
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Synthesis of biocompatible and cost-effective novel nonionic surfactants from renewable resources has been the subject of greater scientific interest for enhancing the bioavailability of less water-soluble drugs. The present study focuses on the synthesis of α-tocopherol-based novel biocompatible nonionic surfactant and its evaluation for forming clarithromycin-loaded niosomal drug delivery system. α-tocopherol was hydrophilically modified through multistep reactions and characterized using mass and 1H NMR spectroscopic techniques. Drug-loaded niosomal vesicles were investigated for entrapment efficiency (%EE), size, polydispersity index (PDI), zeta potential (ζ) and morphology using LC-MS, dynamic light scattering (DLS) and atomic force microscopy (AFM). Blood haemolysis, cell culture and acute toxicity tests were performed to investigate its biocompatibility. In vivo oral bioavailability of clarithromycin loaded in niosomal formulation was studied in rabbits. The vesicles were spherical in shape and entrapped up to 75 ± 2.57% of the drug. They exhibited a homogeneous size distribution with a mean diameter of 245 ± 4.66 nm. The surfactant was quite haemocompatible, low cytotoxic and safe in mice. Improved oral bioavailability of clarithromycin was achieved when carried in α-tocopherol-based niosomes. Results obtained showed that the synthesized amphiphile is biocompatible and has excellent capability for formation of niosomal vesicles and enhancing oral bioavailability of less water-soluble drugs like clarithromycin.
Introduction
Oral route has been the preferable route for administration of drugs and other therapeutic substances due to its cost effectiveness, easy administration and enhanced patient compliance. However, the tough acidic environment of stomach and enzymes in liver degrades most of the drugs before they enter the systemic circulation. Furthermore, drug solubility is one of the core parameters required for achieving their desired concentrations in blood for expected pharmacological and therapeutic responses. Macromolecules and drugs with increased polar nature are less absorbed due to their lipophilic nature, higher molecular weights and poor aqueous solubility [Citation1–3]. Drugs are only absorbed and reach systemic circulation when they are in solution form at the site of absorption. Particle size reduction, salt formation, crystal engineering, complexation, drug delivery systems, chemical and physical modifications are some approaches used for enhancing solubility of poor water-soluble drugs [Citation4].
To increase the bioavailability, minimize drug loss/degradation in gastrointestinal tract (GIT) and prevent unwanted side effects of poor water-soluble drugs, different drug delivery systems have been developed or under development [Citation5]. Owing to their multifunctional properties, nanocarriers are widely investigated as delivery vehicles for improving therapeutic effects and reducing side effects of therapeutic substances [Citation6]. Among them, niosomes are promising devices for carrying a large number of poor water-soluble drugs, especially anticancer and antibiotics [Citation7,Citation8]. They are unilamellar or multilamellar vesicular structures composed of surfactant/amphiphilic molecules capable of self-assembling into bilayers with or without cholesterol [Citation9]. They are preferred over other vesicular systems due to their biodegradability, chemical stability, lower toxicity and nonimmunogenicity [Citation10]. Moreover, their cost effectiveness, ease of production and greater chemical stability make them attractive alternative vesicular vectors for drug delivery [Citation11].
Synthetically derivatized nonionic surfactants are attractive amphiphilic molecules capable to lower interfacial and surface tensions and enhance the solubility and bioavailability of poor water-soluble drugs. At present, used nonionic surfactants are considered to be toxic, structurally complex and expensive [Citation12,Citation13]. Their shortcomings have motivated the synthetic and drug delivery scientists to search for surfactants from renewable sources that are biologically safe, inexpensive, environment friendly and easy to be synthesized [Citation14,Citation15]. Literature shows many reports on the use of tocols and their esters as carriers for enhancing the oral absorption of several classes of drug molecules [Citation16]. Most recently, a vitamin E (α-tocopherol) derivative, the α-tocopheryl succinate, was suggested to be promising material with antitumour activity. In addition, it enhanced anticancer efficiency of doxorubicin by increasing its encapsulation and absorption [Citation17]. Being biocompatible and from renewable sources, it is a versatile molecule to be derivatized for nonionic surfactants for niosomal drug delivery purposes.
Clarithromycin, a broad spectrum macrolide antibiotic, belongs to BCS class II drugs and indicated for treatment of respiratory tract, skin and soft tissue infections. It is also used in treatment regimens of H. pylori and peptic ulcer diseases. But it suffers from poor absorption through gastrointestinal tract and undergoes first-pass metabolism [Citation18]. It also causes undesirable side effects like diarrhoea, vomiting, nausea, headache and abdominal pain. The short terminal half-life of clarithromycin needs its frequent dosing while its poor aqueous solubility limits its oral bioavailability to about 55%. Thus, loading of clarithromycin in niosomal vesicles is desirable for obtaining its enhanced oral bioavailability in a controlled release manner [Citation19]. Here, we are reporting a novel nonionic surfactant synthesized from lipophilic α-tocopherol (vitamin E) through its hydrophilic modification following multistep synthetic route. The synthesized novel surfactant was subjected to biocompatibility studies and then used as niosomal vesicular carrier for model hydrophobic drug clarithromycin.
Experimental
Materials
All the organic solvents used in this study were of HPLC grade. α-Tocopherol, propargyl bromide, sodium azide, CuSO4, Na-ascorbate, clarithromycin, Dulbecco's modified Eagle's medium (DMEM), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), poly-L-lysine (PLL), K2CO3 and foetal bovine serum (FBS) were obtained from Sigma-Aldrich (Deisenhofen, Germany) through local supplier. USP reference standard clarithromycin was purchased from LGC Promochem, India. Cholesterol was bought from BDH (Pool, Dorset, U.K.) and Tween 80 from Merck (Darmstadt, Germany). Clarithromycin commercial suspension (Canter) and tablets (Canter) were obtained from local market (Indus Pharma, Karachi, Pakistan).
Synthesis
Propargyl derivative of vitamin E: (compound 1)
In a 100-ml round bottom (RB) flask, 1.2 mmol (500 mg) vitamin E and 1.5 mmol (208 mg) K2CO3 were added in 30 ml dry acetone and stirred for 1 h at 60 °C. Then, 1.5 mmol (0.15 ml) propargyl bromide was added dropwise to this mixture and was further refluxed for 6 h. Reaction was monitored with TLC under UV lamp. Upon completion, the reaction mixture was concentrated under reduced pressure using rotary evaporator (BUCHI, 131 Rotavapor, Flawil, Switzerland), diluted with distilled water (60 ml) and extracted with dichloromethane (3 × 30 ml). Dichloromethane layer was combined, dried with sodium sulphate (Na2SO4) and concentrated in vacuum. The resultant reaction mixture was then passed through silica gel column to obtain compound 1 as a viscous liquid with pure n-hexane.
Yield: 90%, EI-MS m/z 468.4, 1H NMR (400 MHz CDCl3) δ ppm: 4.57 (s, 2H, OCH2), 2.83 (s, 2H, CH, chromane), 2.48 (s, 1H, CH, acetylene), 2.19 (s, 3H, CH3, aromatic), 2.15 (s, 3H, CH3, aromatic), 2.07 (s, 3H, CH3, aromatic), 0.83–0.85 (4d, 4CH3, phytyl)
Ethyl 2-(2-(2-azidoacetamido) thiazol-4-yl) acetate: (compound 2)
Ethyl 2-(2-(2-chloroacetamido) thiazol-4-yl) acetate 1.2 mmol (315 mg) and equimolar NaN3 (80 mg) were dissolved in 30 ml dry acetone in a 100-ml round bottom flask equipped with a reflux condenser and magnetic stirrer. This reaction mixture was stirred for 6 h at 55–60 °C and monitored with TLC. Upon reaction completion, excess acetone was evaporated with rotary evaporator under reduced pressure. The crude solid mixture was then washed with distilled water and extracted with ethyl acetate (3 × 25 ml). The ethyl acetate portions were combined, dried with Na2SO4 and concentrated with rotary evaporator under reduced pressure to obtain compound 2 as off-white solid.
Yield: 90%, EI-MS m/z 269.0, 1H NMR (300 MHz, DMSO) δ ppm: 12.36 (s, 1H, NHCO), 7.02 (s, 1H, CH), 4.10 (s, 2H, NCH2CO), 4.05 (q, 2H, J = 6.9 Hz, CH2–O), 3.68 (s, 2H, CH2CO), 1.17 (t, 3H, CH3, J = 6.9 Hz).
Final product (compound E1CLK)
A 100-ml round bottom flask equipped with a magnetic stirrer and a reflux condenser was added 1.12 mmol of compound 1 (525 mg), 1.12 mmol ethyl 2-(2-(2-azidoacetamido) thiazol-4-yl) acetate (270 mg), Na-ascorbate (20 mol %) and CuSO4 (5 mol %) in 20 ml DMF. Reaction mixture was stirred for 6 h at 70 °C and monitored with TLC. After completion of the reaction, it was quenched through addition of ice-cooled water and extracted with ethyl acetate (3 × 25 ml). The organic phase was combined, dried with Na2SO4 and concentrated with rotary evaporator. A sticky white solid final product (E1CLK) was purified with silica gel column chromatography using EtOAc: n-hexane (1.5:8.5) as solvent system.
Yield: 87%, m.p. 59–61 °C, EI-MS m/z = 737.1, 1H NMR (300 MHz CDCl3) δ ppm: 7.86 (s, 1H, =CH, triazole), 6.83 (s, 1H, =CH, thiazole), 5.35 (s, 2H, CH2, amide), 4.86 (s, 2H, CH2–CO), 2.19 (s, 3H, CH3), 2.14 (s, 3H, CH3), 2.07 (s, 3H, CH3), 0.83–0.85 (2d, 12H, phytyl 4CH3).
Preparation of E1CLK-based clarithromycin niosomes
Thin film hydration method was used for preparation of clarithromycin-loaded E1CLK-based niosomes following the previously reported methods [Citation20]. Briefly, novel E1CLK (30 mg) and cholesterol (15 mg) were dissolved in 20 ml of methanol/chloroform mixture (1: 2 v/v). Then, 10 mg clarithromycin was dissolved separately in 15 ml methanol and added to the previously prepared lipid phase solution and thoroughly mixed. All the organic solvents were then rotary-evaporated under reduced pressure at 45 °C to dryness. The resulting thin film was further dried overnight under negative pressure for removal of organic solvent traces. The lipid film was rehydrated with PBS (10 ml, pH 7.4) at 50 °C for 30 min. Drug-loaded niosomal vesicle size was reduced using ultrasonicator (LABSONIC L, B. Braun Biotech International, Bethlehem, PA) at 25 °C for 4 min.
Characterization
Size, size distribution, zeta potential and morphology
Mean hydrodynamic diameter and polydispersity index (PDI) of empty and clarithromycin-loaded niosomes were investigated through dynamic light scattering technique using Zetasizer (Zetasizer Nano ZS90, Malvern Instruments, Malvern, UK). Briefly, diluted niosomal formulation was transferred to disposable plastic cuvette with care to avoid bubble formation. The cuvette was then placed in the cell holder of the instrument and reading was taken with 90 degree scattering angle at 25 °C. The medium viscosity, refractive index and dielectric constant were kept 1.0, 1.33 and 80.4 mPa s, respectively. To determine zeta potential (ζ), the diluted niosomal formulation was slowly injected to disposable folded capillary cell (DTS1060), caped and placed in the cell holder of the Zetasizer. Reading was taken at 25 °C using the dispersant as water and material as polystyrene latex. Vesicles were also characterized for shape using AFM (Agilent 5500, Chandler, AZ). A drop of niosomal formulation was placed on mica slide, air-dried at ambient temperature and mounted on microscope. The vesicle shapes were visualized and photographed using noncontact mode.
Drug entrapment efficiency (%EE) determination
E1CLK-based niosomal vesicles were investigated for clarithromycin entrapment efficiency using LC-MS validated method. Niosomal suspension containing clarithromycin (1 mg) was taken and centrifuged (Universal 16, Hitachi, Tuttlingen, Germany) for 25 min at 12,000 rpm for separation of free drug. Sedimented pellets entrapping clarithromycin were washed twice with PBS (pH 7.4), disrupted with methanol (10 ml), vertexed (Heidolph Reax Top; Heidolph Instruments, Schwabach, Germany) and again centrifuged (10 min, 12000 rpm). Supernatant (5 μL) was injected into LC-MS run with mobile phase consisting water: acetonitrile in a v/v ratio of 30:70 with 0.1% formic acid. Following formula was used to determine EE%:
In-vitro release study
Clarithromycin-loaded E1CLK-based niosomal formulation containing 2 mg drug was taken in PBS (pH 2.0, 4.0 and 7.4), wrapped into dialysis membrane (12,000 KDa) and placed in a beaker containing PBS (50 ml) of respective pH values. The beaker was placed in shaker with 100 rpm at 37 °C. At different time intervals, 2 ml media was withdrawn. To prevent drug saturation, 2 ml PBS was added to beaker after each withdrawal. Clarithromycin released from formulation in PBS was quantified through double-beam UV spectrophotometer (Shimadzu, UV-240, Hitachi U-3200, Kyoto, Japan). Wave length of 205 nm was selected for the detection of clarithromycin.
Stability against simulated gastric and intestinal fluids
Simulated gastric and intestinal fluids (SGF and SIF) were prepared according to the previously reported method [Citation21]. For preparation of SGF, water (1 L) was added to the mixture of HCl (7 ml, 37 wt %), NaCl (2 g) and pepsin (3.2 mg/mL). The final pH was adjusted to 2.0 with 1.0 M HCl solution. Similarly, SIF was prepared by taking bile salts and pancreatin in 0.05 M PBS (pH 7.4) at 7 and 1 mg/mL concentrations, respectively. Niosomal suspension containing a specific amount of clarithromycin was diluted with SGF or ISF (1; 10, niosomes: SGF or SIF, v/v) and incubated with agitation speed of 100 rpm at 37 °C. Samples were withdrawn at 10-, 20-, 40- and 60-min time intervals and were investigated for their percent of drug retained following the method used for EE% determination.
Biocompatibility studies
Haemocompatibility
Haemocompatibility of E1CLK was checked using fresh human venous blood. Fresh human blood was centrifuged at 700 g for 10 min for separation of red blood cells (RBCs). The isolated RBC pellets were washed with PBS (pH 7.4) three times and re-centrifuged. To prepare RBC suspension, they were added to PBS in 0.5:5 w/v ratios (erythrocytes: PBS). Then, 4 ml of E1CLK samples (62.5–1000 μg/mL) were added to 0.2 ml of RBC suspension and incubated at 37 °C for 4 h. After incubation, the samples were subjected to centrifugation under the same conditions for removal of nonlysed RBCs. To determine the release of haemoglobin, supernatants were decanted and read at 540 nm using UV spectrophotometer. For 100 and 0% haemolysis, RBCs were incubated with distilled water and PBS, respectively. For comparison, Tween 80 was also investigated. The percent haemolysis was calculated using the following equation:
where Abs, Abs0 and Abs100 denote absorbance of test samples, RBC solution in PBS and distilled water, respectively [Citation22].
In-vitro cell culture toxicity
Novel surfactant E1CLK toxicity was investigated against two different cell cultures using MTT method. HeLa and NIH/3T3 cells were first cultured in DMEM containing antibiotics (penicillin and streptomycin, 50 IU/mL each) and FBS (10%) at 37 °C in CO2 (5%) humidified atmosphere. Both HeLa and 3T3 cells were seeded into 96-well plates at 6 × 104 and 8 × 103 cells per well density, respectively, in 200 μL medium. After 24 h, medium was removed and 200 μL fresh medium containing various concentrations of E1CLK (62.5–1000 μg/mL) was added. Cells were cultured to grow for further 24 and 48 h. Then each well was added 20 μL of MTT solution in PBS (5 mg/mL). Unreacted dye was removed after 4-h incubation from the medium. Then, 200 μL per well of DMSO was added for dissolution of purple formazan crystals and its optical density (OD) was measured at 570 nm using microplate reader (ELx808, BioTek, Winooski, VT). Cells incubated with media only were considered as negative control while for positive control and reference standard, Tween 80 and poly-L-lysine (PLL) were used, respectively. Following formula was then used to determine percent cell viability after 24 and 48 h:
Animals used
All the animals were purchased from Dow University of Health Sciences, OJHA Campus, Karachi, Pakistan. Guidelines of “International Center for Chemical and Biological Sciences (ICCBS), Principles of Institutional Animal Care and Use” were followed for all the studies. The study protocol (Protocol No: 2016–0003) was approved by ethical committee for the use of animals in this research.
Acute toxicity
Acute toxicity of E1CLK was carried out in male Swiss albino mice having an average weight of 21 g. The study was conducted in two phases. Before experiments, all animals were overnight fasted and were maintained in a 12-h dark–light cycle. During 1st phase, animals were divided into three different groups (n = 4) and injected E1CLK intraperitoneally (i.p.) at 1000, 500 and 100 mg/kg body weight doses. All the animals were kept under observation up to 24 h. During 2nd phase of study, two groups of 4 animals in each group were made and given E1CLK by i.p. at doses of 1500 and 2000 mg/kg body weight. The animals were kept under observation for next 24 h and numbers of animals died in each group were recorded. Dose causing deaths in 50% mice (LD50) in each group was calculated according to literature [Citation23].
In-vitro bioavailability study
Eighteen male New Zealand albino rabbits, (average weight of 1.8 kg) were selected for in-vivo bioavailability study. Animals were kept at 25 °C and 12-h dark–light cycle with free access to water and food. Prior to conducting studies, animals were deprived of food but have free access to water. All the animals were divided into three groups (n = 6). Animals in group 1 were given clarithromycin loaded in E1CLK niosomal formulation at 15 mg/kg body weight as a single oral dose. Similarly for reference standards, animals of group 2 and 3 were given clarithromycin commercial tablets and suspension at 15 mg/kg body weight single oral dose, respectively. Then, 800 μL blood samples were collected from each animal through marginal ear vein at time intervals of 0, 0.5, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 18 and 24 h in heparinized tubes. Collected blood was centrifuged at 4000 rpm for 5 min to separate plasma and stored at −80 °C till further analysis. The values of maximum plasma drug concentration (Cmax) and time to reach maximum plasma drug concentration (Tmax) were obtained from plasma drug concentration–time curve directly. Different pharmacokinetic parameters like area under the concentration–time curve to the last measured plasma concentration (AUC0–24), mean residence time (MRT), area under the first moment curve (AUMC0–24) and clearance (Cl) were derived from drug plasma concentrations versus respective time intervals by linear trapezoidal rule using noncompartmental model.
Statistical analysis
All the experiments were performed in triplicate. Values were expressed as mean ± SEM. Data were analyzed for statistical significance using two-way ANOVA followed by Bonferroni test. p-Values ≤ .05 were considered statistically significant.
Results and discussion
Synthesis
Synthetic scheme for the amphiphilic carrier is shown in detail in . Final product (E1CLK), compound 1 and compound 2 all were characterized using mass spectrometry and 1H NMR Spectroscopy. Spectral results are shown in and for the final product. The final product has molecular ion peak at m/z 737.1 () and all the corresponding proton peaks are evident in 1H NMR spectra () that is consistent with theoretical molecular weight and proton peak assignments of the final product E1CLK.
Figure 1. Synthesis scheme of hydrophilically modified α-tocopherol product “E1CLK”.
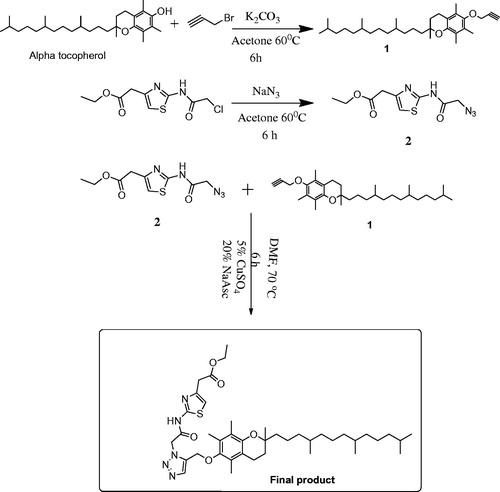
Figure 2. EI-MS mass spectra of synthesized amphiphilic carrier E1CLK.
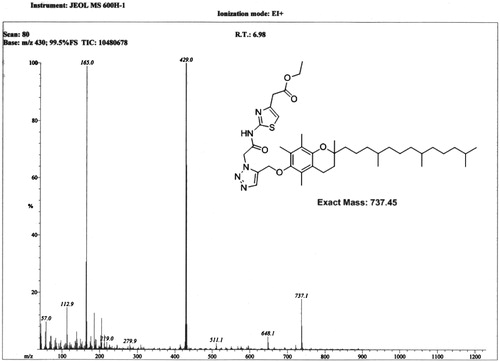
Figure 3. 1H NMR spectra of synthesized amphiphilic carrier (E1CLK).
Size, size distribution, zeta potential and morphology
E1CLK-based empty and clarithromycin-loaded niosomal vesicles were investigated for their mean hydrodynamic diameter using DLS zetasizer. Empty vesicles were of relatively small size as compared to the drug-loaded vesicles. The average size of drug-loaded vesicles was found to be 245.31 ± 4.66 nm as compared to 209.79 ± 5.10 nm of empty vesicles. Polydispersity of clarithromycin-loaded and empty vesicles was found to be 0.28 ± 0.04 and 0.26 ± 0.02, respectively. Zeta potentials of clarithromycin-loaded and empty vesicles were found to be −32 ± 2.22 and −28 ± 1.89 mV, respectively (). AFM images reveal that clarithromycin-loaded vesicles are spherical with smooth surface as shown in . The size of nonionic surfactant-based niosomal vesicles is considered vital as it determines their physical properties and the biological fate of their entrapped drugs. Vesicle size also greatly influences the stability and therapeutic efficacy of niosomal formulations [Citation24]. Small size vesicles have been found less toxic as compared to the larger ones of the same constituents [Citation25]. Similarly, phagocytic uptake of vesicles and drug targeting to solid tumours is affected by their size. Additionally, membrane permeability also depends upon vesicle size [Citation26,Citation27]. Niosomal vesicles in this size range have been reported for enhanced therapeutic efficacy and stability of the formulation [Citation28]. Increased size of the clarithromycin-loaded E1CLK-based vesicles can better be attributed to the increased lipophilicity (due to α-tocopherol moiety presence in E1CLK) of the surfaces of niosomes. This leads to drug interaction with surfactant head groups, resulting in enhanced surfactant bilayers mutual repulsion and thus more drugs get entrapped with a relative large size of the vesicles [Citation29]. Empty vesicles were of relatively small size because of the absence of drug in them and hence there is no interaction between drug and surfactant. The PDI values show that the vesicles population in both empty and drug-loaded formulations was well homogenous. Zeta potential is the estimate of charge on the vesicles surface they gain in the dispersed state. It predicts about the storage stability of the suspension of vesicles. Its measurement also helps in designing dosage form with reduced reticuloendothelial system uptake, thus decreasing the clearance of the drug from body [Citation30]. Here in case of E1CLK-based vesicles, zeta potential lies well in the limits. Size, polydispersity and zeta potential of the vesicles were technically well, indicating the suitability of novel E1CLK surfactant for niosomal drug delivery purposes.
Figure 4. AFM images of clarithromycin-loaded E1CLK-based niosomal vesicles.
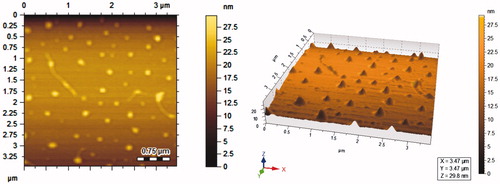
Table 1. Characterization of E1CLK-based niosomal formulation (composition, particle size, PD, EE% and zeta potential).
EE determination
%EE of clarithromycin-loaded E1CLK-based niosomes was determined using LC-MS validated method and was found to be 75.93 ± 2.57% as shown in . This means that 7.6 mg of the total drug (10 mg) used was successfully entrapped in the lipid phase of the formulation. Each 1 mg of the lipid phase (66.66% E1CLK and 33.33% cholesterol) of formulation entrapped 0.17 mg of the drug, indicating increased entrapment efficiency. %EE is one of the important parameters for drug delivery systems. Enhanced oral bioavailability of the drugs in a controlled and sustained manner is a function of increased %EE of the drug delivery system [Citation29,Citation31] . %EE of the niosomes is also greatly affected by HLB value of surfactant used in composition of the formulation. The calculated HLB value of E1CLK (8.3) based on Griffin’s method [Citation32] confirms its effectiveness in increased %EE as reported for other surfactants elsewhere. Increased drug encapsulation of this formulation can also be linked to amount of cholesterol used in the formulation (33.33% of the total organic phase) as it stabilizes the vesicles by preventing the leaking of drugs from vesicles and increasing drug entrapment [Citation33]. Furthermore, as the lipophilicity of the surfactant increases, it causes much drug to be entrapped. Here in case of E1CLK, the lipophilicity gets increased by virtue of α-tocopherol incorporation in the structure, thus resulting in increased %EE of the vesicles. Results justify E1CLK as a versatile surfactant capable of entrapping increased amount of drug
In-vitro release study
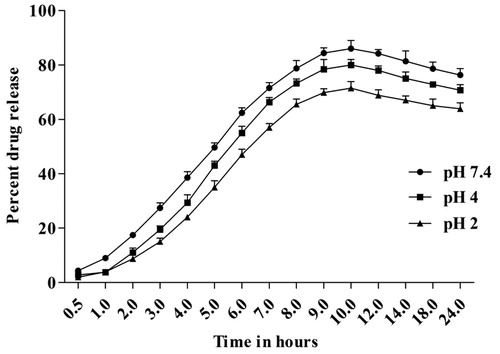
In-vitro release of clarithromycin from E1CLK-based niosomes was studied at three different pH values, i.e. 7.4, 4.0 and 2.0. The drug release from the vesicles was well controlled and programmed but pH dependent. Fifty percent of the drug release was achieved at the 5th h for pH 7.4, while for pH 4.0 and 2.0, it was achieved at the 6th h. Maximum drug release for pH 7.4, 4.0 and 2.0 was 86.12 ± 3.03, 80.00 ± 2.00 and 71.45 ± 2.43, respectively, achieved mostly at the 10th h as shown in . E1CLK-based drug-loaded niosomes showed higher stability at all pH as there was no abrupt release from the formulation at any time interval during in-vitro release study. Interestingly, the vesicles were stable enough even at low pH of 4 and 2 to fulfil the stability criteria of the prepared niosomes.
Figure 5. In-vitro release study of drug-loaded E1CLK niosomal vesicles at pH 7.4, 4.0 and 2.0 (n = 3, mean ± SEM).
Stability against SGF and SIF
Niosomal formulations intended for oral drug delivery are required to be highly stable against SGF and SIF. The novel niosomal formulation proved to be highly stable upon its incubation with SGF and SIF for a time period of 1 h. In case of GSF stability study, the niosomes retained 98.92 ± 0.88, 96.47 ± 2.31, 92.53 ± 0.65 and 89.11 ± 0.48% of the loaded drug after 10-, 20-, 40- and 60-min incubation, respectively. Similarly, they were able to retain 97.56 ± 1.45, 94.70 ± 2.00, 91.39 ± 0.61 and 85.44 ± 1.88% of the loaded drug after 10-, 20-, 40- and 60-min incubation with SIF, respectively. The results confirm that novel drug-loaded niosomal vesicles are capable of withstanding harsh gastric and intestinal environments and do not permit the premature leakage of loaded drug upon exposure to such conditions.
Haemocompatibility
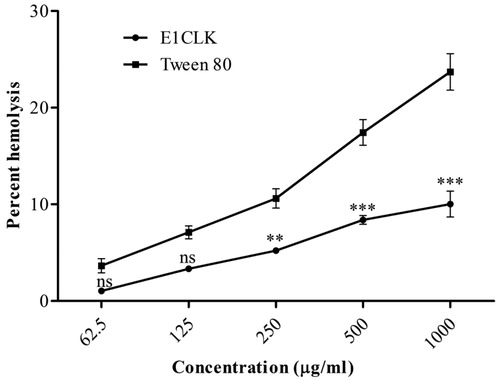
In haemocompatibility screening, our novel α-tocopherol-based surfactant (E1CLK) showed very placid haemolytic effect, exhibiting 1.04 ± 0.06% and 10.03 ± 1.33% haemolysis in RBCs for 62.5 and 1000 μg/mL concentrations, respectively, as shown in . In comparison, Tween 80 caused increased release of haemoglobin from RBCs. It caused 3.65 ± 0.73% and 23.71 ± 1.89% haemolysis in RBCs for 62.5 and 1000 μg/mL concentrations, respectively. Solubilization of lipophilic drugs has been a challenging task for drug delivery scientists. Surfactants are frequently utilized for this purpose, but many amphiphilic molecules have been reported for the induction of haemolysis when they are in contact with RBCs [Citation34]. Haemolysis occurs when hydrophobic portion of amphiphilic surfactant molecules begins to anchor into the cell membrane, resulting in bilayer-to-micelle transition as a function of the surfactant/lipid molar ratio [Citation35]. The haemolytic effect of our novel E1CLK was much lower from that of Tween 80 surfactant used as reference standard and falls well within the allowed limits for haemocompatible biomaterials [Citation36], indicating E1CLK nonhaemolytic in nature and as safe drug carrier.
Figure 6. In-vitro haemolysis study of synthesized amphiphilic carrier (E1CLK) at different concentrations (n = 3, mean ± SEM).
In-vitro cell culture toxicity
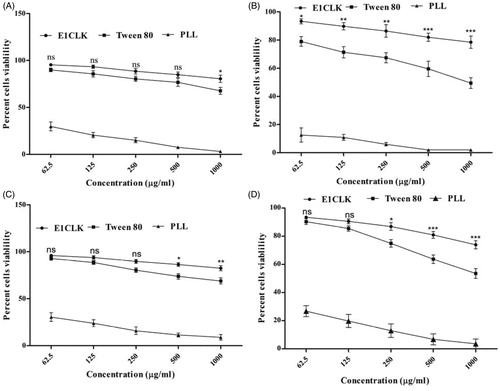
The novel surfactant was screened for cell cytotoxicity using two different cell lines and the results are depicted in . Our novel surfactant exhibited minimal cytotoxicity even after prolonged exposure of cell cultures to its highest concentrations. When incubated with NIH/3T3 cells, the percent cell viability was found to be as high as 80.55 ± 3.80% and 78.49 ± 4.40% at 1000 μg/mL concentration after 24 and 48 h, respectively, as compared to 67.70 ± 3.65% and 49.44 ± 3.81%, respectively, using Tween 80. Similarly, increased percent cell viability (82.44 ± 2.67% and 73.91 ± 2.88) was noted when HeLa cells were exposed to E1CLK as compared to that of Tween 80 (68.88 ± 3.23% and 53.55 ± 3.50%) at 1000 μg/mL concentration after 24 and 48 h, respectively. E1CLK showed decreased cytotoxic effects on both cell cultures. This decrease in the cytotoxicity of the novel surfactant may be linked to its nonionic nature as previously reported by numerous studies [Citation15]. Another possible attribute for the minimal cytotoxicity of novel E1CLK may be its long lipophilic chain (α-tocopherol moiety in its backbone) as short lipophilic chains have been reported for strong disruptive interactions with cell membrane [Citation37].
Figure 7. Cell cytotoxicity study of synthesized surfactant (E1CLK), where (A) and (B) show percent cell viability against NIH/3T3 cell line after 24 and 48 h, respectively, while (C) and (D) show percent cell viability against HeLa cell line after 24 and 48 h, respectively. (n = 3, mean ± SEM). Poly-L-lysine and Tween 80 are used as reference standard and positive control, respectively.
Acute toxicity
Prior to conducting in-vivo pharmacokinetic study, biomaterials are subjected to toxicity studies in animals to ascertain the safe dose range [Citation38]. When screened for acute toxicity study, E1CLK did not reveal any lethality nor did it show any apparent abnormalities in the experimental animals in both phases of the study. E1CLK was found to be safe up to 2 g per kg body weight for 24 h of the study period (). In this case of toxicity study in animals, our novel surfactant was found quite safe.
Table 2. In-vivo acute toxicity of E1CLK in mice.
In-vivo pharmacokinetic study
Following oral administration of clarithromycin encapsulated in E1CLK niosomes or in commercial suspension and tablets, drug plasma concentrations against respective times are represented in , while its derived pharmacokinetic parameters are documented in . When administered in E1CLK niosomes, the maximum drug plasma concentration (Cmax) was achieved (5.21 ± 0.40 μg/mL) after 3 h of administration. Similarly, Cmax achieved for commercial suspension and tablets was 3.10 ± 0.53 and 3.93 ± 0.51 μg/mL, respectively, after 2 h of their administration. Interestingly, E1CLK-based niosomes showed increased MRT (9.56 h) and decreased Cl (0.28 L/h.kg) values of the drug in comparison with 0.46 and 0.58 L/h.kg clearance shown by commercial tablets and suspension, respectively. Data indicate that higher Cmax of the drug was achieved when administered in E1CLK-based niosomes as compared to other commercial formulations. The mean diameter of the drug carrier vesicles in nanorange has been attributed for the enhanced absorption of the drug through GIT epithelium and hence increased oral bioavailability [Citation29]. Moreover, when the drug was given in E1CLK-based vesicles, it remained in the body for extended period as indicated from its increased MRT value. This depicts the slow release of the drug from niosomal vesicles. This slow release can also be judged from the plasma drug concentration maintained over extended period of the time. This may be due to the presence of cholesterol in niosomal formulation. Cholesterol decreases the bilayer fluidity of vesicles by lowering the movement of lipophilic chain and enhancing the packing of lipid head groups. Thus, this decreases the encapsulated drug release from drug-loaded vesicles [Citation24].
Figure 8. Plasma drug concentration of clarithromycin when given orally as E1CLK niosomal formulation, commercial suspension and tablets at 15 mg/kg body weight dose (n = 6, mean ± SE).
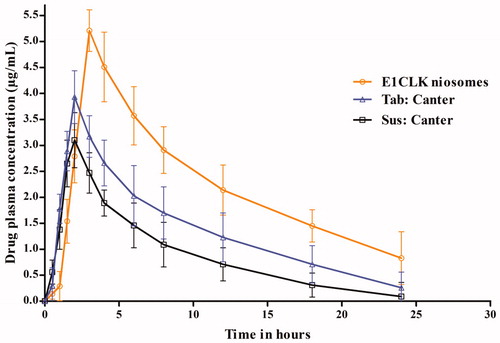
Table 3. In-vivo oral pharmacokinetics of clarithromycin when administered as E1CLK-based niosomal formulation, commercial suspension and tablets.
Conclusion
Owing to the limitations and safety concerns of presently used nonionic surfactants, synthesis of novel biocompatible and efficient surfactants from renewable sources has got much scientific attention. In continuation of our previous work on the synthesis of nonionic amphiphilic compounds for niosomal preparations, herein we synthesized a novel nonionic surfactant by the hydrophilic modification of α-tocopherol (vitamin E) through multistep synthetic route. The surfactant demonstrated to be nontoxic and was capable of forming niosomal vesicles of nanorange size with excellent morphology. Novel niosomal vesicles showed elevated oral bioavailability of clarithromycin with a slow release pattern when entrapped as model drug. Results of the study suggest that this novel surfactant can be used as a promising biomaterial for delivery of less soluble drugs in order to achieve their increased bioavailability.
Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors. However, Mr. Shafiullah thanks Higher Education Commission (HEC), Pakistan, for providing PhD scholarship (PIN. NO 213–61444-2BM2–050).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Amidon GL, Lennernäs H, Shah VP, et al. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420.
- Avdeef A. Absorption and drug development: solubility, permeability, and charge state. Hoboken (NJ): John Wiley & Sons; 2012.
- Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25:103–128.
- Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012;2012:195727.
- Safari J, Zarnegar Z. Advanced drug delivery systems: nanotechnology of health design. A review. J Saudi Chem Soc. 2014;18:85–99.
- Ye L, Zhang Y, Yang B, et al. Zwitterionic-modified starch-based stealth micelles for prolonging circulation time and reducing macrophage response. ACS Appl Mater Interfaces. 2016;8:4385–4398.
- Rajera R, Nagpal K, Singh SK, et al. Niosomes: a controlled and novel drug delivery system. Biol Pharm Bull. 2011;34:945–953.
- Singh D, Pradhan M, Nag M, et al. Vesicular system: versatile carrier for transdermal delivery of bioactives. Artif Cells Nanomed Biotecnol. 2015;43:282–290.
- Escudero I, Geanta RM, Ruiz MO, et al. Formulation and characterization of Tween 80/cholesterol niosomes modified with tri-n-octylmethylammonium chloride (TOMAC) for carboxylic acids entrapment. Colloids Surf A. 2014;461:167–177.
- Tavano L, Muzzalupo R, Picci N, et al. Co-encapsulation of lipophilic antioxidants into niosomal carriers: percutaneous permeation studies for cosmeceutical applications. Colloids Surf B Biointerfaces. 2014;114:144–149.
- Uchegbu IF, Vyas SP. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int J Pharm. 1998;172:33–70.
- Albert J, Verweij J, Loos WJ, et al. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin Pharmacokinet. 2003;42:665–685.
- Lim SB, Banerjee A, Önyüksel H. Improvement of drug safety by the use of lipid-based nanocarriers. J Control Release. 2012;163:34–45.
- Santos DKF, Rufino RD, Luna JM, et al. Biosurfactants: multifunctional biomolecules of the 21st century. IJMS. 2016;17:401.
- Ullah S, Shah MR, Shoaib M, et al. Development of a biocompatible creatinine-based niosomal delivery system for enhanced oral bioavailability of clarithromycin. Drug Deliv. 2016;23:3480–3491.
- Constantinides PP, Han J, Davis SS. Advances in the use of tocols as drug delivery vehicles. Pharm Res. 2006;23:243–255.
- Oliveira MS, Mussi SV, Gomes DA, et al. α-Tocopherol succinate improves encapsulation and anticancer activity of doxorubicin loaded in solid lipid nanoparticles. Colloids Surf B Biointerfaces. 2016;140:246–253.
- Morakul B, Suksiriworapong J, Leanpolchareanchai J, et al. Precipitation-lyophilization-homogenization (PLH) for preparation of clarithromycin nanocrystals: influencing factors on physicochemical properties and stability. Int J Pharm. 2013;457:187–196.
- Shilakari Asthana G, Sharma PK, Asthana A. In vitro and in vivo evaluation of niosomal formulation for controlled delivery of clarithromycin. Scientifica. 2016;2016:6492953.
- Gupta M, Vaidya B, Mishra N, et al. Effect of surfactants on the characteristics of fluconazole niosomes for enhanced cutaneous delivery. Artif Cells Blood Substitutes Biotechnol. 2011;39:376–384.
- Liang R, Chen L, Yokoyama W, et al. Niosomes consisting of Tween-60 and cholesterol improve the chemical stability and antioxidant activity of (-)-epigallocatechin gallate under intestinal tract conditions. J Agric Food Chem. 2016;64:9180–9188.
- Cenni E, Granchi D, Avnet S, et al. Biocompatibility of poly(D,L-lactide-co-glycolide) nanoparticles conjugated with alendronate . Biomaterials. 2008;29:1400–1411.
- Akuodor G, Usman MI, Ibrahim J, et al. Anti-nociceptive, anti-inflammatory and antipyretic effects of the methanolic extract of Bombax buonopozense leaves in rats and mice. Afr J Biotechnol. 2011;10:3191–3196.
- Essa E. Effect of formulation and processing variables on the particle size of sorbitan monopalmitate niosomes. Asian J Pharm. 2010;4:227.
- Szoka F, Milholland D, Barza M. Effect of lipid composition and liposome size on toxicity and in vitro fungicidal activity of liposome-intercalated amphotericin B. Antimicrob Agents Chemother. 1987;31:421–429.
- Nagayasu A, Shimooka T, Kinouchi Y, et al. Effects of fluidity and vesicle size on antitumor activity and myelosuppressive activity of liposomes loaded with daunorubicin. Biol Pharm Bull. 1994;17:935–939.
- Shi B, Fang C, Pei Y. Stealth PEG-PHDCA niosomes: effects of chain length of PEG and particle size on niosomes surface properties, in vitro drug release, phagocytic uptake, in vivo pharmacokinetics and antitumor activity. J Pharm Sci. 2006;95:1873–1887.
- Kaur P, Garg T, Rath G, et al. Development, optimization and evaluation of surfactant-based pulmonary nanolipid carrier system of paclitaxel for the management of drug resistance lung cancer using Box-Behnken design. Drug Deliv. 2014;23:1912–1925.
- Imran M, Shah MR, Ullah F, et al. Glycoside-based niosomal nanocarrier for enhanced in-vivo performance of cefixime. Int J Pharm. 2016;505:122–132.
- Müller RH. Colloidal carriers for controlled drug delivery and targeting: modification, characterization and in vivo distribution. Boca Raton (FL): Taylor & Francis, CRC Press; 1991.
- Arzani G, Haeri A, Daeihamed M, et al. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int J Nanomed. 2015;10:4797.
- GriYn W. Calculation of HLB values of non-ionic surfactants. J Soc Cosmet Chem. 1954;5:249–355.
- Ag Seleci D, Seleci M, Walter J-G, et al. Niosomes as nanoparticular drug carriers: fundamentals and recent applications. J Nanomater. 2016;2016:7372306.
- Tao L, Uhrich KE. Novel amphiphilic macromolecules and their in vitro characterization as stabilized micellar drug delivery systems. J Colloid Interface Sci. 2006;298:102–110.
- Partearroyo MA, Urbaneja M, Goñi FM. Effective detergent/lipid ratios in the solubilization of phosphatidylcholine vesicles by Triton X-100. FEBS Lett. 1992;302:138–140.
- Amin K, Dannenfelser RM. In vitro hemolysis: guidance for the pharmaceutical scientist. J Pharm Sci. 2006;95:1173–1176.
- Ménard N, Tsapis N, Poirier C, et al. Drug solubilization and in vitro toxicity evaluation of lipoamino acid surfactants. Int J Pharm. 2012;423:312–320.
- Kong M, Park H, Feng C, et al. Construction of hyaluronic acid noisome as functional transdermal nanocarrier for tumor therapy. Carbohydr Polym. 2013;94:634–641.