iran J Biotechnol. 2006;4(1):1–16.
- Ban N, Takahashi Y, Takayama T, et al. Transfection of glutathione S-transferase (GST)-pi antisense complementary DNA increases the sensitivity of a colon cancer cell line to adriamycin, cisplatin, melphalan, and etoposide. Cancer Res. 1996;56:3577–3582.
- Morrow CS, Smitherman PK, Diah SK, et al. Coordinated action of glutathione S-transferases (GSTs) and multidrug resistance protein 1 (MRP1) in antineoplastic drug detoxification. Mechanism of GST A1-1- and MRP1-associated resistance to chlorambucil in MCF7 breast carcinoma cells. Biol Chem. 1998;273:20114–20120.
- Ruzza P, Calderan A. Glutathione transferase (GST)-activated prodrugs. Pharmaceutics. 2013;5:220–231.
- Adler V, Yin Z, Fuchs SY, et al. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–1334.
- Asakura T, Sasagawa A, Takeuchi H, et al. Conformational change in the active center region of GST P1-1, due to binding of a synthetic conjugate of DXR with GSH, enhanced JNK-mediated apoptosis. Apoptosis. 2007;12:1269–1280.
- Mulder TP, Peters WH, Wobbes T, et al. Measurement of glutathione S-transferase P1-1 in plasma: pitfalls and significance of screening and follow-up of patients with gastrointestinal carcinoma. Cancer. 1997;80:873–880.
- Oakley AJ, Lo Bello M, Battistoni A, et al. The structures of human glutathione transferase P1-1 in complex with glutathione and various inhibitors at high resolution. J Mol Biol. 1997;274:84–100.
- Koeplinger KA, Zhao Z, Peterson T, et al. Activated sulfonamides are cleaved by glutathione-S-transferases. Drug Metab Dispos. 1999;27:986–991.
- Kodera Y, Akiyama S, Isobe K, et al. Expression of pi-glutathione s-transferase gene (GSTP1) in gastric cancer: lack of correlation with resistance against cis-diamminedichloroplatinum (II). Eur J Cancer. 1994;30A(14):2158–2162.
- Axarli I, Labrou NE, Petrou C, et al. Sulphonamide-based bombesin prodrug analogues for glutathione transferase, useful in targeted cancer chemotherapy. Eur J Med Chem. 2009;44:2009–2016.
- Musdal Y, Ertan-Bolelli T, Bolelli K, et al. Inhibition of human glutathione transferase P1-1 by novel benzazole derivatives. Turk J Biochem. 2012;37:431–436.
- Ertan-Bolelli T, Musdal Y, Bolelli K, et al. Synthesis and biological evaluation of 2-substituted-5-(4-nitrophenylsulfonamido)benzoxazoles as human GST P1-1 inhibitors, and description of the binding site features. ChemMedChem. 2014;9:984–992.
- Oakley AJ, Rossjohn J, Lo Bello M, et al. The three-dimensional structure of the human Pi class glutathione transferase P1-1 in complex with the inhibitor ethacrynic Acid and Its glutathione conjugate. Biochemistry. 1997;36:576–585.
- Dassault Systèmes BIOVIA [Internet]. San Diego (CA): BIOVIA Discovery Studio Modeling Environment; c2002–2017 [cited 2017 Feb 18]. Available from: http://accelrys.com/products/collaborative-science/biovia-discovery-studio/
- Wu G, Robertson DH, Brooks CL, et al. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-A CHARMm-based MD docking algorithm. J Comput Chem. 2003;24:1549–1562.
- Yildiz-Oren I, Tekiner-Gulbas B, Yalcin I, et al. Synthesis and antimicrobial activity of new 2-[p-substituted-benzyl]-5-[substitutedcarbonylamino]-benzoxazoles. Arch Pharm. 2004;337(7):402–410.
- Wynne GM, Wren SP, Johnson PD, et al. Summit Corporation Plc. Treatment of Duchenne muscular dystrophy. Patent no. WO2007091106 A2, 2007 Aug 16.
- Kolm RH, Stenberg G, Widersten M, et al. High-level bacterial expression of human glutathione transferase P1-1 encoded by semisynthetic DNA. Protein Expr Purif. 1995;6:265–271.
- Mannervik B, Guthenberg C. Glutathione transferase (human placenta). Meth Enzymol. 1981;77:231–235.
- Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249:7130–7139.
- Quesada-Soriano I, Parker LJ, Primavera A, et al. Influence of the H-site residue 108 on human glutathione transferase P1-1 ligand binding: structure-thermodynamic relationships and thermal stability. Protein Sci. 2009;18:2454–2470.
- Sener E, Yalcin I, Ozden S, et al. Synthesis and antimicrobial activities of 5-amino-2-(p-substitutedphenyl)benzoxazole derivatives. Turk J Med Pharm. 1987;11(3):391–396.